Methylation-Based Classification of Cervical Squamous Cell Carcinoma into Two New Subclasses Differing in Immune-Related Gene Expression


Abstract
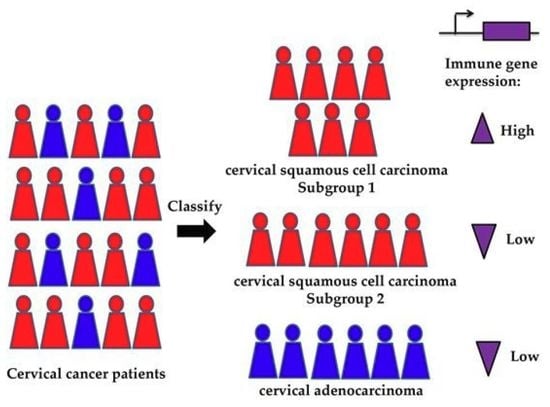
:
1. Introduction
2. Results
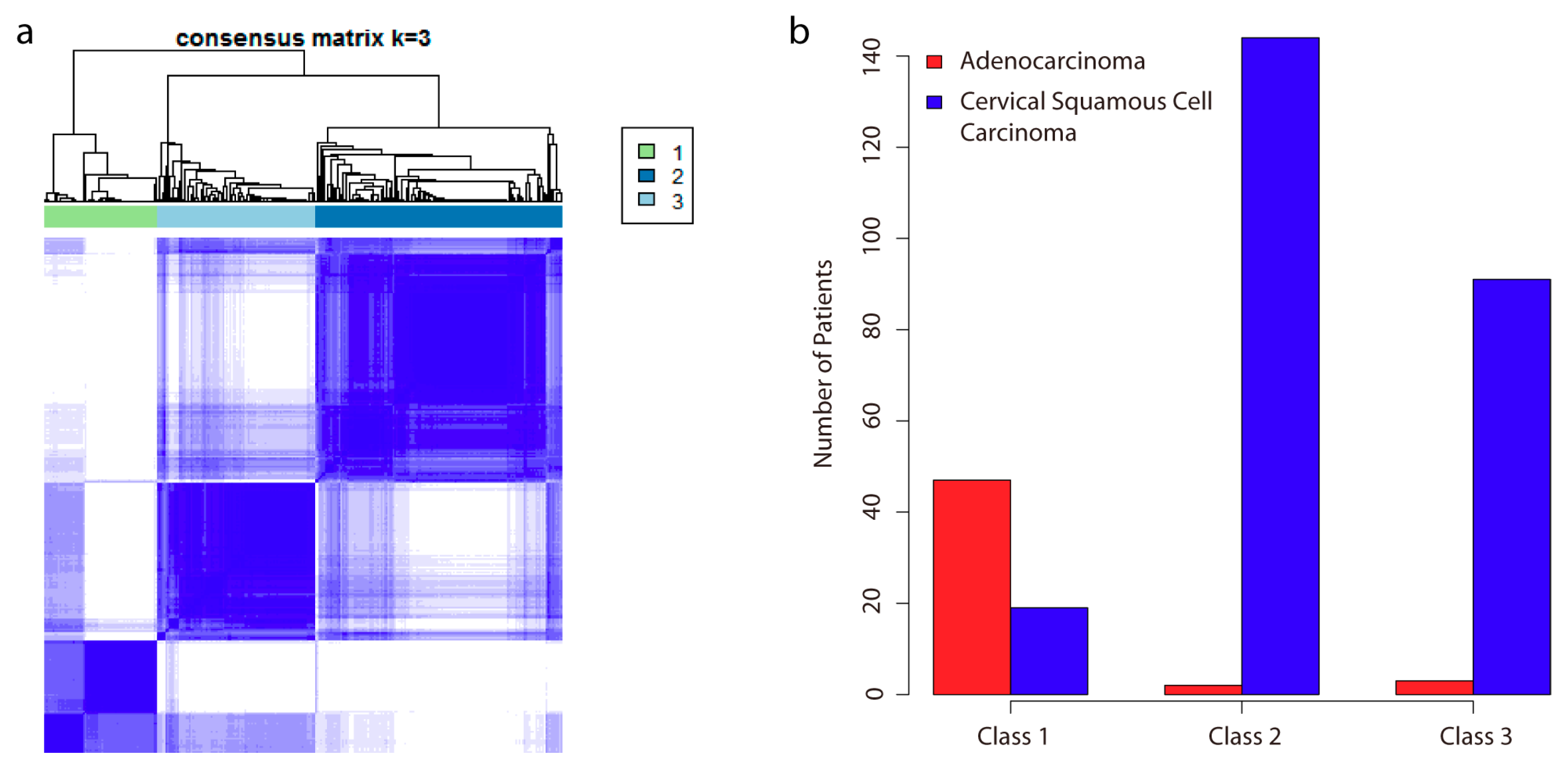
2.1. Analysis of DNA Methylation Identifies Two New Subclasses of CSCC
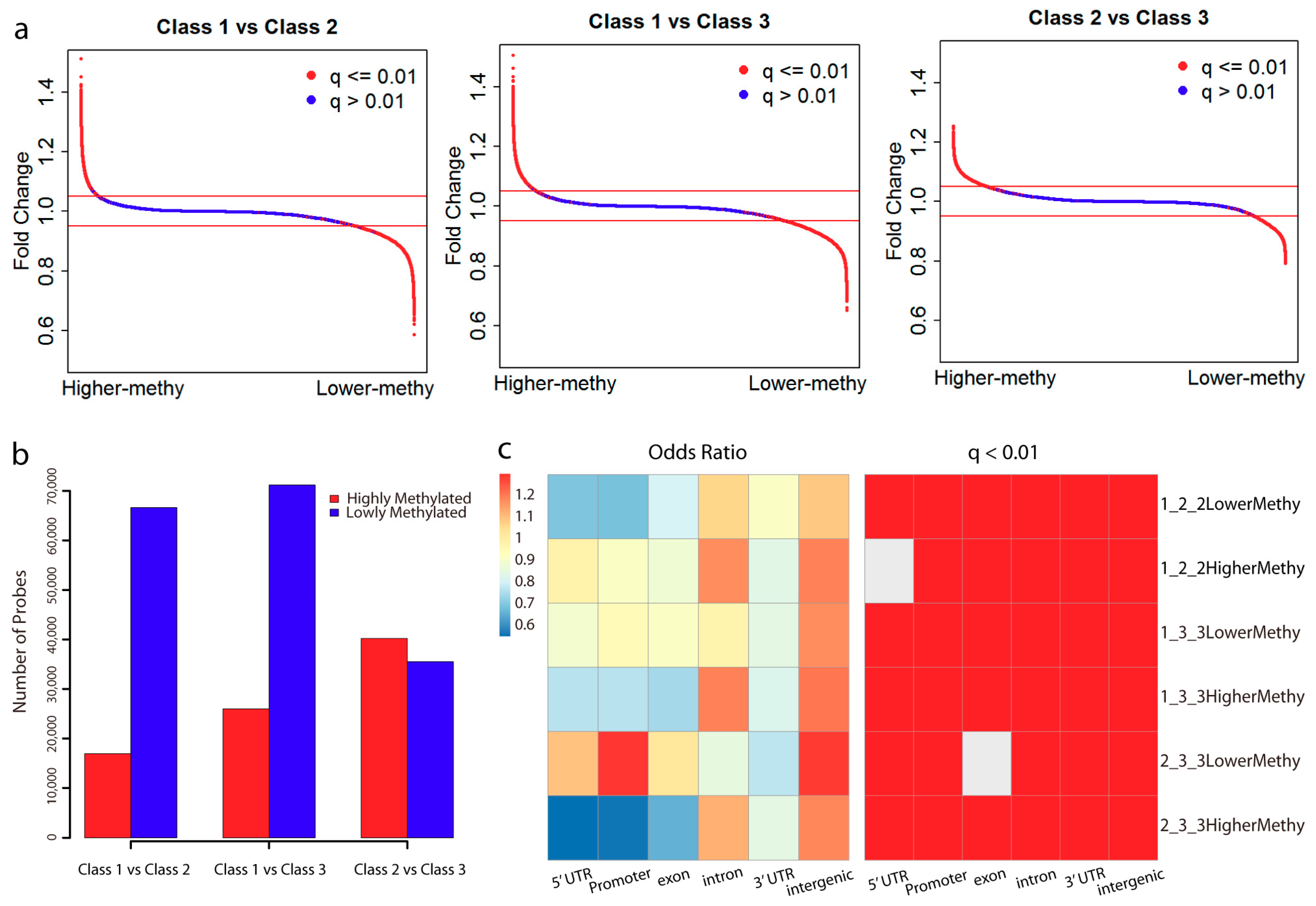
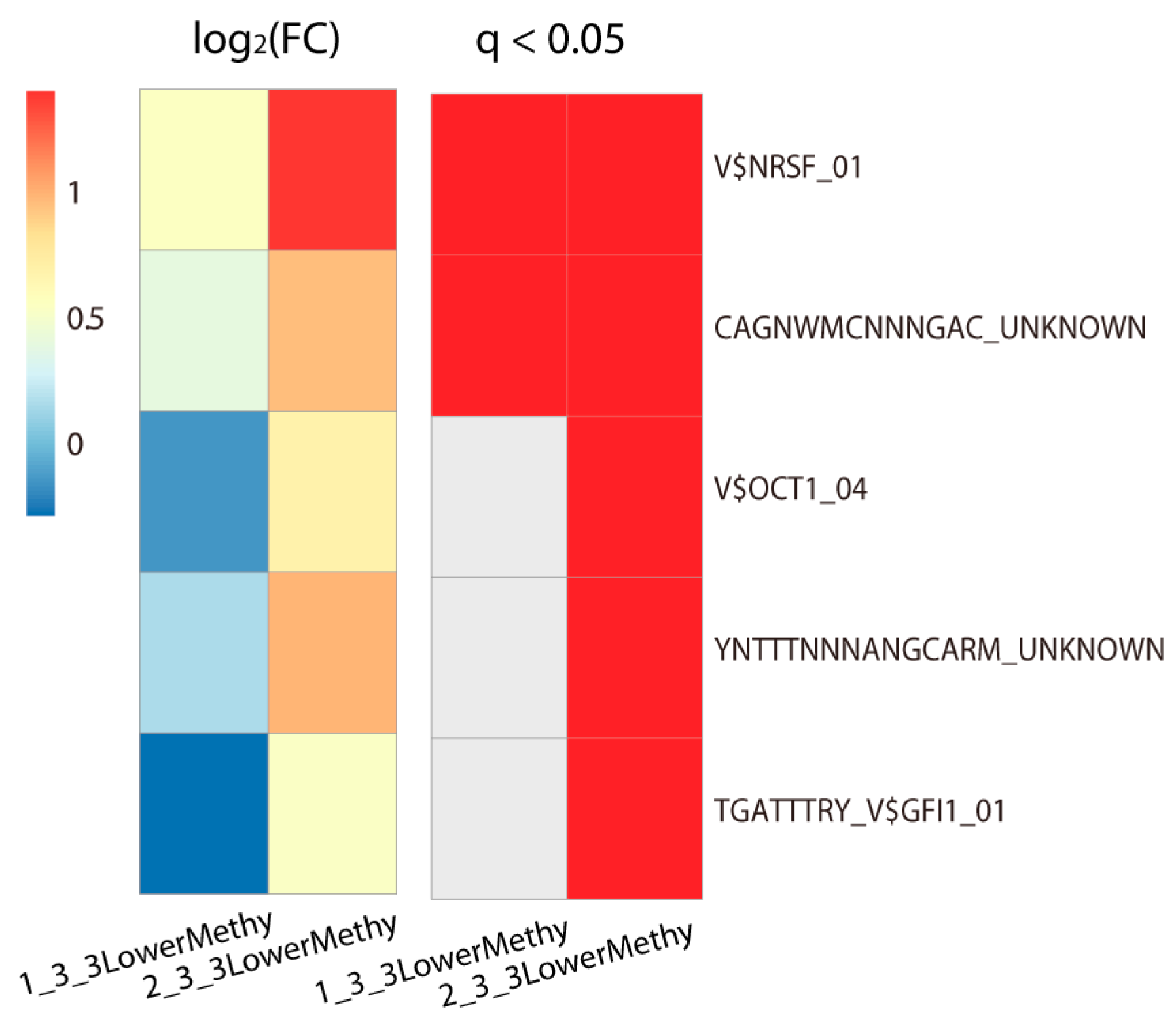
2.2. Lowly Methylated Promoters in Class 3 Show Enrichment in the Binding of 5 TFs
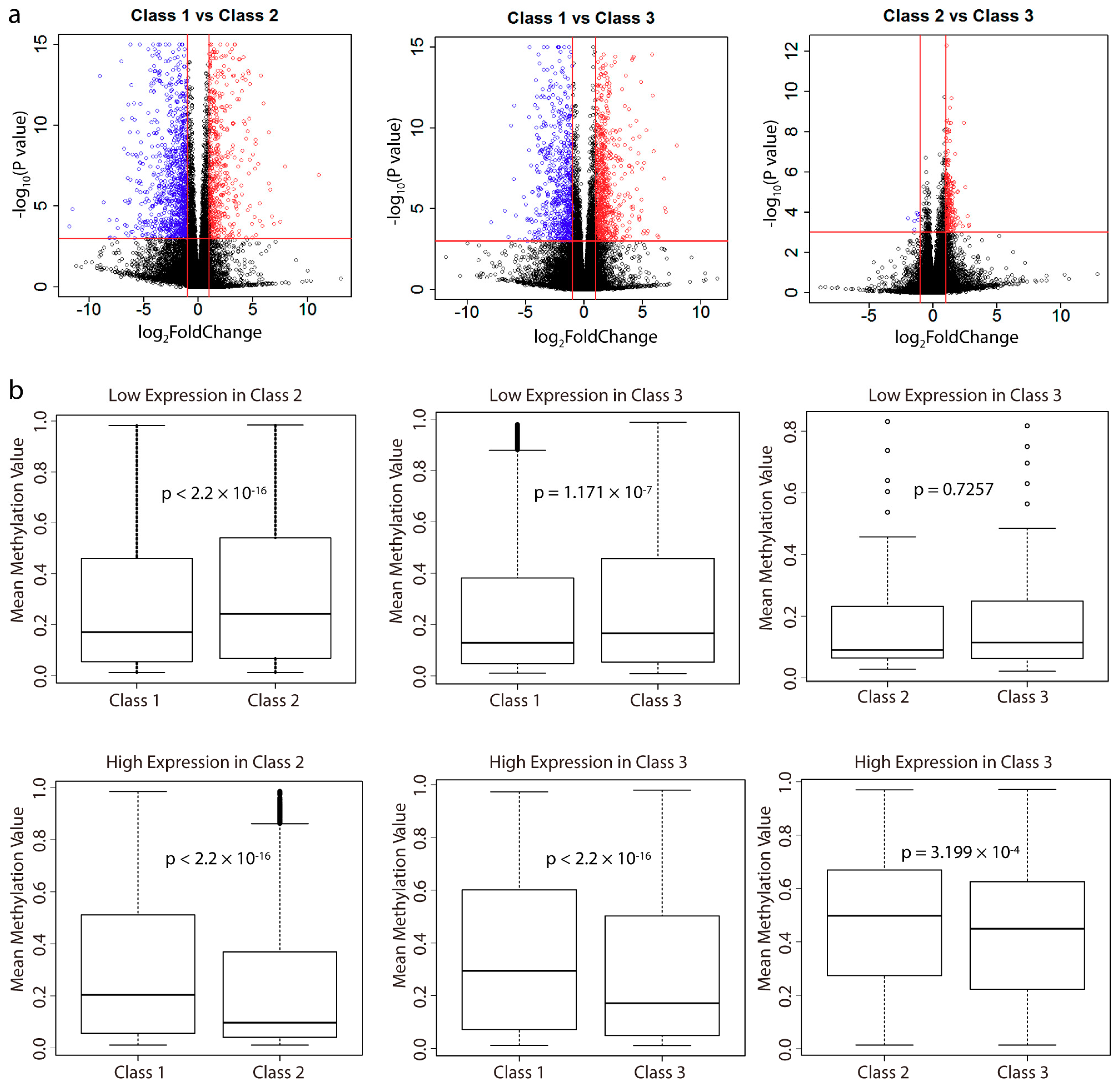
2.3. Analysis of Differentially Expressed Genes between Subclasses and Their Correlation with Methylation
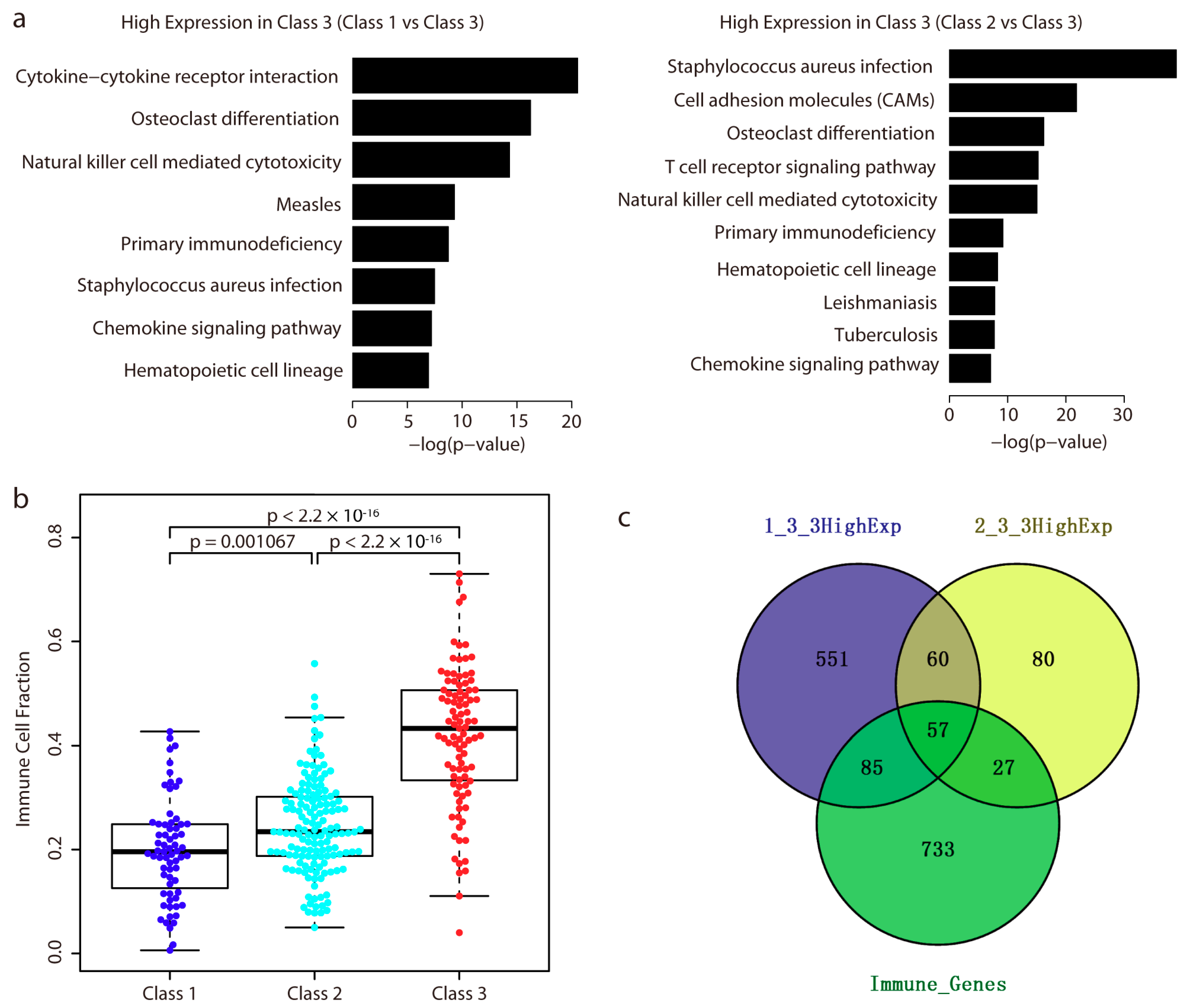
2.4. Immune-Related Genes are Highly Expressed in Class 3
2.5. Correlation of 24 Immune-Related Genes’ Expression with Predicted Neo-Epitope Burden
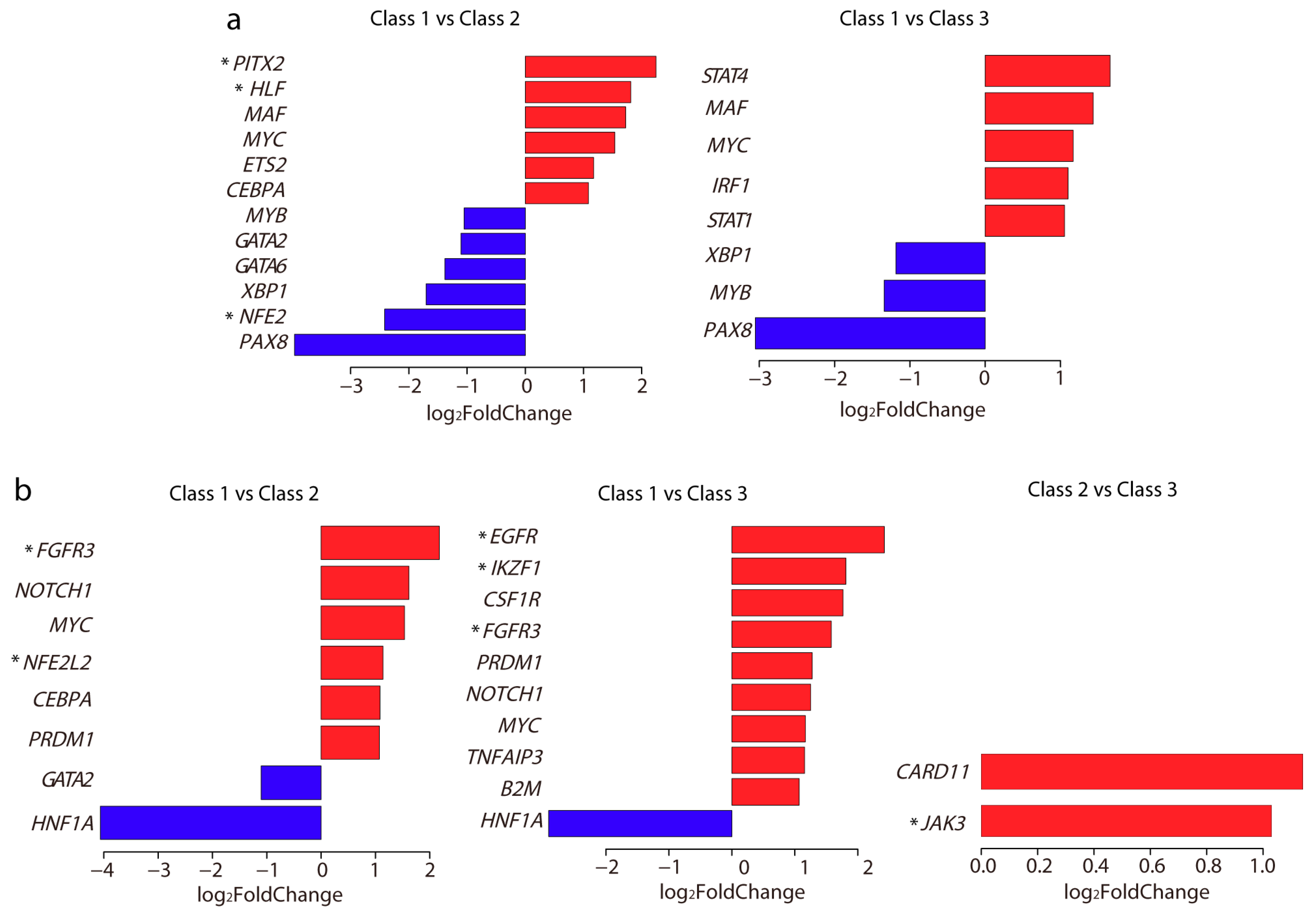
2.6. Differentially Expressed TFs and Driver Genes between Each Subclass
3. Discussion
4. Materials and Methods
4.1. Summary of Samples
4.2. Consensus Clustering
4.3. Differential Methylation Analysis
4.4. Genomic Region Enrichment
4.5. TFs Binding Enrichment and Target Gene Function
4.6. Analysis of Differentially Expressed Genes and Gene Function
4.7. Immune Cell Fraction Calculation
4.8. Correlation Analysis of Gene Expression with Predicted Neo-Epitope Burden
4.9. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCC | cervical squamous cell carcinoma |
| CA | cervical adenocarcinoma |
| TCGA | The Cancer Genome Atlas |
| TFs | transcription factors |
| MAD | median absolute deviation |
| DAVID | Database for Annotation, Visualization and Integrated Discovery |
| MHC-I | the major histocompatibility complex class I molecules |
| HPV | human papilloma virus |
| GDC | Genomic Data Commons |
| UTR | untranslated region |
| MSigDB | Molecular Signatures Database |
| NRSF | Neuron-Restrictive Silencer Factor |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| JAK | Janus kinases |
| STAT | Signal Transducer and Activator of Transcription proteins |
| CIMP | CpG island methylator phenotype |
| UCSC | the University of California, Santa Cruz |
References
- Irie, T.; Kigawa, J.; Minagawa, Y.; Itamochi, H.; Sato, S.; Akeshima, R.; Terakawa, N. Prognosis and clinicopathological characteristics of Ib-IIb adenocarcinoma of the uterine cervix in patients who have had radical hysterectomy. Eur. J. Surg. Oncol. 2000, 26, 464–467. [Google Scholar] [CrossRef] [PubMed]
- International Collaboration of Epidemiological Studies of Cervical Cancer. Comparison of risk factors for invasive squamous cell carcinoma and adenocarcinoma of the cervix: Collaborative reanalysis of individual data on 8097 women with squamous cell carcinoma and 1374 women with adenocarcinoma from 12 epidemiological studies. Int. J. Cancer 2007, 120, 885–891. [Google Scholar]
- Smith, H.O.; Tiffany, M.F.; Qualls, C.R.; Key, C.R. The rising incidence of adenocarcinoma relative to squamous cell carcinoma of the uterine cervix in the United States—A 24-year population-based study. Gynecol. Oncol. 2000, 78, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.P.; Morley, G.W. A comparison of adenocarcinoma and squamous cell carcinoma of the cervix. Obstet. Gynecol. 1991, 77, 912–917. [Google Scholar] [CrossRef]
- Wright, A.A.; Howitt, B.E.; Myers, A.P.; Dahlberg, S.E.; Palescandolo, E.; Van Hummelen, P.; MacConaill, L.E.; Shoni, M.; Wagle, N.; Jones, R.T.; et al. Oncogenic mutations in cervical cancer: Genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 2013, 119, 3776–3783. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Nishimura, R.; Nogawa, T.; Hatae, M.; Takehara, K.; Yamada, H.; Kurachi, H.; Yokoyama, Y.; Sugiyama, T.; Kigawa, J. Comparison of the outcome between cervical adenocarcinoma and squamous cell carcinoma patients with adjuvant radiotherapy following radical surgery: SGSG/TGCU Intergroup Surveillance. Mol. Clin. Oncol. 2013, 1, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar] [PubMed]
- Bachtiary, B.; Boutros, P.C.; Pintilie, M.; Shi, W.; Bastianutto, C.; Li, J.H.; Schwock, J.; Zhang, W.; Penn, L.Z.; Jurisica, I.; et al. Gene expression profiling in cervical cancer: An exploration of intratumor heterogeneity. Clin. Cancer Res. 2006, 12, 5632–5640. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.E.; West, C.M.; Roberts, S.A.; Hendry, J.H.; Hunter, R.D. Radiosensitivity testing of primary cervical carcinoma: Evaluation of intra- and inter-tumour heterogeneity. Radiother. Oncol. 1990, 18, 349–356. [Google Scholar] [CrossRef]
- Grigsby, P.W.; Watson, M.; Powell, M.A.; Zhang, Z.; Rader, J.S. Gene expression patterns in advanced human cervical cancer. Int. J. Gynecol. Cancer 2006, 16, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, H.; Guan, Y.; Gong, Y.; He, C.Y.; Yi, X.; Qi, M.; Chen, Z.Y. Whole-exome sequencing predicted cancer epitope trees of 23 early cervical cancers in Chinese women. Cancer Med. 2017, 6, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Kidd, E.A.; Grigsby, P.W. Intratumoral metabolic heterogeneity of cervical cancer. Clin. Cancer Res. 2008, 14, 5236–5241. [Google Scholar] [CrossRef] [PubMed]
- Ronneberg, J.A.; Fleischer, T.; Solvang, H.K.; Nordgard, S.H.; Edvardsen, H.; Potapenko, I.; Nebdal, D.; Daviaud, C.; Gut, I.; Bukholm, I.; et al. Methylation profiling with a panel of cancer related genes: Association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol. Oncol. 2011, 5, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Chambwe, N.; Kormaksson, M.; Geng, H.; De, S.; Michor, F.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Godley, L.A.; Gascoyne, R.D.; et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 2014, 123, 1699–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevaert, O.; Tibshirani, R.; Plevritis, S.K. Pancancer analysis of DNA methylation-driven genes using MethylMix. Genome Biol. 2015, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Koike, F.; Satoh, J.; Miyake, S.; Yamamoto, T.; Kawai, M.; Kikuchi, S.; Nomura, K.; Yokoyama, K.; Ota, K.; Kanda, T.; et al. Microarray analysis identifies interferon beta-regulated genes in multiple sclerosis. J. Neuroimmunol. 2003, 139, 109–118. [Google Scholar] [CrossRef]
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol. Cancer 2005, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Zhang, H.; Jin, S. Epigenetics and cervical cancer: From pathogenesis to therapy. Tumour Biol. 2014, 35, 5083–5093. [Google Scholar] [CrossRef] [PubMed]
- Szalmas, A.; Konya, J. Epigenetic alterations in cervical carcinogenesis. Semin. Cancer Biol. 2009, 19, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Widschwendter, M.; Teschendorff, A.E. A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 2014, 30, 2360–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.C.; Webster, A.P.; Dong, D.; Feber, A.; Graham, D.G.; Sullivan, R.; Jevons, S.; Lovat, L.B.; Beck, S.; Widschwendter, M.; et al. A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics 2018, 10, 925–940. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Emerging role of mutations in epigenetic regulators including MLL2 derived from The Cancer Genome Atlas for cervical cancer. BMC Cancer 2017, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; O′Shea, J.J. Jaks and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jones, A.; Fasching, P.A.; Ruebner, M.; Beckmann, M.W.; Widschwendter, M.; Teschendorff, A.E. The integrative epigenomic-transcriptomic landscape of ER positive breast cancer. Clin. Epigenetics 2015, 7, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [PubMed]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catani, L.; Vianelli, N.; Amabile, M.; Pattacini, L.; Valdre, L.; Fagioli, M.E.; Poli, M.; Gugliotta, L.; Moi, P.; Marini, M.G.; et al. Nuclear factor-erythroid 2 (NF-E2) expression in normal and malignant megakaryocytopoiesis. Leukemia 2002, 16, 1773–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhang, J.; Liu, S.; Huang, Y.; Chen, B.; Wang, D. Nrf2 knockdown by shRNA inhibits tumor growth and increases efficacy of chemotherapy in cervical cancer. Cancer Chemother. Pharmacol. 2012, 69, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Fung, F.K.; Chan, D.W.; Liu, V.W.; Leung, T.H.; Cheung, A.N.; Ngan, H.Y. Increased expression of PITX2 transcription factor contributes to ovarian cancer progression. PLoS ONE 2012, 7, e37076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.X.; Tong, Z.T.; Yang, L.; Wang, F.; Chai, H.P.; Zhang, F.; Xie, M.R.; Zhang, A.L.; Wu, L.M.; Hong, H.; et al. PITX2: A promising predictive biomarker of patients′ prognosis and chemoradioresistance in esophageal squamous cell carcinoma. Int. J. Cancer 2013, 132, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Landhuis, E.; Dose, M.; Hazan, I.; Zhang, J.; Naito, T.; Jackson, A.F.; Wu, J.; Perotti, E.A.; Kaufmann, C.; et al. Transcriptional regulation of the Ikzf1 locus. Blood 2013, 122, 3149–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordhuis, M.G.; Eijsink, J.J.; Ten Hoor, K.A.; Roossink, F.; Hollema, H.; Arts, H.J.; Pras, E.; Maduro, J.H.; Reyners, A.K.; de Bock, G.H.; et al. Expression of epidermal growth factor receptor (EGFR) and activated EGFR predict poor response to (chemo)radiation and survival in cervical cancer. Clin. Cancer Res. 2009, 15, 7389–7397. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Chung, J.Y.; Kim, J.H.; Kim, B.G.; Hewitt, S.M. Expression of fibroblast growth factor receptor family members is associated with prognosis in early stage cervical cancer patients. J. Transl. Med. 2016, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Punt, S.; Houwing-Duistermaat, J.J.; Schulkens, I.A.; Thijssen, V.L.; Osse, E.M.; de Kroon, C.D.; Griffioen, A.W.; Fleuren, G.J.; Gorter, A.; Jordanova, E.S. Correlations between immune response and vascularization qRT-PCR gene expression clusters in squamous cervical cancer. Mol. Cancer 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Widschwendter, M.; Teschendorff, A.E. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinform. 2012, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, X.; Wang, Q.F.; Ning, Y. Differential principal component analysis of ChIP-seq. Proc. Natl. Acad. Sci. USA 2013, 110, 6789–6794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Event Status | Events No. | p Value | Adjusted p value 1 | Hazard Ratio | 95% Confidence Interval | p Value 2 | Adjusted p Value 3 |
|---|---|---|---|---|---|---|---|---|
| CCR5 | High Expression | 60 | 0.0268 | 0.0804 | 0.3279 | 0.116–0.925 | 0.00194 | 0.00418 |
| Low Expression | 58 | |||||||
| CXCL9 | High Expression | 61 | 0.0892 | 0.0931 | 0.4483 | 0.1734–1.159 | 0.00686 | 0.0087 |
| Low Expression | 58 | |||||||
| CXCR3 | High Expression | 59 | 0.0806 | 0.0912 | 0.4847 | 0.2114–1.1114 | 0.00305 | 0.0048 |
| Low Expression | 59 | |||||||
| IL10RA | High Expression | 59 | 0.0705 | 0.0912 | 0.4541 | 0.1893–1.0892 | 0.002 | 0.0042 |
| Low Expression | 55 | |||||||
| IL18RAP | High Expression | 61 | 0.0102 | 0.0612 | 0.3637 | 0.1629–0.8123 | 0.00627 | 0.0084 |
| Low Expression | 58 | |||||||
| ITGAX | High Expression | 61 | 0.063 | 0.0912 | 0.4837 | 0.2212–1.0573 | 0.0249 | 0.0249 |
| Low Expression | 56 | |||||||
| CCR2 | High Expression | 59 | 0.0549 | 0.0912 | 0.3987 | 0.1509–1.0532 | 0.000628 | 0.0023 |
| Low Expression | 57 | |||||||
| DOCK2 | High Expression | 61 | 0.078 | 0.0912 | 0.486 | 0.2143–1.1025 | 0.0184 | 0.0201 |
| Low Expression | 55 | |||||||
| WAS | High Expression | 60 | 0.0836 | 0.0912 | 0.5171 | 0.2417–1.1063 | 0.00209 | 0.0042 |
| Low Expression | 57 | |||||||
| BTK | High Expression | 61 | 0.0949 | 0.0949 | 0.4938 | 0.2123–1.149 | 0.00251 | 0.0046 |
| Low Expression | 56 | |||||||
| CD3D4 | High Expression | 60 | 0.0648 | 0.0912 | 0.4393 | 0.179–1.078 | 8.46 × 10−5 | 0.0016 |
| Low Expression | 57 | |||||||
| CD3E4 | High Expression | 60 | 0.009 | 0.0612 | 0.3262 | 0.1347–0.7899 | 1.62 × 10−4 | 0.0016 |
| Low Expression | 57 | |||||||
| CD74 | High Expression | 60 | 0.0361 | 0.0912 | 0.4499 | 0.209–0.969 | 5.51 × 10−4 | 0.0023 |
| Low Expression | 58 | |||||||
| CD48 | High Expression | 60 | 0.046 | 0.0912 | 0.4262 | 0.18–1 | 0.0017 | 0.0042 |
| Low Expression | 57 | |||||||
| CD247 | High Expression | 60 | 0.0654 | 0.0912 | 0.5043 | 0.24–1.0593 | 0.00321 | 0.0048 |
| Low Expression | 58 | |||||||
| CTLA4 | High Expression | 58 | 0.0696 | 0.0912 | 0.4556 | 0.191–1.0882 | 0.00913 | 0.011 |
| Low Expression | 57 | |||||||
| GZMB | High Expression | 58 | 0.0664 | 0.0912 | 0.4189 | 0.1609–1.091 | 0.0028 | 0.0048 |
| Low Expression | 58 | |||||||
| ITGAL | High Expression | 60 | 0.0216 | 0.0804 | 0.3823 | 0.1634–0.8945 | 0.00426 | 0.006 |
| Low Expression | 57 | |||||||
| ITK | High Expression | 59 | 0.0253 | 0.0804 | 0.4266 | 0.1978–0.92 | 6.57 × 10−4 | 0.0023 |
| Low Expression | 57 | |||||||
| KLRK1 | High Expression | 60 | 0.00449 | 0.05388 | 0.3 | 0.125–0.7236 | 2.65 × 10−4 | 0.0016 |
| Low Expression | 57 | |||||||
| PIK3CG | High Expression | 60 | 0.0819 | 0.0912 | 0.46 | 0.1879–1.126 | 0.0201 | 0.021 |
| Low Expression | 54 | |||||||
| PTPRC | High Expression | 60 | 0.0175 | 0.0804 | 0.3662 | 0.1546–0.8681 | 0.0124 | 0.0142 |
| Low Expression | 56 | |||||||
| SELL4 | High Expression | 62 | 0.000698 | 0.0168 | 0.2366 | 0.096–0.5824 | 2.25 × 10−4 | 0.0016 |
| Low Expression | 57 | |||||||
| SPN | High Expression | 61 | 0.0508 | 0.0912 | 0.4424 | 0.1909–1.0256 | 0.00163 | 0.0042 |
| Low Expression | 56 |
| 24 Prognostic Genes | Spearman’s Rho | p-Value |
|---|---|---|
| CD247 | 0.18011804 | 0.011746072222 |
| ITGAL | 0.23202640 | 0.001098899936 |
| ITGAX | 0.18054878 | 0.011543220339 |
| ITK | 0.11669343 | 0.104244914224 |
| PIK3CG | 0.10671102 | 0.137596629299 |
| SPN | 0.18914430 | 0.008091618099 |
| CD3D | 0.21162724 | 0.002978712632 |
| CD3E | 0.22309120 | 0.001719277379 |
| CD48 | 0.17823018 | 0.012672607086 |
| IL10RA | 0.18267087 | 0.010588223639 |
| DOCK2 | 0.16089662 | 0.024639133354 |
| CD7 | 0.21193808 | 0.002935714814 |
| PTPRC | 0.17346131 | 0.015304390326 |
| GZMB | 0.26446319 | 0.000186953723 |
| CTLA4 | 0.23369399 | 0.001008922888 |
| WAS | 0.17836121 | 0.012606283349 |
| SELL | 0.06245491 | 0.385736421301 |
| KLRK1 | 0.21769835 | 0.002234159280 |
| IL18RAP | 0.15330837 | 0.032376073551 |
| CXCL9 | 0.32242557 | 0.000004289406 |
| CXCR3 | 0.24277477 | 0.000626961434 |
| CCR2 | 0.17927831 | 0.012150572695 |
| CCR5 | 0.24935640 | 0.000439124718 |
| BTK | 0.13588272 | 0.058212535746 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Cai, Y. Methylation-Based Classification of Cervical Squamous Cell Carcinoma into Two New Subclasses Differing in Immune-Related Gene Expression. Int. J. Mol. Sci. 2018, 19, 3607. https://doi.org/10.3390/ijms19113607
Li X, Cai Y. Methylation-Based Classification of Cervical Squamous Cell Carcinoma into Two New Subclasses Differing in Immune-Related Gene Expression. International Journal of Molecular Sciences. 2018; 19(11):3607. https://doi.org/10.3390/ijms19113607
Chicago/Turabian StyleLi, Xia, and Yunpeng Cai. 2018. "Methylation-Based Classification of Cervical Squamous Cell Carcinoma into Two New Subclasses Differing in Immune-Related Gene Expression" International Journal of Molecular Sciences 19, no. 11: 3607. https://doi.org/10.3390/ijms19113607