Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
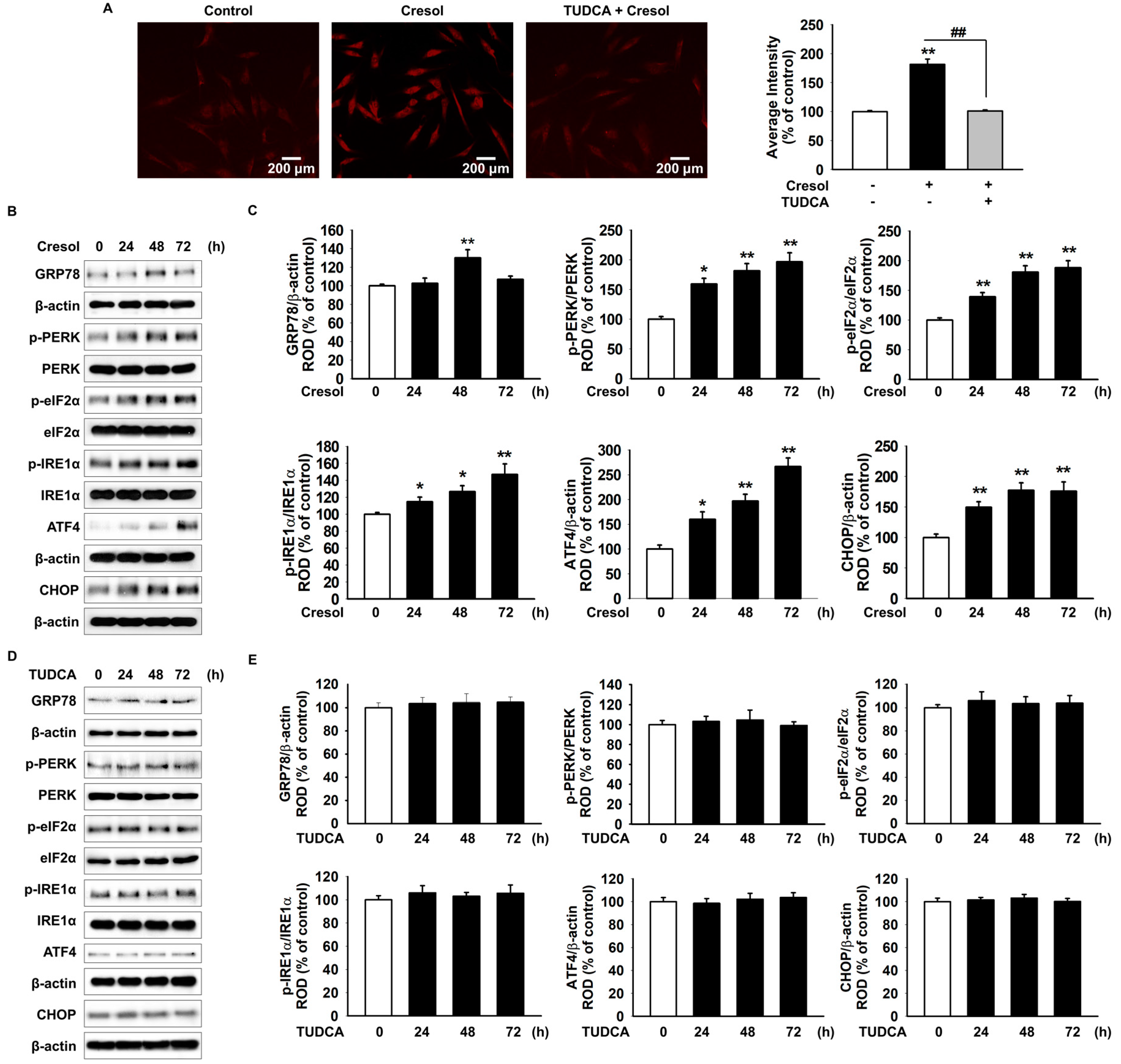
2.1. TUDCA Inhibits PC-Induced ER Stress by Suppressing ROS Generation
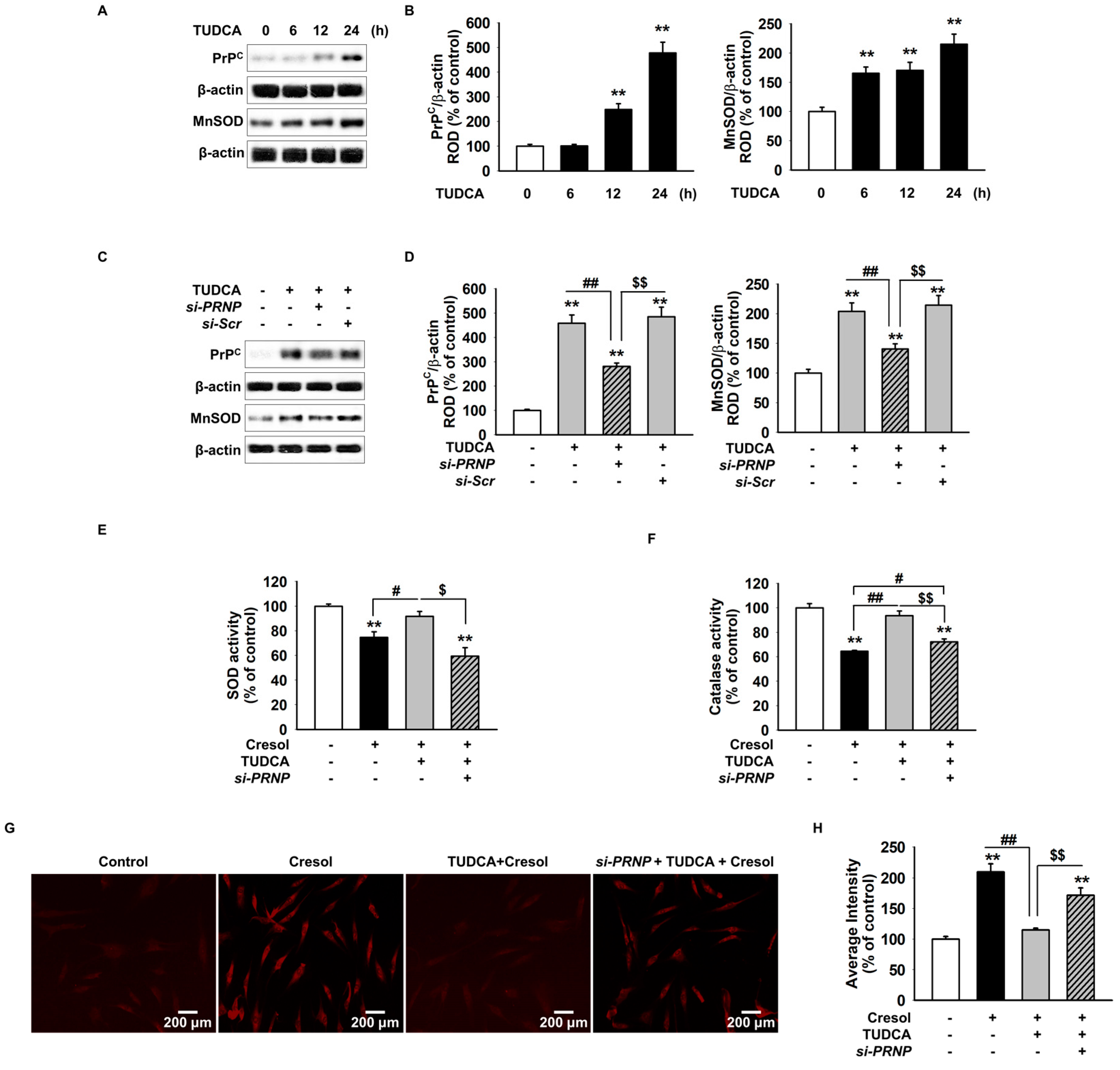
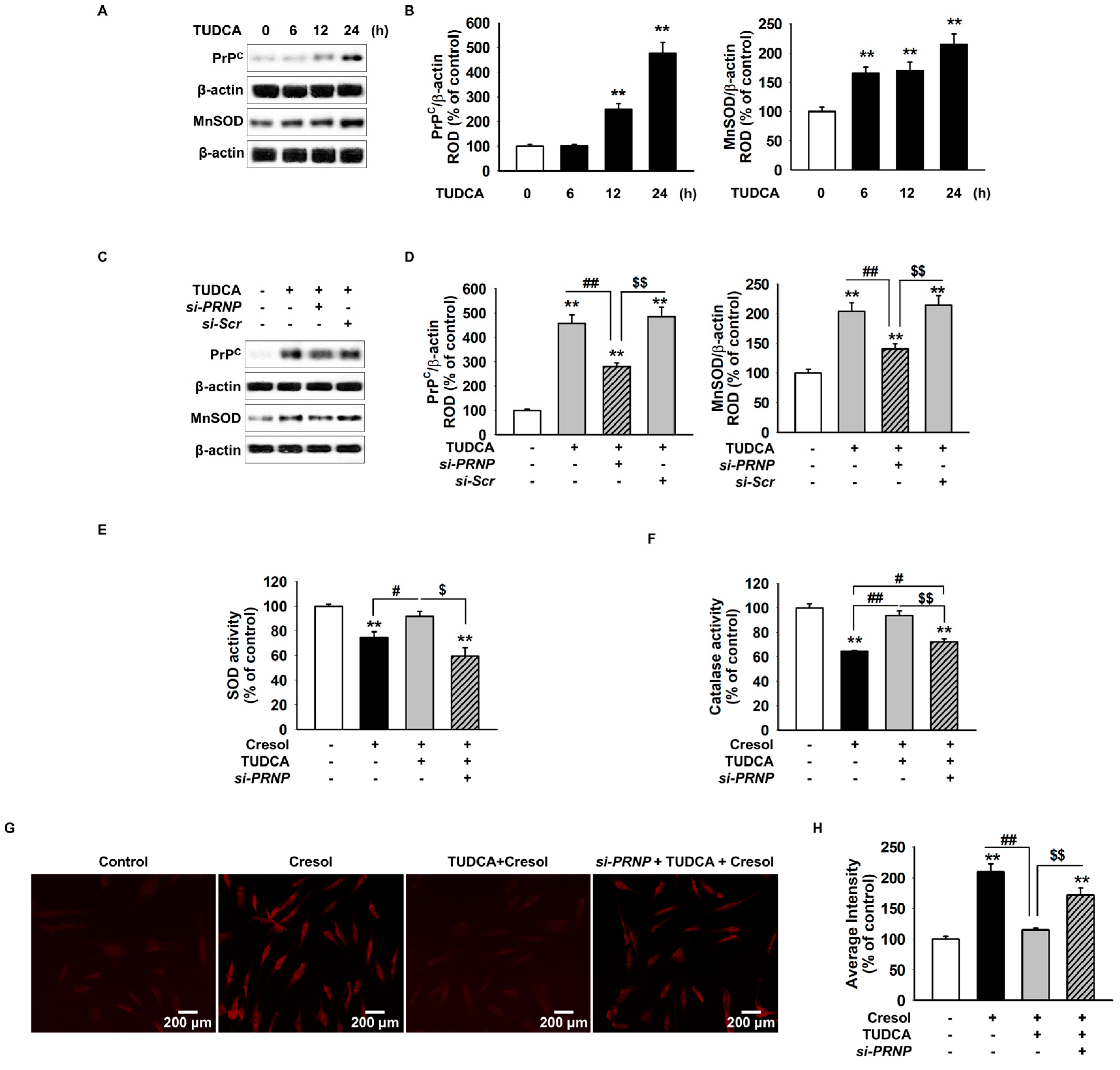
2.2. TUDCA Inhibits PC-Induced ROS Generation via Cellular Prion Protein-Dependent Catalase and SOD Activation
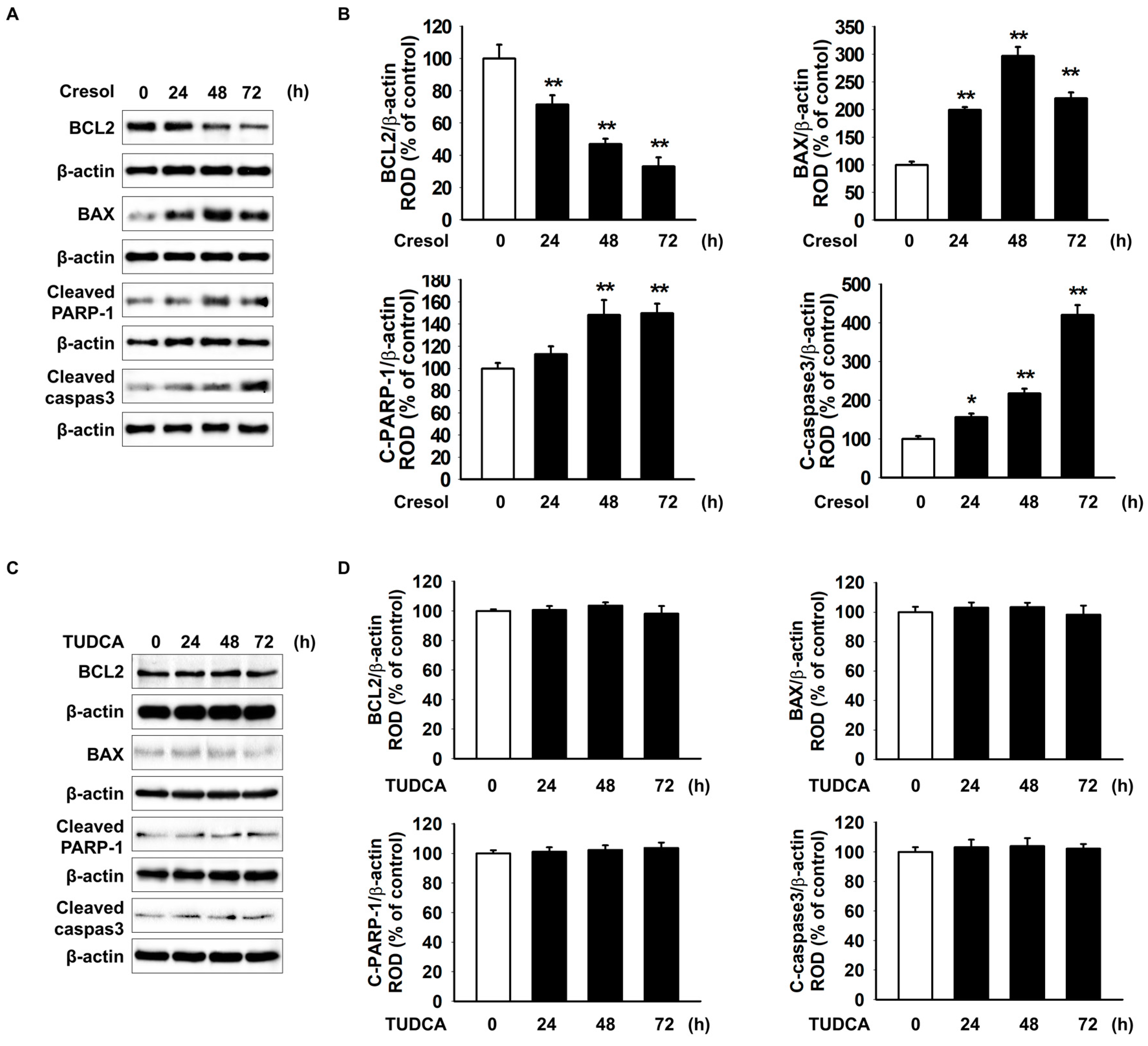
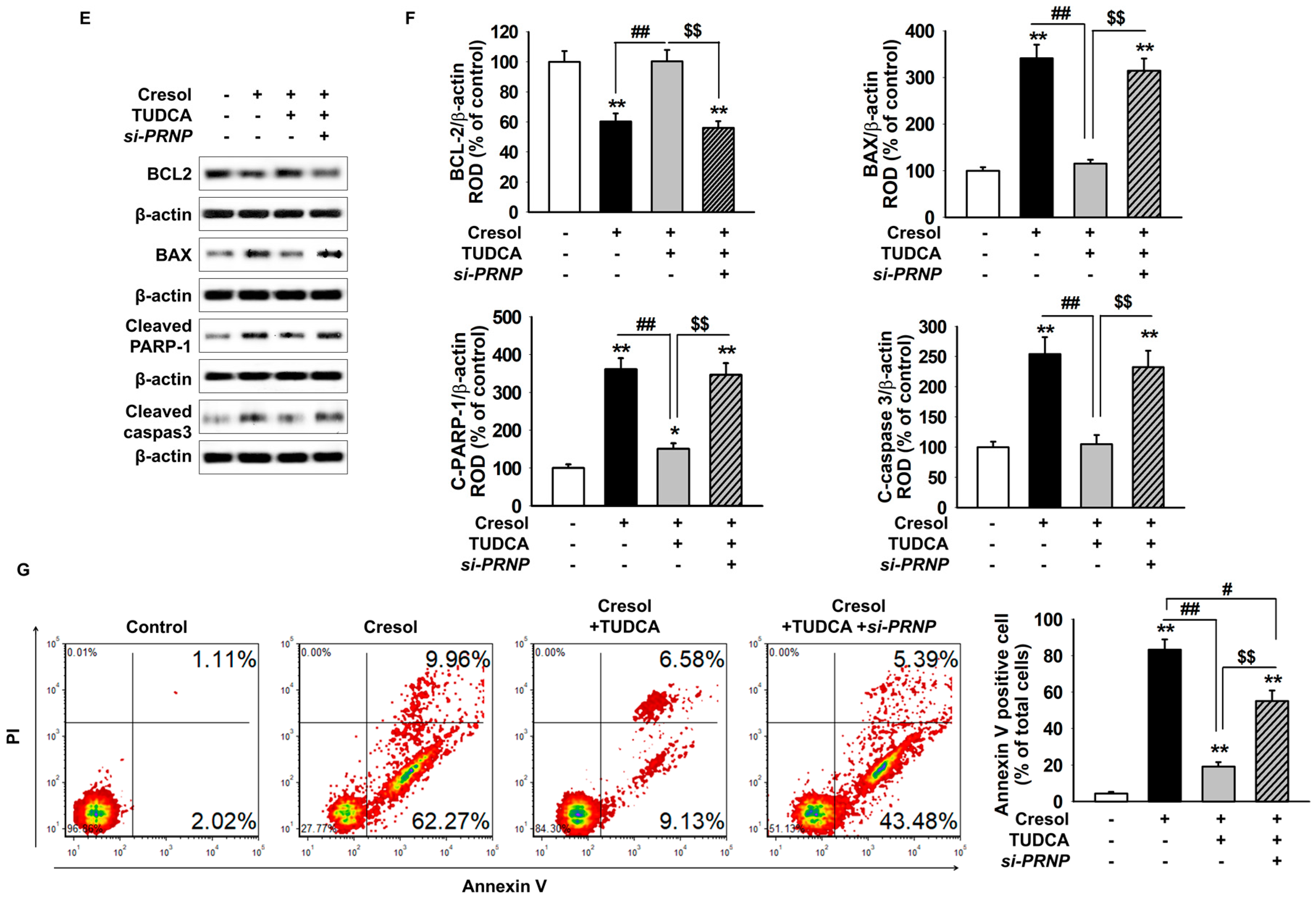
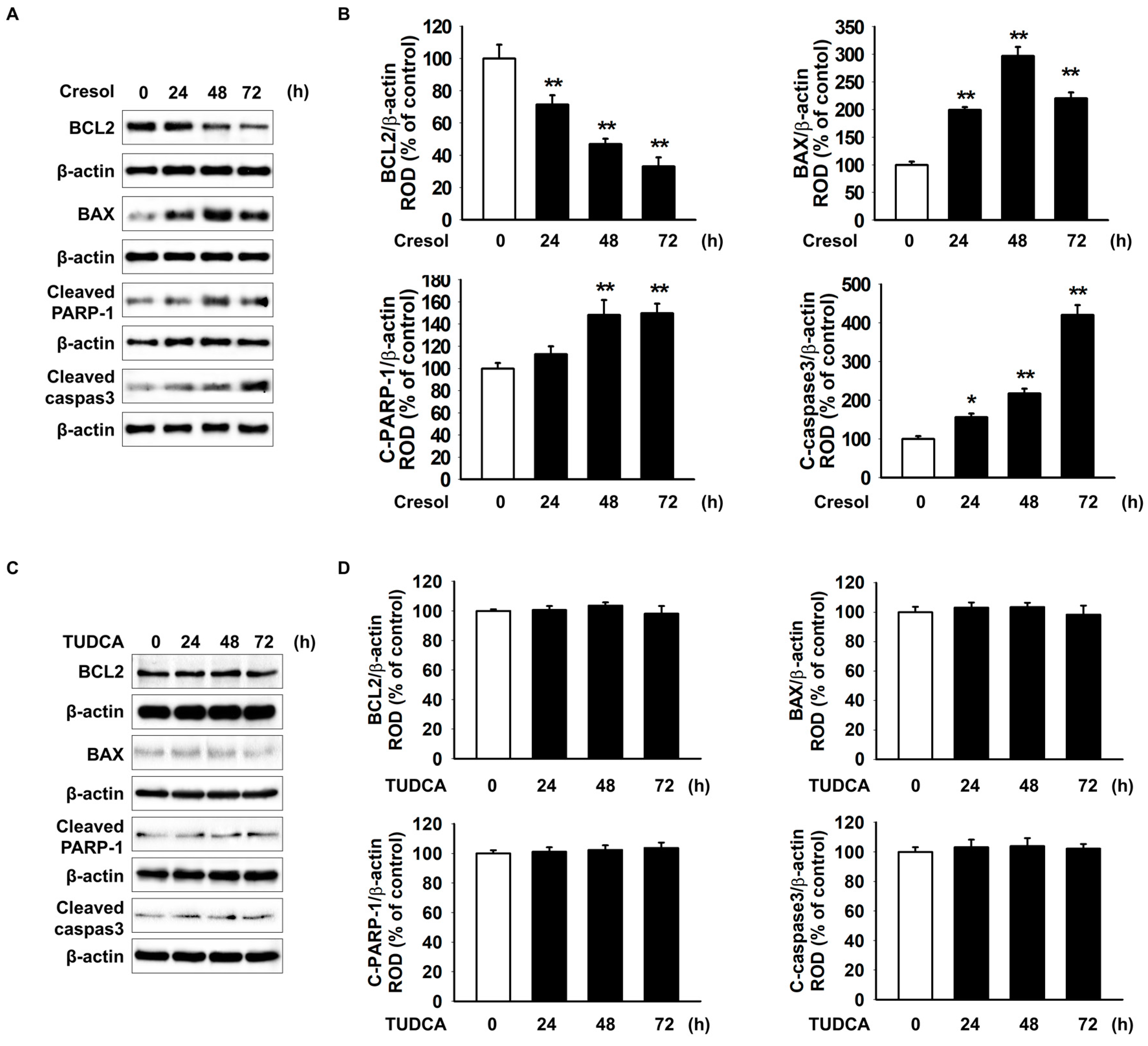
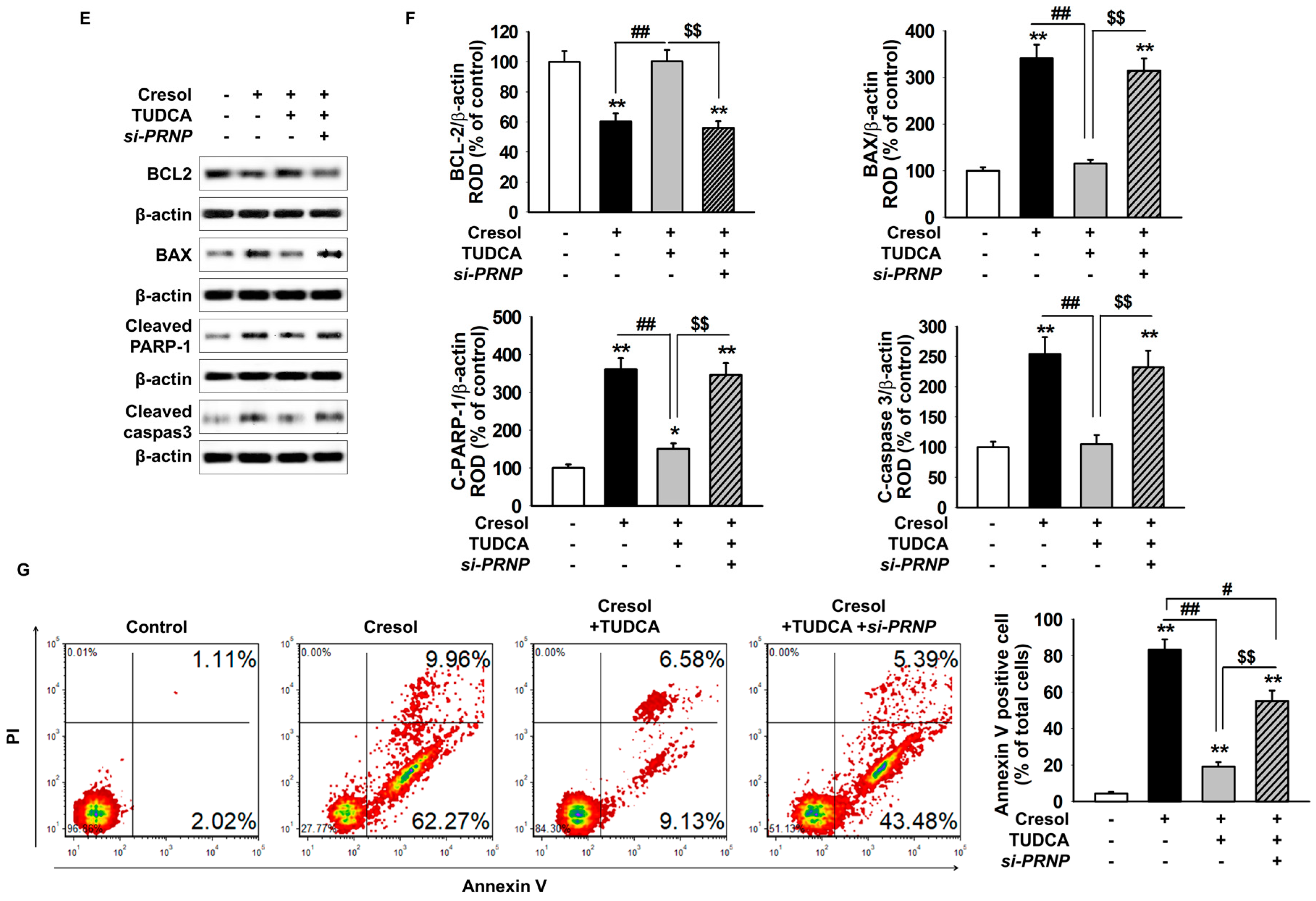
2.3. TUDCA Exerts Protective Effect on PC-Induced Apoptosis of MSCs
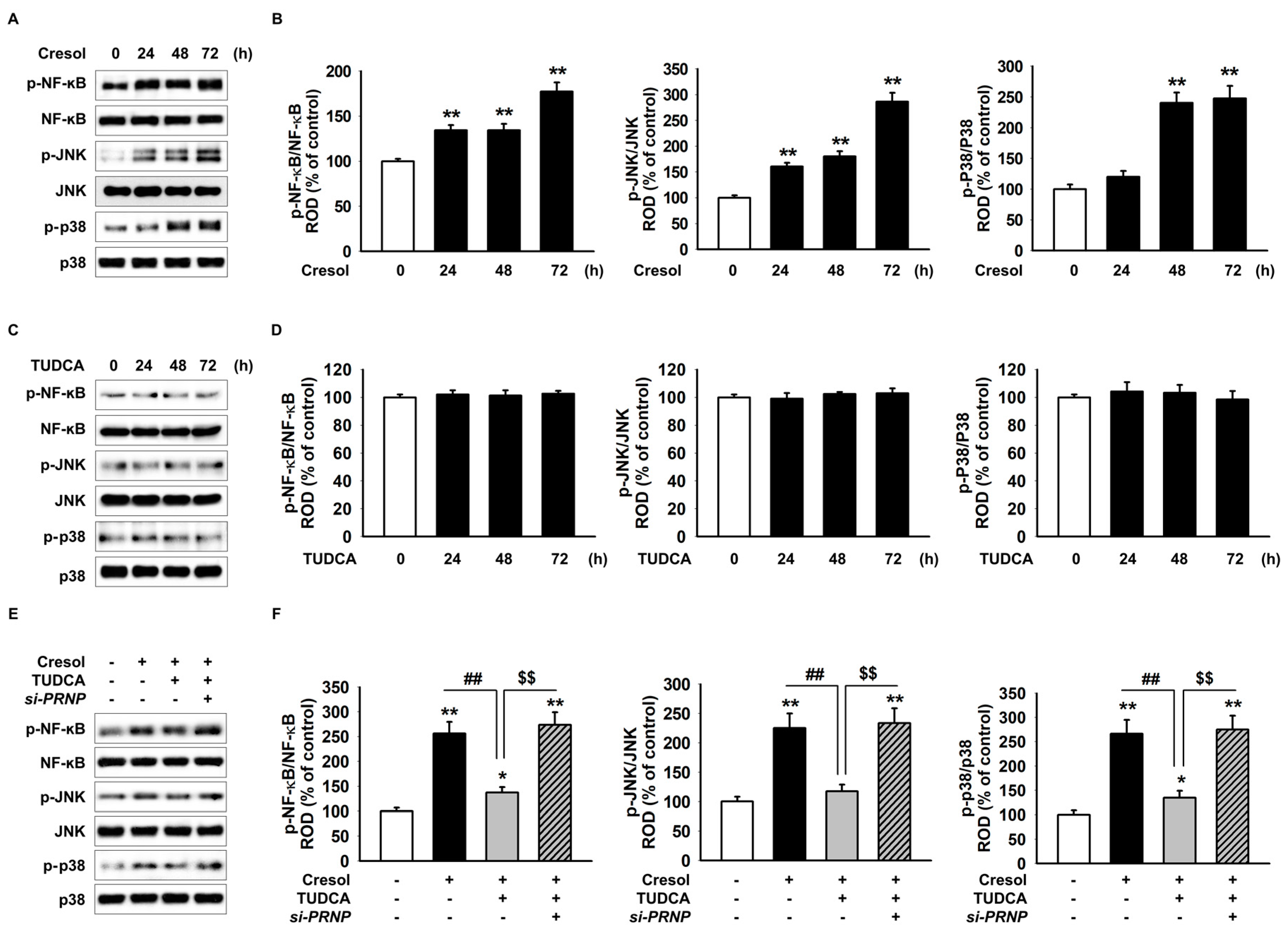
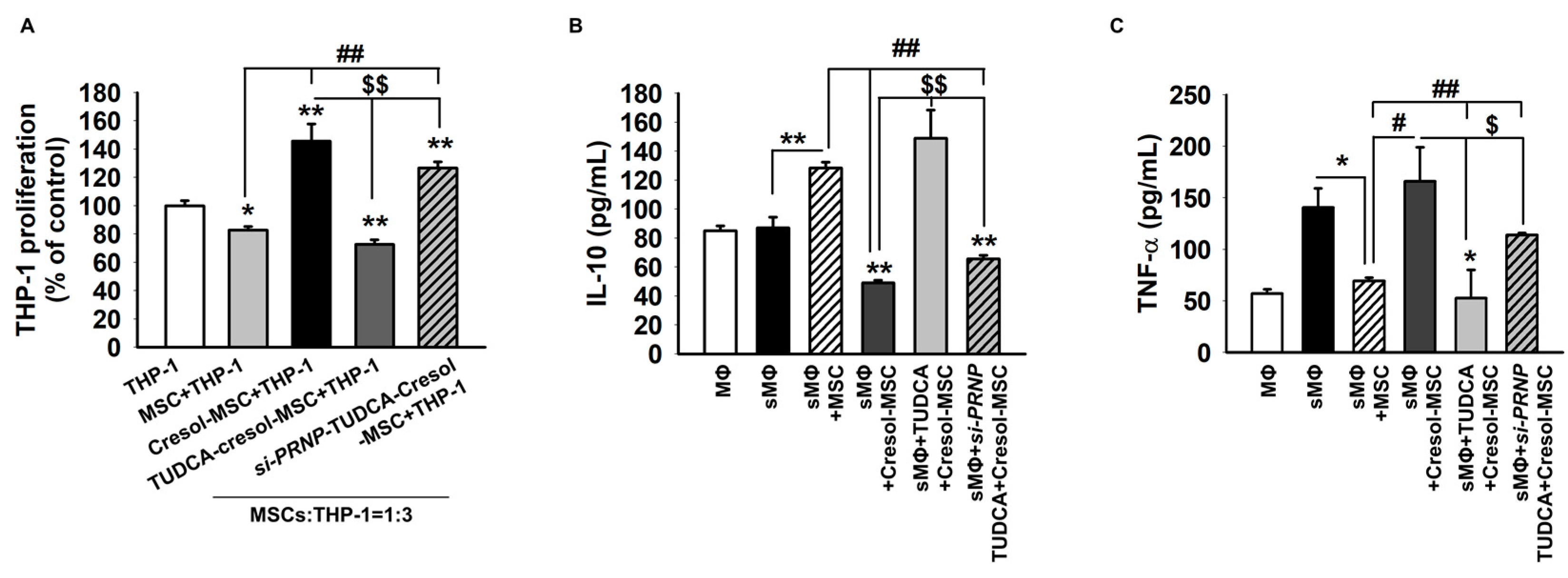
2.4. TUDCA Prevents PC-Induced Inflammation via the Expression of PrPC
3. Discussion
4. Materials and Methods
4.1. MSC Culture
4.2. Chemical Treatments
4.3. Dihydroethidium (DHE) Staining
4.4. Western Blot Assay
4.5. Inhibition of PrPC Expression by RNA Interference
4.6. SOD Activity
4.7. Catalase Activity
4.8. Annexin V/PI Assay and Flow Cytometry-Based Analysis
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- D’Hooge, R.; Van de Vijver, G.; Van Bogaert, P.P.; Marescau, B.; Vanholder, R.; De Deyn, P.P. Involvement of voltage- and ligand-gated Ca2+ channels in the neuroexcitatory and synergistic effects of putative uremic neurotoxins. Kidney Int. 2003, 63, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, S. Role of uremic toxin indoxyl sulfate in the progression of cardiovascular disease. Life Sci. 2017, 185, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Osonoi, T.; Terauchi, Y. Relationship between urinary sodium excretion and pioglitazone-induced edema. J. Diabetes Investig. 2010, 1, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Neirynck, N.; Glorieux, G. An update on protein-bound uremic retention solutes. J. Ren. Nutr. 2012, 22, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Cao, W.; Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 2014, 15, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Chen, X.; Dong, F.; Li, W.; Ren, X.; Zhang, Y.; Shi, Y. Concise review: Mesenchymal stem cells and translational medicine: Emerging issues. Stem Cells Transl. Med. 2012, 1, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Yue, Y.; Dong, N.; Xiong, S. Endoplasmic Reticulum Stress Aggravates Viral Myocarditis by Raising Inflammation Through the IRE1-Associated NF-kappaB Pathway. Can. J. Cardiol. 2015, 31, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Dell, A.; Morris, H.R. Glycoprotein structure determination by mass spectrometry. Science 2001, 291, 2351–2356. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Tsenkova, R.N.; Iordanova, I.K.; Toyoda, K.; Brown, D.R. Prion protein fate governed by metal binding. Biochem. Biophys. Res. Commun. 2004, 325, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, R.M.; Viana, R.J.; Low, W.C.; Steer, C.J.; Rodrigues, C.M. Bile acids and apoptosis modulation: An emerging role in experimental Alzheimer’s disease. Trends Mol. Med. 2008, 14, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cao, Y.; Yang, X.; Shan, P.; Liu, H. Tauroursodeoxycholic acid suppresses endoplasmic reticulum stress in the chondrocytes of patients with osteoarthritis. Int. J. Mol. Med. 2015, 36, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Sawitza, I.; Kordes, C.; Gotze, S.; Herebian, D.; Haussinger, D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci. Rep. 2015, 5, 13320. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, J.; Konopka, C.J.; Schuh, S.; Bouvin, H.; Cole, J.A.; Huntsman, H.D.; Kilian, K.A.; Dobrucki, I.T.; Boppart, M.D.; Dobrucki, L.W. Multimodal Assessment of Mesenchymal Stem Cell Therapy for Diabetic Vascular Complications. Theranostics 2017, 7, 3876–3888. [Google Scholar] [CrossRef] [PubMed]
- Despeyroux, A.; Duret, C.; Gondeau, C.; Perez-Gracia, E.; Chuttoo, L.; de Boussac, H.; Briolotti, P.; Bony, C.; Noel, D.; Jorgensen, C.; et al. Mesenchymal stem cells seeded on a human amniotic membrane improve liver regeneration and mouse survival after extended hepatectomy. J. Tissue Eng. Regener. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.H.; Mo, X.M.; Han, Z.C.; Zhou, B. Stem cell engraftment and survival in the ischemic heart. Ann. Thorac. Surg. 2011, 92, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, J.T., Jr.; Dalboni, M.A.; Watanabe, R.; Peres, A.T.; Goes, M.A.; Manfredi, S.R.; Canziani, M.E.; Cendoroglo, G.S.; Guimaraes-Souza, N.; Batista, M.C.; et al. Effects of spermidine and p-cresol on polymorphonuclear cell apoptosis and function. Artif. Organs 2011, 35, E27–E32. [Google Scholar] [CrossRef] [PubMed]
- Brocca, A.; Virzi, G.M.; de Cal, M.; Cantaluppi, V.; Ronco, C. Cytotoxic effects of p-cresol in renal epithelial tubular cells. Blood Purif. 2013, 36, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Cerini, C.; Brunet, P.; Guilianelli, C.; Moal, V.; Grau, G.; De Smet, R.; Vanholder, R.; Sampol, J.; Berland, Y. P-cresol, a uremic toxin, decreases endothelial cell response to inflammatory cytokines. Kidney Int. 2002, 62, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.P.; Sui, X.L.; Zhang, A.S.; Ye, L.; Gu, F.J.; Chen, J.H. Monocytes, endoplasmic reticulum stress and metabolomics in dogs with multiple organ dysfunction syndrome treated by continuous venovenous hemodiafiltration. Oncotarget 2017, 8, 34992–35008. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis Int. J. Prog. Cell Death 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bahr, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Lee, J.H.; Jung, J.S.; Noh, H.; Baek, M.J.; Ryu, J.M.; Yoon, Y.M.; Han, H.J.; Lee, S.H. Fucoidan protects mesenchymal stem cells against oxidative stress and enhances vascular regeneration in a murine hindlimb ischemia model. Int. J. Cardiol. 2015, 198, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.S.; Jeong, S.O.; Pae, H.O.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Takahashi, T.; Niinuma, Y.; Nomura, Y. Manganese superoxide dismutase is induced by endoplasmic reticulum stress through IRE1-mediated nuclear factor (NF)-kappaB and AP-1 activation. Biol. Pharm. Bull. 2004, 27, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.L.; Lebeau, J.; Guillaumot, P.; Petrilli, V.; Malek, M.; Chilloux, J.; Fauvet, F.; Payen, L.; Kfoury, A.; Renno, T.; et al. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol. Cell 2013, 49, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Grechowa, I.; Horke, S.; Wallrath, A.; Vahl, C.F.; Dorweiler, B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J. 2017, 31, 3868–3881. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, S.P.; Yoon, Y.M.; Lee, J.H.; Kook, M.; Han, Y.-S.; Jung, S.K.; Lee, S.H. Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein. Int. J. Mol. Sci. 2018, 19, 352. https://doi.org/10.3390/ijms19020352
Yun SP, Yoon YM, Lee JH, Kook M, Han Y-S, Jung SK, Lee SH. Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein. International Journal of Molecular Sciences. 2018; 19(2):352. https://doi.org/10.3390/ijms19020352
Chicago/Turabian StyleYun, Seung Pil, Yeo Min Yoon, Jun Hee Lee, Minjee Kook, Yong-Seok Han, Seo Kyung Jung, and Sang Hun Lee. 2018. "Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein" International Journal of Molecular Sciences 19, no. 2: 352. https://doi.org/10.3390/ijms19020352