Aberrant DOCK2, GRASP, HIF3A and PKFP Hypermethylation has Potential as a Prognostic Biomarker for Prostate Cancer

 ,
,
Abstract
:1. Introduction
2. Results
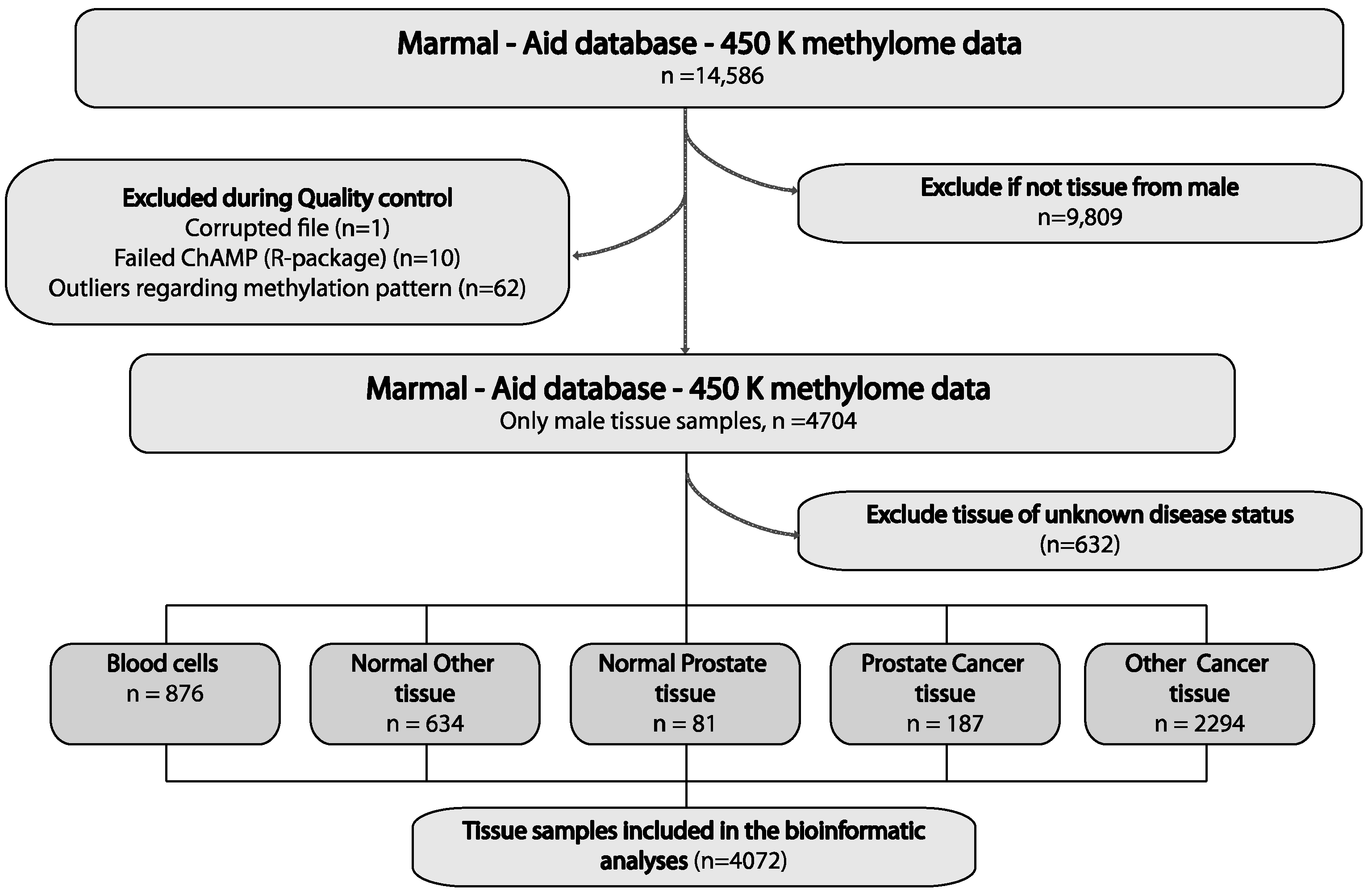
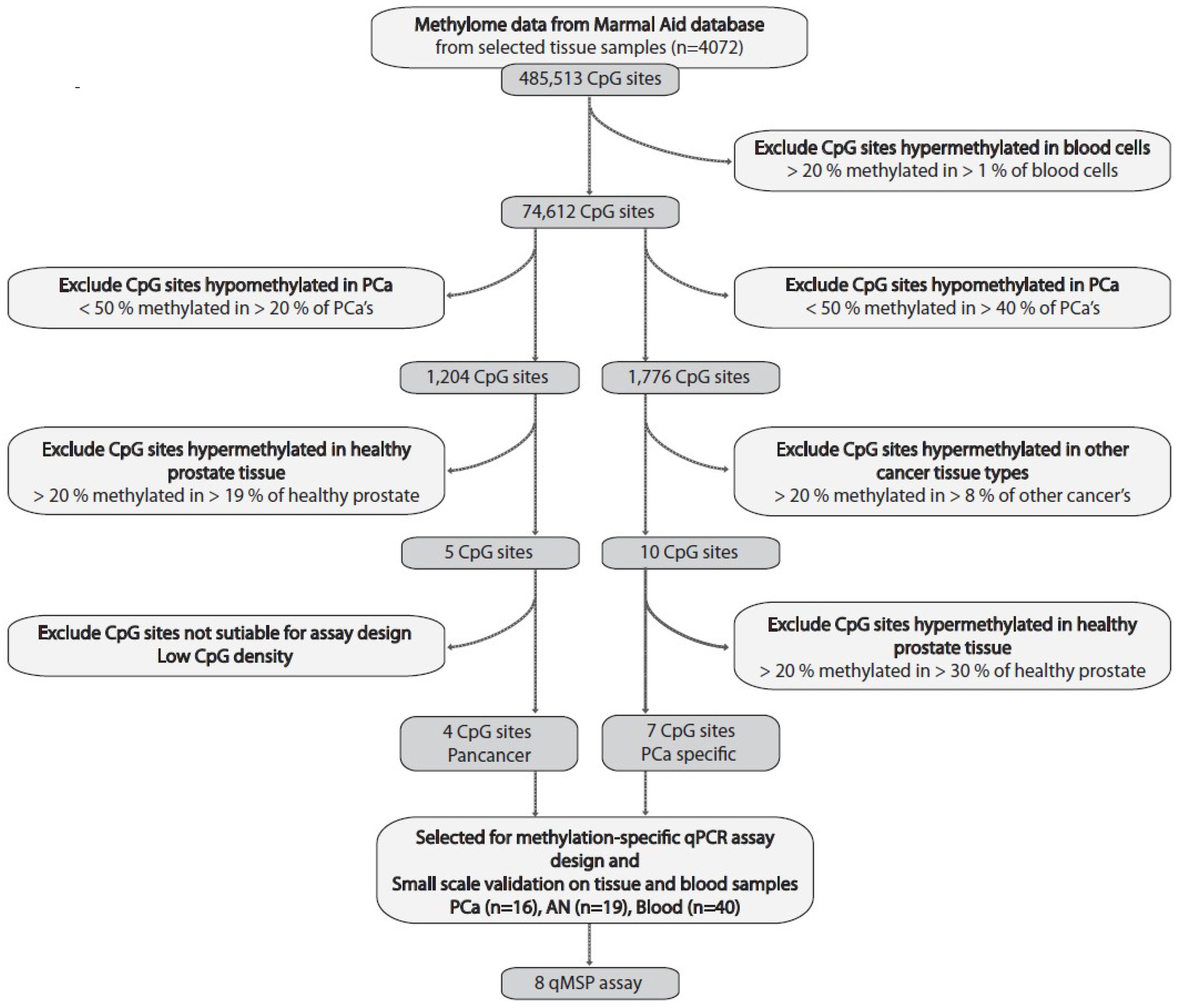
2.1. Large-Scale Bioinformatics Analysis for the Discovery of PCa-Specific Candidate DNA Methylation Markers
2.2. Independent Experimental Validation of Top Candidate Hypermethylation Markers
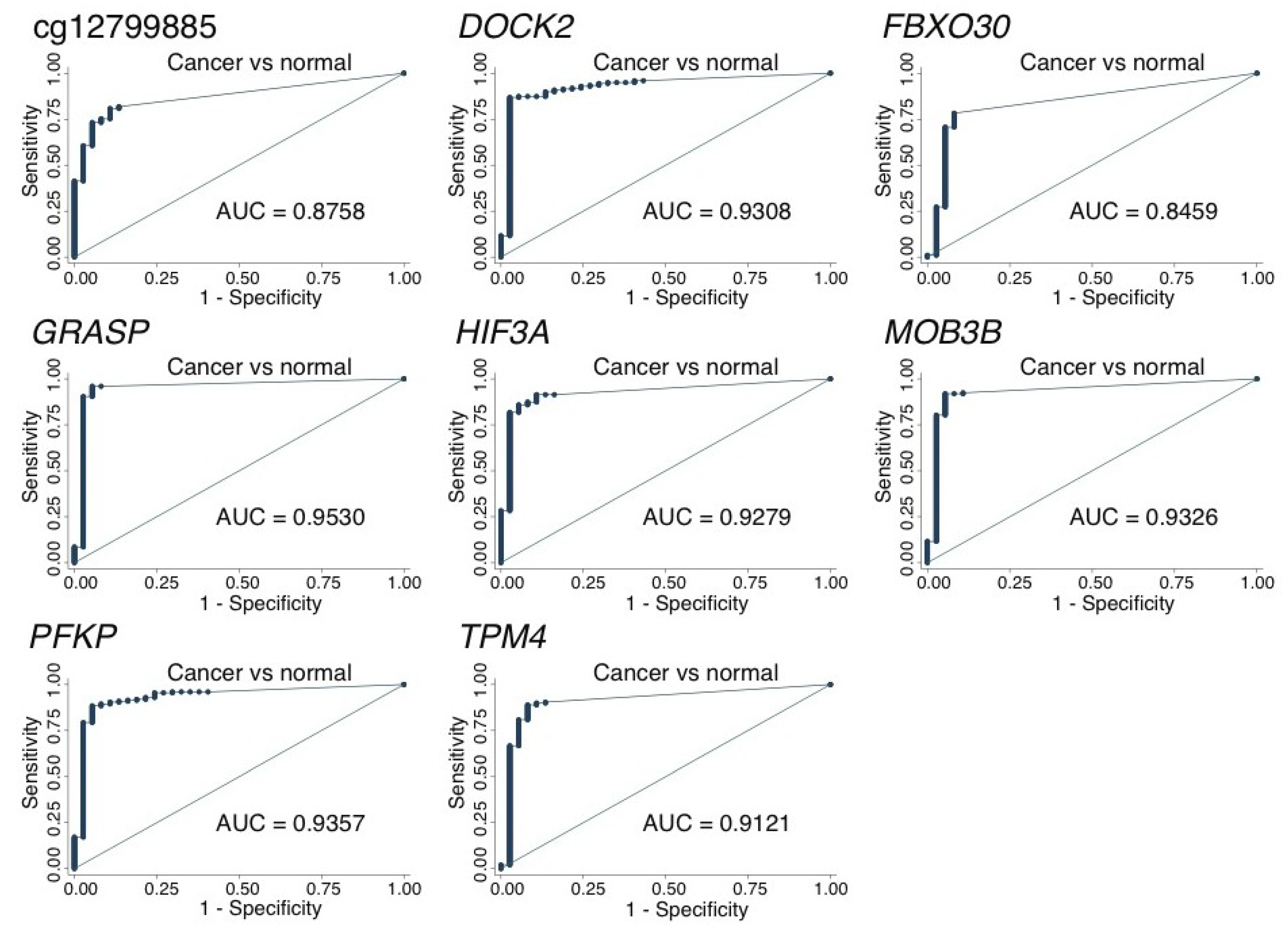
2.3. Large-Scale Validation of Diagnostic Potential
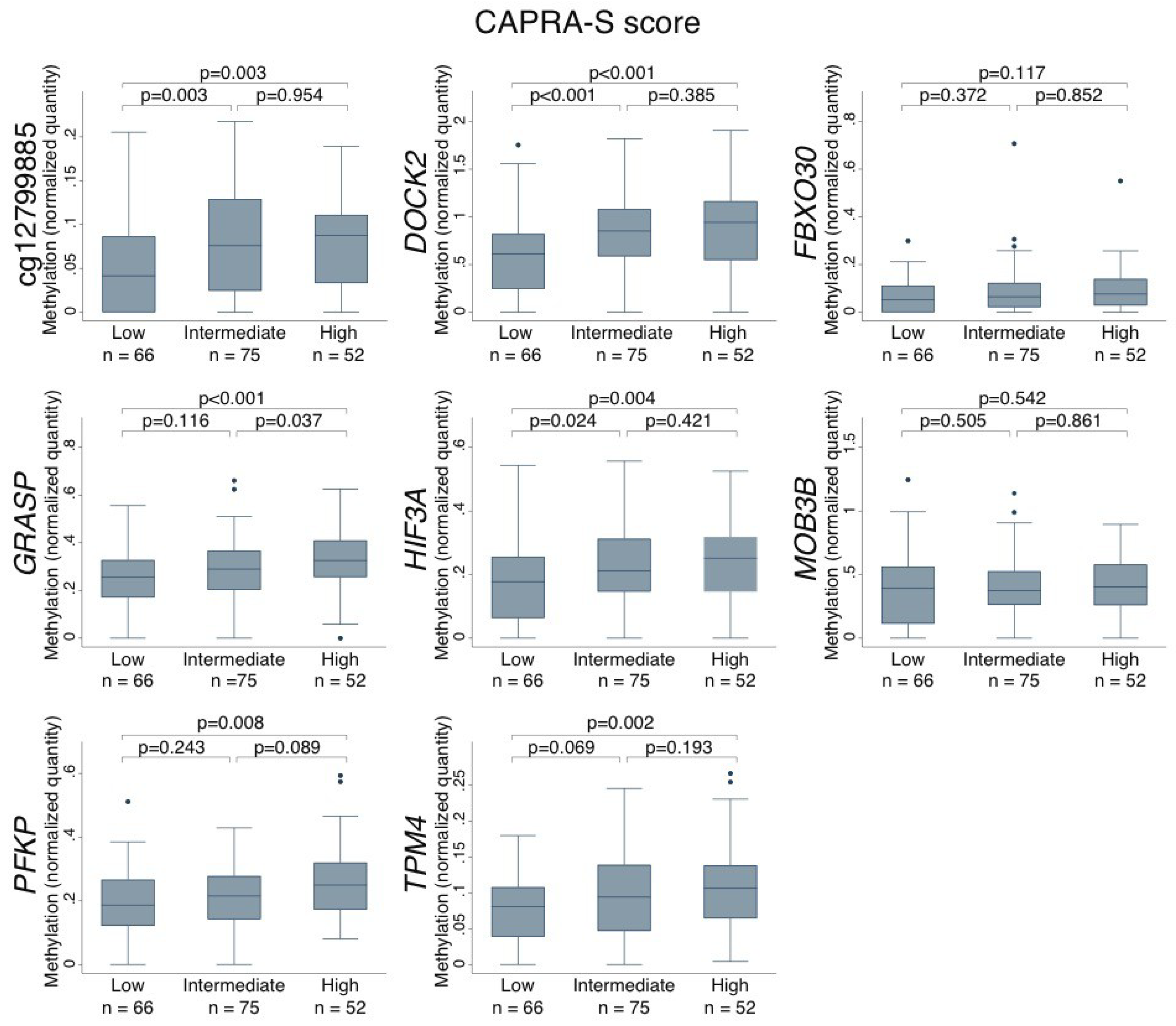
2.4. Association Between DNA Methylation Levels in PCa Tumors and Clinicopathological Variables
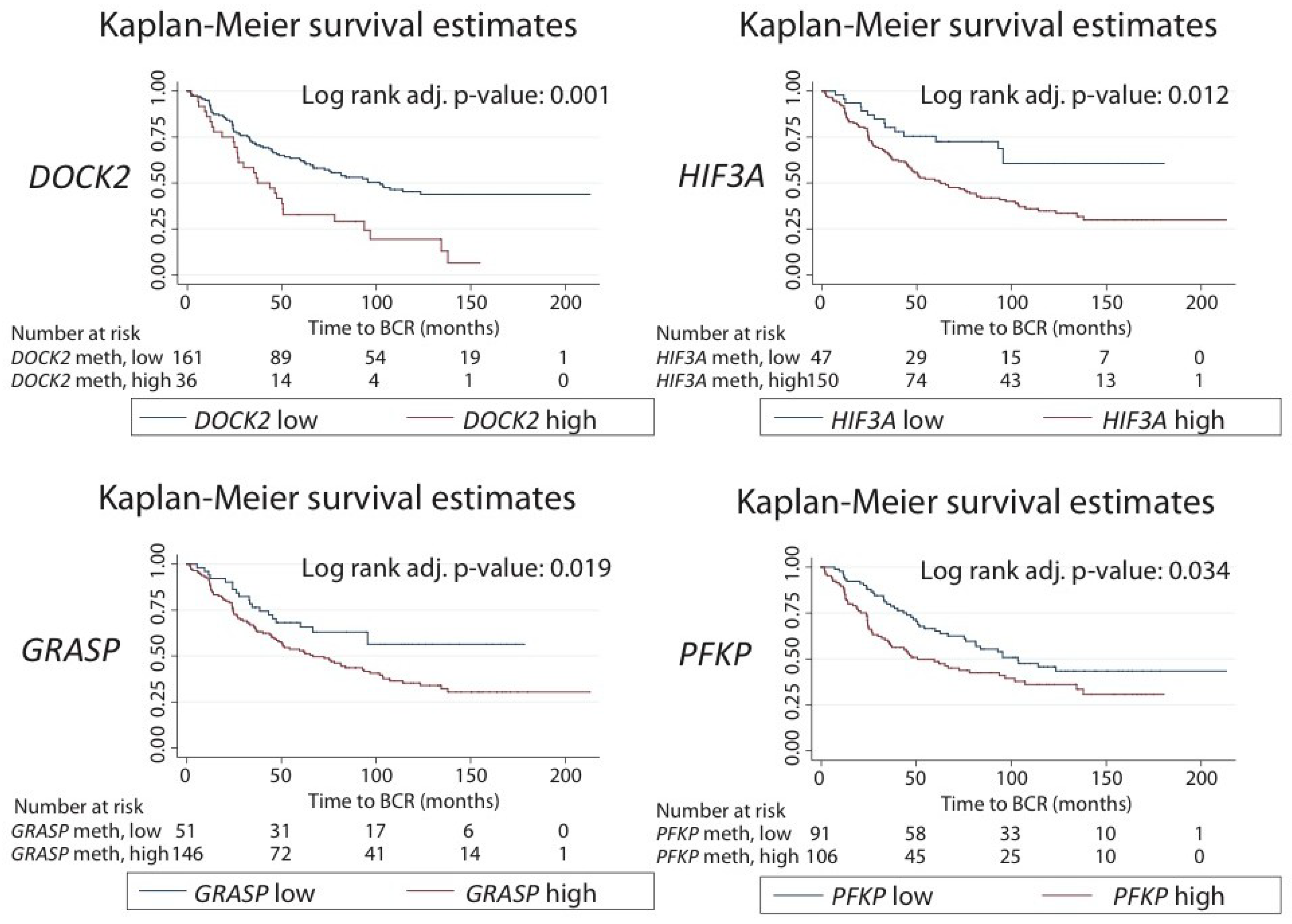
2.5. Prognostic Potential in Relation to Post-Operative Biochemical Recurrence Risk
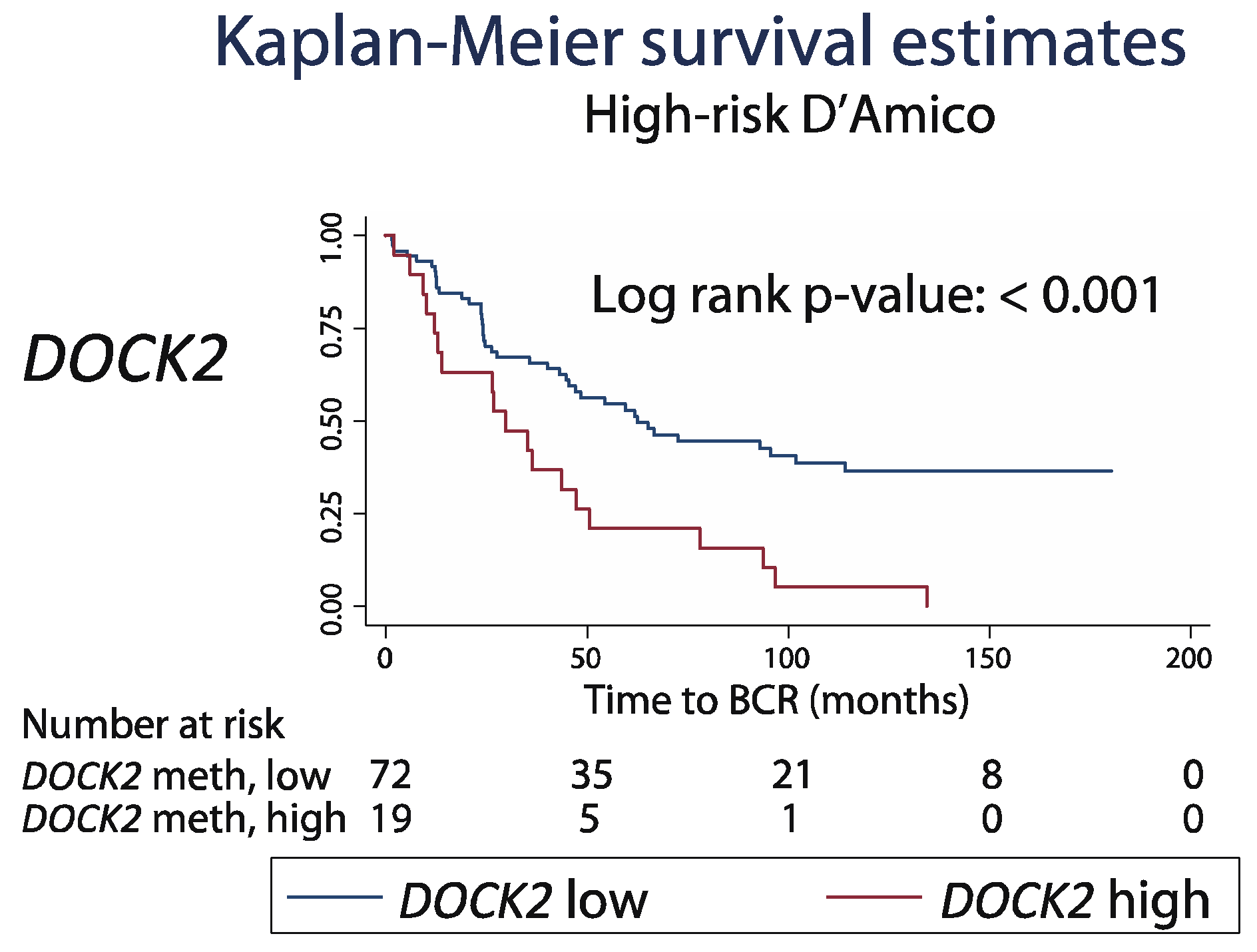
2.6. Prognostic Potential of DOCK2 Hypermethylation in the Pre-Operative Setting
3. Discussion
3.1. Major Findings
3.2. Diagnostic and Prognostic Potential of Eight Top Candidate Hypermethylation Markers
3.3. Known Molecular Functions and Relations to Cancer.
3.4. Study Limitations
4. Materials and Methods
4.1. Biomarker Discovery
4.2. Patients and Tissue Samples
4.2.1. Patient Samples for Small-Scale Experimental Validation
4.2.2. Clinical Cohort for Large-Scale Validation
4.3. DNA Extraction, Bisulfite Conversion and Quantitative Methylation-Specific PCR Analysis (qMSP)
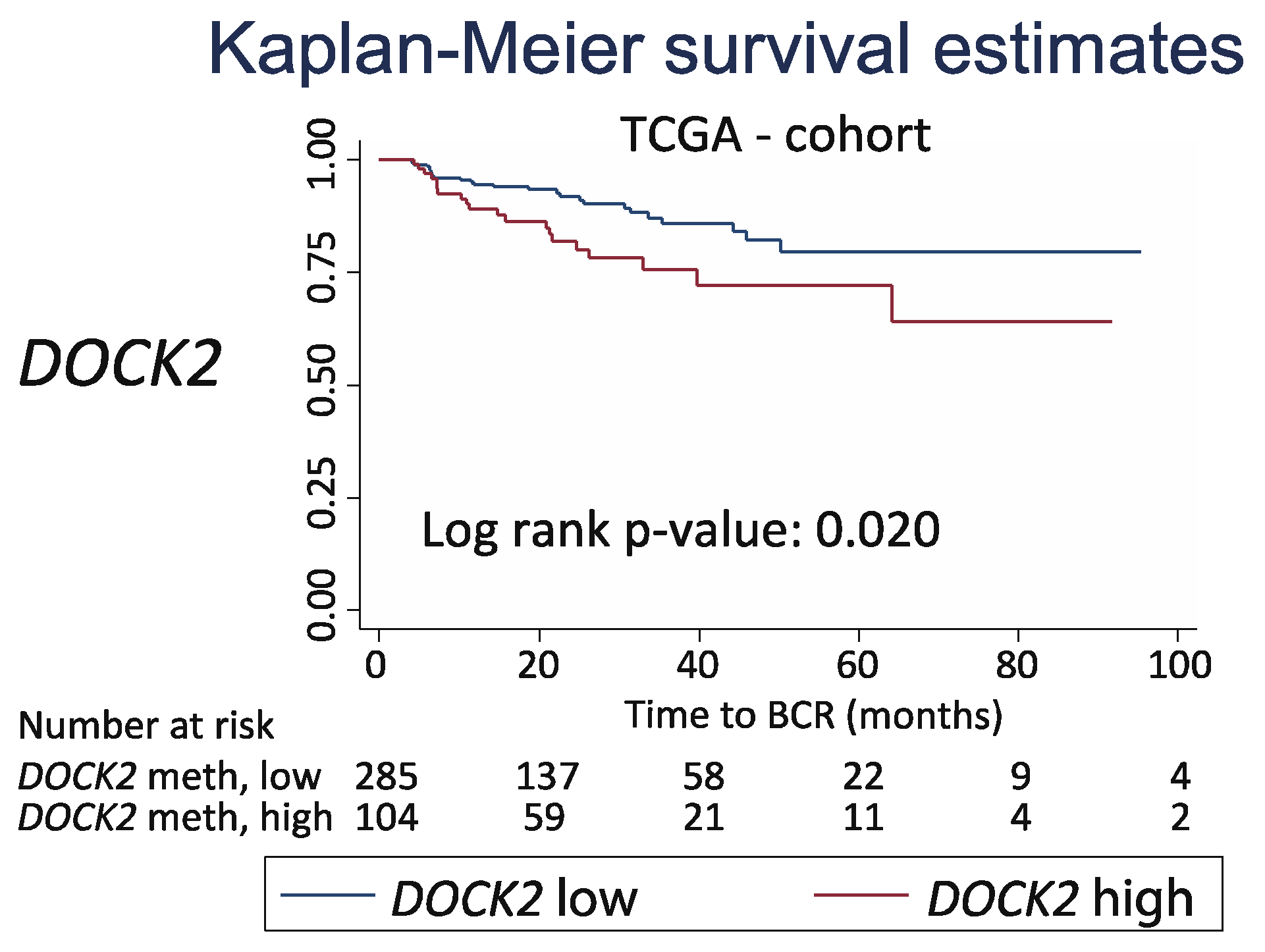
4.4. TCGA Data Used for External Validation of DOCK2 Prognostic Potential
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 450K | Illumina 450K DNA methylation array |
| ADT | Androgen deprivation therapy |
| AN | Adjacent normal |
| AUC | Area under the curve |
| BCR | Biochemical recurrence |
| BPH | Benign prostatic hyperplasia |
| cfDNA | Cell-free DNA |
| CI | Confidence interval |
| CT | Cycle threshold |
| cT | Clinical T-stage |
| ctDNA | Circulating tumor DNA |
| DRE | Digital rectal examination |
| FFPE | Formalin-fixed paraffin embedded |
| HR | Hazard ratio |
| GS | Gleason Score |
| n | Number |
| N | N-stage |
| PBC | Peripheral blood cells |
| PCa | Prostate cancer |
| PSA | Prostate specific antigen |
| pT | Pathological T-stage |
| qMSP | Quantitative methylation specific PCR |
| qPCR | Quantitative PCR |
| RNAseq | RNA sequencing |
| ROC | Receiver operating characteristics |
| RP | Radical prostatectomy |
| RT | Radiotherapy |
| SM | Surgical margin |
| T-stage | Tumor stage |
| TCGA | The Cancer Genome Atlas |
| TRUS | Trans-rectal ultrasound |
| TURP | Transurethral resection of the prostate |
| WGA | Whole-genome amplified DNA |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.M.; Joniau, S.; et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part 1: Screening, diagnosis, and local treatment with curative intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.J. Clinical practice. Prostate-specific-antigen testing for early diagnosis of prostate cancer. N. Engl. J. Med. 2001, 344, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Bangma, C.H.; Venderbos, L.D.F.; Roobol, M.J. Individual and Population-Based screening. In Management of Prostate Cancer a Multidisciplinary Approach, 2nd ed.; Bolla, M., Van Poppel, H., Eds.; Springer: Basel, Switzerland, 2017; pp. 43–55. [Google Scholar]
- Pettersson, A.; Gerke, T.; Fall, K.; Pawitan, Y.; Holmberg, L.; Giovannucci, E.L.; Kantoff, P.W.; Adami, H.O.; Rider, J.R.; Mucci, L.A.; et al. The ABC model of prostate cancer: A conceptual framework for the design and interpretation of prognostic studies. Cancer 2017, 123, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, F.B.; Brasso, K.; Klotz, L.H.; Roder, M.A.; Berg, K.D.; Iversen, P. Active surveillance for clinically localized prostate cancer–A systematic review. J. Surg. Oncol. 2014, 109, 830–835. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Ploussard, G.; Isbarn, H.; Suardi, N.; De Visschere, P.J.; Futterer, J.J.; Ghadjar, P.; Massard, C.; Ost, P.; Sooriakumaran, P.; et al. What is the optimal definition of misclassification in patients with very low-risk prostate cancer eligible for active surveillance? Results from a multi-institutional series. Urol. Oncol. 2015, 33, 164.e1–164.e9. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Broering, J.M.; Carroll, P.R. Time trends and local variation in primary treatment of localized prostate cancer. J. Clin. Oncol. 2010, 28, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Switnicki, M.; Moller, M.; Haldrup, C.; Storebjerg, T.M.; Hedegaard, J.; Nordentoft, I.; Hoyer, S.; Borre, M.; Pedersen, J.S.; et al. RHCG and TCAF1 promoter hypermethylation predicts biochemical recurrence in prostate cancer patients treated by radical prostatectomy. Oncotarget 2017, 8, 5774–5788. [Google Scholar] [CrossRef] [PubMed]
- Haldrup, C.; Mundbjerg, K.; Vestergaard, E.M.; Lamy, P.; Wild, P.; Schulz, W.A.; Arsov, C.; Visakorpi, T.; Borre, M.; Hoyer, S.; et al. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J. Clin. Oncol 2013, 31, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, H.; Haldrup, C.; Strand, S.; Mundbjerg, K.; Mortensen, M.M.; Thorsen, K.; Ostenfeld, M.S.; Wild, P.J.; Arsov, C.; Goering, W.; et al. Hypermethylation of the GABRE~miR-452~miR-224 promoter in prostate cancer predicts biochemical recurrence after radical prostatectomy. Clin. Cancer Res. 2014, 20, 2169–2181. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Orntoft, T.F.; Sorensen, K.D. Prognostic DNA methylation markers for prostate cancer. Int J. Mol. Sci. 2014, 15, 16544–16576. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.O. DNA methylation changes in prostate cancer: Current developments and future clinical implementation. Expert Rev. Mol. Diagn 2009, 9, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Rakyan, V.K. Marmal-aid--a database for Infinium HumanMethylation450. BMC Bioinformatics 2013, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Hilton, J.F.; Carroll, P.R. The CAPRA-S score: A straightforward tool for improved prediction of outcomes after radical prostatectomy. Cancer 2011, 117, 5039–5046. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Klee, E.W.; Young, C.Y.; Sun, Z.; Jimenez, R.E.; Klee, G.G.; Tindall, D.J.; Donkena, K.V. Global methylation profiling for risk prediction of prostate cancer. Clin. Cancer Res. 2012, 18, 2882–2895. [Google Scholar] [CrossRef] [PubMed]
- Vasiljevic, N.; Ahmad, A.S.; Carter, P.D.; Fisher, G.; Berney, D.M.; Foster, C.S.; Cuzick, J.; Lorincz, A.T. DNA methylation of PITX2 predicts poor survival in men with prostate cancer. Biomark. Med. 2014, 8, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Raymond, E.; O’Callaghan, M.E.; Vincent, A.D.; Beckmann, K.R.; Roder, D.; Evans, S.; McNeil, J.; Millar, J.; Zalcberg, J.; et al. Optimum Tools for Predicting Clinical Outcomes in Prostate Cancer Patients Undergoing Radical Prostatectomy: A Systematic Review of Prognostic Accuracy and Validity. Clin. Genitourin. Cancer 2017, 15, e827–e834. [Google Scholar] [CrossRef] [PubMed]
- Briganti, A.; Wiegel, T.; Joniau, S.; Cozzarini, C.; Bianchi, M.; Sun, M.; Tombal, B.; Haustermans, K.; Budiharto, T.; Hinkelbein, W.; et al. Early salvage radiation therapy does not compromise cancer control in patients with pT3N0 prostate cancer after radical prostatectomy: Results of a match-controlled multi-institutional analysis. Eur. Urol. 2012, 62, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.; Karnes, R.J.; Rangel, L.J.; Pagliaro, L.C. Outcomes and Prognostic Factors in Men Receiving Androgen Deprivation Therapy for Prostate Cancer Recurrence after Radical Prostatectomy. J. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Meng, F.; Wang, B.; He, L.; Liu, Y.; Liu, Z. Dock2 in the development of inflammation and cancer. Eur. J. Immunol. 2018, 48, 915–922. [Google Scholar] [CrossRef] [PubMed]
- El-Haibi, C.P.; Singh, R.; Sharma, P.K.; Singh, S.; Lillard, J.W., Jr. CXCL13 mediates prostate cancer cell proliferation through JNK signalling and invasion through ERK activation. Cell Prolif. 2011, 44, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.M.; Ross, J.P.; Drew, H.R.; Ho, T.; Brown, G.S.; Saunders, N.F.; Duesing, K.R.; Buckley, M.J.; Dunne, R.; Beetson, I.; et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Yasui, K.; Dohi, O.; Gen, Y.; Tomie, A.; Kitaichi, T.; Iwai, N.; Mitsuyoshi, H.; Sumida, Y.; Moriguchi, M.; et al. Genome-wide DNA methylation analysis in hepatocellular carcinoma. Oncol. Rep. 2016, 35, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Kopantzev, E.P.; Monastyrskaya, G.S.; Vinogradova, T.V.; Zinovyeva, M.V.; Kostina, M.B.; Filyukova, O.B.; Tonevitsky, A.G.; Sukhikh, G.T.; Sverdlov, E.D. Differences in gene expression levels between early and later stages of human lung development are opposite to those between normal lung tissue and non-small lung cell carcinoma. Lung Cancer 2008, 62, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Rostila, A.; Puustinen, A.; Toljamo, T.; Vuopala, K.; Lindstrom, I.; Nyman, T.A.; Oksa, P.; Vehmas, T.; Anttila, S.L. Peroxiredoxins and tropomyosins as plasma biomarkers for lung cancer and asbestos exposure. Lung Cancer 2012, 77, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Beer, L.A.; Tanyi, J.L.; Zhang, R.; Liu, Q.; Speicher, D.W. Protein isoform-specific validation defines multiple chloride intracellular channel and tropomyosin isoforms as serological biomarkers of ovarian cancer. J. Proteomics 2013, 89, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Zheng, G.; Ren, D.; Chen, C.; Zeng, C.; Lu, W.; Li, H. The clinical significance and biological function of tropomyosin 4 in colon cancer. Biomed. Pharmacother. 2018, 101, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thang, N.D.; Yajima, I.; Nakagawa, K.; Tsuzuki, T.; Kumasaka, M.Y.; Ohgami, N.; Ly, T.B.; Iwamoto, T.; Watanabe, D.; Kato, M. A novel hairless mouse model for malignant melanoma. J. Dermatol. Sci. 2012, 65, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.A.; Kim, Y.H.; Kang, H.W.; Yoon, H.Y.; Kim, W.T.; Kim, Y.J.; Yun, S.J.; Moon, S.K.; Choi, Y.H.; Kim, I.Y.; et al. Lower Levels of Human MOB3B Are Associated with Prostate Cancer Susceptibility and Aggressive Clinicopathological Characteristics. J. Korean Med. Sci. 2015, 30, 937–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Cards-Human Gene Database. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=FBXO30 (accessed on 3 September 2018).
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Jia, X.; Hou, L.; Liu, X. Screening of Differently Expressed miRNA and mRNA in Prostate Cancer by Integrated Analysis of Transcription Data. Urology 2016, 94, 313.e1–313.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mei, Q.; Ai, Y.Q.; Li, R.Q.; Chang, L.; Li, Y.F.; Xia, Y.X.; Li, W.H.; Chen, Y. Identification of lung cancer oncogenes based on the mRNA expression and single nucleotide polymorphism profile data. Neoplasma 2015, 62, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas. Available online: https://cancergenome.nih.gov/ (accessed on 1 June 2018).
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Pound, C.R.; Partin, A.W.; Eisenberger, M.A.; Chan, D.W.; Pearson, J.D.; Walsh, P.C. Natural history of progression after PSA elevation following radical prostatectomy. JAMA 1999, 281, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, P.C. Observational studies and the natural history of screen-detected prostate cancer. Curr. Opin. Urol. 2015, 25, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y. Chip Analysis Methylation Pipeline for Illumina HumanMethylation450. Available online: https://git.bioconductor.org/packages/ChAMP (accessed on 16 September 2015).
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 15 April 2018).
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E., Jr.; Lee, K.L.; Mark, D.B. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RP Cohort | PCa | |
|---|---|---|
| N | 197 | |
| Age years, median (range) | 64 (49–77) | |
| Preoperative PSA | ||
| PSA ng/mL, median (range) | 12.9 (2.1–61.0) | |
| D’Amico Risk classification | ||
| Low risk, N (%) | 23 (11.7) | |
| Intermediate risk, N (%) | 80 (40.6) | |
| High risk, N (%) | 91 (46.2) | |
| Unknown, N (%) | 3 (1.5) | |
| Pathological Gleason score | ||
| <7, N (%) | 63 (32.0) | |
| =7, N (%) | 98 (49.7) | |
| >7, N (%) | 36 (18.3) | |
| Pathological T-stage | ||
| <pT2c, N (%) | 42 (21.3) | |
| =pT2c, N (%) | 85 (43.1) | |
| >pT2c, N (%) | 70 (35.5) | |
| Surgical margin status | ||
| Negative, N (%) | 137 (69.5) | |
| Positive, N (%) | 60 (30.5) | |
| Lymph node status | ||
| Positive N1, N (%) | 5 (2.5) | |
| Negative N0, N (%) | 164 (83.2) | |
| Unknown NX, N (%) | 28 (14.2) | |
| Follow-up | ||
| Follow-up months, median (range) | 128 (7–219) | |
| PSA recurrence, N (%) | 107 (54) | |
| Controls | AN | BPH |
| N | 28 | 9 |
| Age years, median (range) | 64 (56–73) | 70 (57–81) |
| Variable | Univariate Cox Regression | |||
|---|---|---|---|---|
| HR (95% CI) | p-value | Adjusted p-value | C-Index | |
| cg12799885 | 34.73 (1.57–769.04) | 0.025* | 0.100 | 0.570 |
| DOCK2 | 2.50 (1.59–3.94) | <0.001* | 0.001* | 0.615 |
| FBXO30 | 7.52 (0.78–72.42) | 0.081 | 0.162 | 0.528 |
| GRASP | 13.87 (3.16–60.80) | <0.001* | 0.003* | 0.606 |
| HIF3A | 6.46 (1.77–23.56) | 0.005* | 0.024* | 0.587 |
| MOBKL2B | 1.22 (0.59–2.52) | 0.599 | 0.599 | 0.510 |
| PFKP | 14.63 (2.95–72.50) | 0.001* | 0.006* | 0.598 |
| TPM4 | 40.03 (1.21–1317.12) | 0.038* | 0.114 | 0.547 |
| Surgical margin (negative vs. positive) | 3.24 (2.20–4.76) | <0.001* | <0.001* | 0.635 |
| Pre-op PSA dichotomized (< 10 vs ≥ 10) | 1.99 (1.25–3.15) | 0.004* | 0.004* | 0.572 |
| Pathological GS (6-7 vs. 8-10) | 2.33 (1.51–3.59) | <0.001* | <0.001* | 0.56 |
| Pathological T-stage (pT2a-T2b vs. pT2c-pT4) | 1.80 (1.07–3.03) | 0.026* | 0.026* | 0.552 |
| Pathological N-stage | 2.83 (1.13–7.04) | 0.026* | 0.026* | 0.514 |
| Age at diagnosis | 0.96 (0.93–1.00) | 0.054 | 0.054 | 0.554 |
| Variable | Multivariate Cox Regression | Final Multivariate Cox Regression | |||||
| HR (95% CI) | p-value | HR (95% CI) | p-value | Adj p-value | C-indexa | C-indexb | |
| DOCK2-continuous | 1.95 (1.20–3.17) | 0.004* | 1.96 (1.24–3.10) | 0.004* | 0.016* | 0.719a | - |
| Surgical margin (negative vs. positive) | 2.48 (1.60–3.84) | <0.001* | 2.53 (1.70–3.75) | <0.001* | <0.001* | 0.692b | |
| Preoperative PSA (<10 vs ≥10) | 1.82 (1.14–2.90) | 0.012* | 1.82 (1.14–2.89) | 0.012* | 0.012* | ||
| Path. Gleason score (6–7 vs. 8–10) | 1.69 (1.09–2.64) | 0.020* | 1.69 (1.09–2.64) | 0.019* | 0.019* | ||
| Path. T-stage (pT2a-T2b vs. pT2c-pT4) | 0.96 (0.54–1.70) | 0.880 | - | - | - | - | - |
| Variable | Multivariate Cox Regression | Final Multivariate Cox Regression | |||||
| HR (95% CI) | p-value | HR (95% CI) | p-value | Adj p-value | C-indexa | C-indexb | |
| HIF3A-continuous | 4.73 (1.14–19.5) | 0.032* | 4.73 (1.19–18.78) | 0.027* | 0.037* | 0.713a | - |
| Surgical margin (negative vs. positive) | 2.58 (1.67–3.99) | < 0.001* | 2.69 (1.81–3.98) | <0.001* | <0.001* | 0.692b | |
| Preoperative PSA (<10 vs ≥10) | 1.82 (1.14–2.90) | 0.012* | 1.83 (1.15–2.92) | 0.011* | 0.011* | ||
| Path. Gleason score (6–7 vs. 8–10) | 1.69 (1.08–2.65) | 0.022* | 1.70 (1.09–2.66) | 0.019* | 0.019* | ||
| Path. T-stage (pT2a-T2b vs. pT2c-pT4) | 0.99 (0.56–1.76) | 0.979 | - | - | - | - | - |
| Variable | Multivariate Cox Regression | Final Multivariate Cox Regression | |||||
| HR (95% CI) | p-value | HR (95% CI) | p-value | Adj p-value | C-indexa | C-indexb | |
| GRASP-continuous | 5.21 (1.04–26.0) | 0.044* | 5.24 (1.11–24.9) | 0.037* | 0.037* | 0.708 a | - |
| Surgical margin (negative vs. positive) | 2.39 (1.54–3.71) | < 0.001* | 2.53 (1.69–3.77) | <0.001* | <0.001* | 0.692b | |
| Preoperative PSA (<10 vs ≥10) | 1.75 (1.10–2.78) | 0.019* | 1.74 (1.10–2.77) | 0.019* | 0.019* | ||
| Path. Gleason score (6–7 vs. 8–10) | 1.70 (1.09–2.67) | 0.018* | 1.71 (1.10–2.67) | 0.017* | 0.017* | ||
| Path T-stage (pT2a-T2b vs. pT2c-pT4) | 1.03 (0.58–1.81) | 0.931 | - | - | - | - | - |
| Variable | Multivariate Cox Regression | Final Multivariate Cox Regression | |||||
| HR (95% CI) | p-value | HR (95% CI) | p-value | Adj p-value | C-indexa | C-indexb | |
| PFKP-continuous | 7.47 (1.27–43.9) | 0.026* | 6.65 (1.23–36.1) | 0.028* | 0.037* | 0.710a | - |
| Surgical margin (negative vs. positive) | 2.44 (1.58–3.79) | < 0.001* | 2.60 (1.75–3.86) | <0.001* | <0.001* | 0.692b | |
| Preoperative PSA (<10 vs ≥10) | 1.84 (1.15–2.96) | 0.012* | 1.85 (1.16–2.97) | 0.010* | 0.010* | ||
| Path.Gleason score (6-7 vs. 8-10) | 1.62 (1.03–2.56) | 0.033* | 1.65 (1.05–2.59) | 0.030* | 0.030* | ||
| Path. T-stage (pT2a-T2b vs. pT2c-pT4) | 1.01 (0.57–1.79) | 0.977 | - | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjerre, M.T.; Strand, S.H.; Nørgaard, M.; Kristensen, H.; Rasmussen, A.K.; Mortensen, M.M.; Fredsøe, J.; Mouritzen, P.; Ulhøi, B.; Ørntoft, T.; et al. Aberrant DOCK2, GRASP, HIF3A and PKFP Hypermethylation has Potential as a Prognostic Biomarker for Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1173. https://doi.org/10.3390/ijms20051173
Bjerre MT, Strand SH, Nørgaard M, Kristensen H, Rasmussen AK, Mortensen MM, Fredsøe J, Mouritzen P, Ulhøi B, Ørntoft T, et al. Aberrant DOCK2, GRASP, HIF3A and PKFP Hypermethylation has Potential as a Prognostic Biomarker for Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(5):1173. https://doi.org/10.3390/ijms20051173
Chicago/Turabian StyleBjerre, Marianne T., Siri H. Strand, Maibritt Nørgaard, Helle Kristensen, Anne KI Rasmussen, Martin Mørck Mortensen, Jacob Fredsøe, Peter Mouritzen, Benedicte Ulhøi, Torben Ørntoft, and et al. 2019. "Aberrant DOCK2, GRASP, HIF3A and PKFP Hypermethylation has Potential as a Prognostic Biomarker for Prostate Cancer" International Journal of Molecular Sciences 20, no. 5: 1173. https://doi.org/10.3390/ijms20051173