Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer


1
Department of Surgery, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105, USA
2
Molecular Oncology Laboratories, Department of Oncology, Weatherall Institute of Molecular Medicine, University of Oxford, Oxford OX3 9DS, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(1), 240; https://doi.org/10.3390/ijms19010240
Submission received: 19 December 2017
/
Revised: 8 January 2018
/
Accepted: 9 January 2018
/
Published: 13 January 2018
(This article belongs to the Special Issue Molecular Pathways of Estrogen Receptor Action)
{kind=link}
{kind=link}
Abstract
:Hormones play an important role in pathophysiology. The hormone receptors, such as estrogen receptor alpha and androgen receptor in breast cancer and prostate cancer, are critical to cancer cell proliferation and tumor growth. In this review we focused on the cross-talk between hormone and hypoxia pathways, particularly in breast cancer. We delineated a novel signaling pathway from estrogen receptor to hypoxia-inducible factor 1, and discussed the role of this pathway in endocrine therapy resistance. Further, we discussed the estrogen and hypoxia pathways converging at histone demethylase KDM4B, an important epigenetic modifier in cancer.
1. Introduction
A solid tumor is a heterogeneous mass that is comprised of not only genetically and epigenetically distinct clones, but also of areas with varying degree of hypoxia that result from rapid cancer cell proliferation that outgrows its blood supply. To survive in hostile hypoxic environments, cancer cells decelerate their proliferation rate, alter metabolism and cellular pH, and induce angiogenesis [1]. These responses of cells to hypoxia are largely coordinated by hypoxia-inducible factors HIF-1 and HIF-2 [2], which drive the expression of a plethora of target genes, such as vascular endothelial growth factor (VEGF), carbonic anhydrase IX (CA9), and glucose transporter 1 (Glut-1) [2]. HIF is a heterodimer composed of an alpha subunit (HIF-1α or HIF-2α) and a β subunit (HIF-1β) [3]. HIF-1β is constitutively expressed, whereas HIF-α is regulated by oxygen availability. Under conditions of normoxia, HIF-α is hydroxylated at conserved prolyl residues by oxygen-dependent prolyl hydroxylases (PHDs), resulting in binding of the von Hippel-Lindau protein (VHL), an E3 ubiquitin ligase, for ubiquitin-mediated degradation [4,5,6]. Under conditions of hypoxia, however, HIFα is stabilized through inhibition of hydroxylation, leading to transactivation of its target genes. Preclinical and clinical studies show that the hypoxia/HIF-1 pathway plays an important role in promoting local tumor invasion and distal metastasis, as well as negatively influencing the responses to radiotherapy and chemotherapy [2,7,8,9,10,11]. The hypoxia/HIF-1 pathway is also involved in immunosuppression and resistance to immunotherapy [12]. In addition to oxygen-mediated degradation of HIFα through VHL, the hypoxia signaling pathway is regulated by a variety of oncogenes (e.g., ERK [13], HER2 [14], mTOR [15], Ras [16,17]) tumor suppressors (e.g., LKB1 [18], PML [19], PTEN [20,21], p53 [22], SDHB [23]), and metabolites (e.g., 2-hydroxyglutarate [24], succinate [25], and fumarate [26]). Here, we focus on the cross-talk between hypoxia and estrogen-mediated pathways, which converge to regulate epigenetic modulators in breast cancer, one of the most common cancers with 450,000 deaths each year worldwide.
2. The Cross-Talk between Hypoxia and Estrogen
Fifty years ago, Mirand et al. reported that in rodents, estradiol cyclopentylpropionate (ECP) was able to inhibit erythropoiesis by suppressing the production of erythropoiesis stimulating factor (ESF), which is now known as erythropoietin (EPO) [27], a direct target of hypoxia inducible factor. A subsequent study in 1973 by Gordon et al. further confirmed that estrogen inhibits the production of EPO in female rats exposed to various degrees of hypoxia [28]. Paradoxically, estrogen increases splenic erythropoiesis that is accompanied with elevated plasma EPO levels [29]. Interestingly, the expression of EPO mRNA is stimulated by both estradiol and hypoxia in the mouse uterus [30], and the hypoxic induction of EPO requires the presence of estradiol [30]. In 1999, Ruohola et al. reported that estradiol caused an increase of the HIF target VEGF mRNA in MCF-7 breast cancer cells, which was blocked by antiestrogen ICI 182780, suggesting that the effect was mediated by the estrogen receptor [31]. Subsequent studies further demonstrated the dual regulation of VEGF by hypoxia and estrogen [32,33,34]. These data indicate that estrogen and hypoxia pathways are connected. A later study showed that 17-β estradiol attenuates the hypoxic induction of HIF-1α and EPO in Hep3B cells [35]. However, in estrogen receptor-positive breast cancer cells, estrogen induces activation of HIF-1α [34] and co-operates with hypoxia to regulate the expression of a subgroup of genes [36]. Estrogen receptor antagonists (e.g., tamoxifen, raloxifene, or bazedoxifene) all suppress HIF-1α protein accumulation in osteoclast precursor cells [37]. Therefore, estrogen-mediated signaling can either negatively or positively affect the hypoxia pathway in different cellular contexts.
Estrogen receptor alpha (ERα) is an estrogen-dependent nuclear transcription factor that is not only critical for mammary epithelial cell division, but also breast cancer progression [38,39]. Despite the multiple molecular subtypes that have been classified based on transcriptomic and genetic features [40], ERα is one of the most important biomarkers directing breast cancer treatment. It is recommended that all patients with ERα positivity should have adjuvant endocrine therapy. ERα is expressed in approximately 70% of breast tumors [41], the majority of which depend on estrogen signaling, thereby providing the rationale for using anti-estrogens as adjuvant therapy to treat breast cancer [42]. Endocrine therapy drugs for breast cancer include selective ER modulators, such as tamoxifen, antagonists such as fulvestrant, and aromatase inhibitors such as anastrozole. Tamoxifen is a first-generation selective ER modulator (SERM) and has been widely used in breast cancer prevention and treatment [42]. It antagonizes ERα function in breast cancer cells by competing with estrogen for ERα binding while preserving its activating and estrogen-like functions in the bone [43]. Although now replaced by aromatase inhibitors (AI) as first-line treatment in post-menopausal women, tamoxifen still remains important in premenopausal breast cancer and after failure of AIs. The antagonist fulvestrant leads to ERα protein degradation [44], while aromatase inhibitors block the conversion of androgens to estrogens thereby reducing overall estrogen levels [45]. The application of endocrine therapies has led to a significant reduction in breast cancer mortality [46]. However, not all ER-positive patients respond to endocrine therapies and nearly all women with advanced cancer will eventually die from metastatic disease [47,48], as resistance often develops [49]. Many mechanisms have been proposed to account for endocrine therapy resistance [50,51], including loss of ERα expression or expression of truncated ER isoforms, posttranslational modification of ERα, deregulation of ERα co-activators, and increased receptor tyrosine kinase signaling. Recent studies further indicate that somatic ERα mutation [52,53], as well as genomic amplification of distant ER response elements [54] could contribute to hormone therapy resistance. Hypoxia is also involved in endocrine therapy resistance. Clinical studies have shown that HIF-1α expression is associated with an aggressive phenotype of breast cancer, i.e., large tumor size, high grade, high proliferation rate, and lymph node metastasis [55]. Increased HIF-1α is also associated with ERα positivity [55], whilst HIF-1β, the partner of HIF-1α, has been shown to function as a potent co-activator of ER-dependent transcription [56]. Importantly, HIF-1α protein expression was associated with tamoxifen resistance in neoadjuvant, primary therapy of ERα-positive breast cancers [57], as well as resistance to chemoendocrine therapy [58].
The exact nature of the relationship between hypoxia and estrogen pathways was a puzzle until our recent findings showing that the HIF-1α gene is a direct target of ERα [59]. In this study, we analyzed the global gene expression profile in response to hypoxia and the ERα antagonist fulvestrant and found a subgroup of genes that were dually responsive to the hormone and to oxygen. These genes were upregulated by hypoxia but the ERα antagonist fulvestrant significantly reduced their expression. These data were consistent with previous studies that showed some genes, such as KDM4B, STC2, and VEGF, bear both a hypoxia response element and estrogen response element [60,61,62,63,64,65,66]. Most interestingly, we found that ERα signaling directly regulates HIF-1α expression. When MCF7 cells were grown without estrogen and then placed in hypoxia or treated with the hypoxia mimetic deferoximine, estradiol greatly enhanced HIF-1α expression and this was reversed by fulvestrant and ERα depletion. By analyzing the HIF-1α genomic sequence that bears 15 exons and 14 introns, we identified a canonical estrogen response element (ERE) located in the first intron (Figure 1A). Interestingly, there is also a FOXA1 binding site that is 64 nucleotides downstream of ERE, further supporting it as a bona fide ERα binding element, because FOXA1 is a pioneer factor that facilitates ERα recruitment [67]. Actually, one study has shown that overexpression of FOXA1 in ER-positive breast cancer cell lines promotes resistance to tamoxifen and to estrogen deprivation [68]. We further validated our findings by chromatin immunoprecipitation-PCR and a luciferase reporter assay, showing that ERα directly binds to this locus, driving HIF-1α gene expression. This finding not only explains the early findings that estrogen and hypoxia pathways crosstalk, but also indicates that overactive HIF-1α function may partially compensate for estrogen signaling when ERα function is compromised, such as in the circumstance of hormone therapy, leading to hormone therapy resistance (Figure 1B,C).
3. The Hypoxia and Estrogen Signaling Pathways Converge on Histone Demethylases
Genetic abnormalities that drive tumorigenesis are usually coupled with epigenetic alterations that engage multiple important biological processes such as DNA replication, DNA repair, and gene expression [69,70,71,72,73]. One such aberrant chromatin modification is histone lysine methylation [69,70], which was believed to be irreversible until the discovery of lysine-specific demethylase 1 (LSD1) [74]. Subsequent studies identified another family of histone demethylases, the Jumonji C (JmjC) domain–containing demethylases [75], which require iron and 2-oxoglutarate (2-OG) for their activities. The JmjC histone lysine demethylase family (KDMs) is composed of 17 members and is responsible for reversing most of the histone methyl marks in the human genome. Dysregulated histone lysine methylation is commonly seen in various cancers [76], which is consistent with observed genetic alterations and/or dysregulation of histone methyltransferases and KDMs [75,77,78,79,80,81]. Interestingly, we and others have shown that many JmjC histone demethylases are hypoxia-inducible [61,82], some of which, including KDM3A [60,61,83], KDM4B [60,61,63], and KDM4C [84], are direct targets of HIFs.
The KDM4 subfamily of histone demethylases consists of four members. KDM4A, KDM4B, and KDM4C share high sequence homology in their catalytic domains, and they remove methyl groups from H3K9me2/me3 and H3K36me2/me3 [75]. KDM4A-4C members also bear other similar functional domains that include two PHD domains and two Tudor domains. However, KDM4D is less conserved and removes methyl groups only from H3K9me2/me3. KDM4B plays important roles in the self-renewal of embryonic stem cells and the conversion of induced pluripotent stem cells [85,86], and is linked to many forms of cancer [87]. KDM4B is amplified in medulloblastoma [88] and malignant peripheral nerve sheath tumors [89], and is overexpressed in many other cancers [90,91,92]. KDM4B regulates the expression of key oncogenes, such as C-MYC [93,94,95,96] and CDK6 [97], and is involved in cancer invasiveness, metastasis, and therapeutic resistance [98,99,100]. Interestingly, KDM4B is a direct target of p53, exerting its DNA repair function in response DNA damage [101,102,103]. Recently, we showed that KDM4B is involved in neuroblastoma growth and tumor maintenance [104]. The expression of KDM4B was highly correlated with that of the MYCN oncogene in neuroblastoma, and it formed a complex with N-Myc protein, thereby facilitating its function by maintaining low levels of repressive H3K9me2/me3 marks at Myc-binding sites. In breast cancer we have shown that HIF-1α and ERα can coordinate expression of genes, such as KDM4B, whose expression is driven by both ERα and HIF-1α and epigenetically regulates the G2/M phase of cell cycle progression in breast cancer cells [63] and other cancer cell lines (unpublished data), as the expression of several key cell cycle genes is correlated with changes in the KDM4B substrate, H3K9me3 [63,104]. Similar to other dual responsive genes such as VEGF and STC2 that are regulated by both estrogens and hypoxia, the genomic locus of KDM4B bears both HIF-1α and ERα binding elements [60,63] (Figure 2A). The cross-talk between HIF-1α and ERα converges at KDM4B, which is important for cell cycle progression and tumor growth in ER positive breast cancer [63,93]. Importantly, in endocrine therapy-resistant breast cancer cells, the regulation of KDM4B by HIF-1α and ERα is intact and KDM4B is still required for G2/M phase progression [59] (Figure 2B). In addition, KDM4B is not only required for enhancing androgen receptor (AR) transcriptional activity through histone modification, but it also enhances AR protein stability via inhibition of AR ubiquitination [105], demonstrating the functional connection between AR and KDM4B in prostate cancer. Therefore, HIF-1α plays an important role in modulating anti-androgen responses via KDM4B in prostate cancer.
4. Future Prospects
Many transcriptional factors, such as Myc, ERα, and AR, exert oncogenic functions to drive cancer cell proliferation. Directly targeting these oncogenic transcription factors is either technically challenging or leads to therapeutic resistance. Therefore, new approaches need to be developed to overcome these obstacles. Transcription factors need to complex with other cofactors to drive gene expression and many of these cofactors are histone modifiers. Thus, development of small molecules to target the histone modifiers, such as KDM4B, may provide an opportunity to enhance the efficacy of standard chemotherapeutics or to overcome drug resistance. Recently, efforts have been made by us and other groups to identify and develop KDM4B inhibitors for cancer treatment [106,107,108,109]. By using a chemoinformatics in combination with high-content imaging approach we identified ciclopirox as a novel histone demethylase inhibitor. Ciclopirox targeted KDM4B, inhibited Myc signaling, resulting in suppression of neuroblastoma cell viability and tumor growth associated with an induction of differentiation [107]. We also found that MCF7 cells (ERα-positive) were much more sensitive to MDA-MB-231 cells (ERα-negative) (Jun Yang, St Jude Children’s Research Hospital, Memphis, TN, USA. unpublished data), suggesting ERα-positive breast cancer cells are more addicted to KDM4B. Chu et al. identified a KDM4B inhibitor that significantly blocked the viability of cultured prostate cancer cells, which was accompanied by transcriptional silencing of growth-related genes, a substantial portion of which were AR-responsive [106]. Recently, a more potent and selective KDM4 inhibitor was developed by Cellgene [108,109], which was efficacious in breast and colon cancer models. Although whether these KDM4B inhibitors are able to reverse endocrine therapy resistance needs to be tested, we believe specific and potent KDM4B inhibitors hold a promise for overcoming endocrine therapy resistance to breast cancer and prostate cancer.
Acknowledgments
Andrew M. Davidoff and Jun Yang was supported by the Assisi Foundation of Memphis, the American Lebanese Syrian Associated Charities (ALSAC), the US Public Health Service Childhood Solid Tumor Program Project grant no. CA23099, and the Cancer Center Support grant no. 21766 from the National Cancer Institute. Jun Yang was also supported by American Cancer Society-Research Scholar 130421-RSG-17-071-01-TBG, the National Cancer Institute of the National Institutes of Health under award number R03CA212802.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; El-Deiry, W.S. HIF-1 signaling in drug resistance to chemotherapy. Curr. Med. Chem. 2014, 21, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor hypoxia, drug resistance, and metastases. J. Natl. Cancer Inst. 1990, 82, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Giaccia, A.J. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006, 66, 10238–10241. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1α) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1α) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Pino, M.S.; Zeng, M.; Shirasawa, S.; Chung, D.C. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1α and -2α in colon cancer. Cancer Res. 2009, 69, 8499–8506. [Google Scholar] [CrossRef] [PubMed]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouyssegur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Vasquez, D.S.; Corbeil, J.; Wu, S.; Leblanc, M.; Wu, C.L.; Vera, D.R.; Shaw, R.J. mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 11137–11142. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Yang, J.; Ahmed, A.; Poon, E.; Perusinghe, N.; de Haven Brandon, A.; Box, G.; Valenti, M.; Eccles, S.; Rouschop, K.; Wouters, B.; et al. Small-molecule activation of p53 blocks hypoxia-inducible factor 1α and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol. Cell. Biol. 2009, 29, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mirand, E.A.; Gordon, A.S. Mechanism of estrogen action in erythropoiesis. Endocrinology 1966, 78, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Peschle, C.; Rappaport, I.A.; Sasso, G.F.; Condorelli, M.; Gordon, A.S. The role of estrogen in the regulation of erythropoietin production. Endocrinology 1973, 92, 358–362. [Google Scholar] [PubMed]
- Anagnostou, A.; Zander, A.; Barone, J.; Fried, W. Mechanism of the increased splenic erythropoiesis in mice treated with estradiol benzoate. J. Lab. Clin. Med. 1976, 88, 700–706. [Google Scholar] [PubMed]
- Yasuda, Y.; Masuda, S.; Chikuma, M.; Inoue, K.; Nagao, M.; Sasaki, R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J. Biol. Chem. 1998, 273, 25381–25387. [Google Scholar] [CrossRef] [PubMed]
- Ruohola, J.K.; Valve, E.M.; Karkkainen, M.J.; Joukov, V.; Alitalo, K.; Harkonen, P.L. Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol. Cell. Endocrinol. 1999, 149, 29–40. [Google Scholar] [CrossRef]
- Bausero, P.; Ben-Mahdi, M.; Mazucatelli, J.; Bloy, C.; Perrot-Applanat, M. Vascular endothelial growth factor is modulated in vascular muscle cells by estradiol, tamoxifen, and hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2033–H2042. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Sall, W.; Koch, C.J.; Oprysko, P.R.; Evans, S.M. Low pO2 and β-estradiol induce VEGF in MCF-7 and MCF-7-5C cells: Relationship to in vivo hypoxia. Breast Cancer Res. Treat. 2001, 67, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Koos, R.D. Estrogen-induced activation of hypoxia-inducible factor-1α, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology 2007, 148, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, H.; Kanagy, N.L.; Resta, T.C. 17-beta estradiol attenuates hypoxic induction of HIF-1α and erythropoietin in Hep3B cells. J. Cardiovasc. Pharmacol. 2004, 44, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Seifeddine, R.; Dreiem, A.; Tomkiewicz, C.; Fulchignoni-Lataud, M.C.; Brito, I.; Danan, J.L.; Favaudon, V.; Barouki, R.; Massaad-Massade, L. Hypoxia and estrogen co-operate to regulate gene expression in T-47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 104, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Sato, Y.; Iwasaki, R.; Kobayashi, T.; Watanabe, R.; Oike, T.; Miyamoto, K.; Toyama, Y.; Matsumoto, M.; Nakamura, M.; et al. Selective estrogen receptor modulators suppress Hif1α protein accumulation in mouse osteoclasts. PLoS ONE 2016, 11, e0165922. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Brown, M. Estrogen receptor target gene: An evolving concept. Mol. Endocrinol. 2006, 20, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Norris, J.D. Connections and regulation of the human estrogen receptor. Science 2002, 296, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Third annual William L. McGuire Memorial Lecture. "Studies on the estrogen receptor in breast cancer"–20 years as a target for the treatment and prevention of cancer. Breast Cancer Res. Treat. 1995, 36, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Jaiyesimi, I.A.; Buzdar, A.U.; Decker, D.A.; Hortobagyi, G.N. Use of tamoxifen for breast cancer: Twenty-eight years later. J. Clin. Oncol. 1995, 13, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Osborne, C.K.; Morris, C.; Wakeling, A.E. ICI 182,780 (Faslodex): Development of a novel, “pure” antiestrogen. Cancer 2000, 89, 817–825. [Google Scholar]
- Osborne, C.K. Aromatase inhibitors in relation to other forms of endocrine therapy for breast cancer. Endocr. Relat. Cancer 1999, 6, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Buzdar, A.; Cuzick, J.; Forbes, J.; Houghton, J.; Howell, A.; Sahmoud, T.; Group, A.T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: Results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer 2003, 98, 1802–1810. [Google Scholar] [PubMed]
- Massarweh, S.; Osborne, C.K.; Jiang, S.; Wakeling, A.E.; Rimawi, M.; Mohsin, S.K.; Hilsenbeck, S.; Schiff, R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006, 66, 8266–8273. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; DeFriend, D.; Robertson, J.; Blamey, R.; Walton, P. Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer. Lancet 1995, 345, 29–30. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Welch, J.N.; Skaar, T.C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 2001, 53, 25–71. [Google Scholar] [PubMed]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Hsu, H.K.; Lan, X.; Juan, L.; Yan, P.S.; Labanowska, J.; Heerema, N.; Hsiao, T.H.; Chiu, Y.C.; Chen, Y.; et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell 2013, 24, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1 α during breast carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Brunnberg, S.; Pettersson, K.; Rydin, E.; Matthews, J.; Hanberg, A.; Pongratz, I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 6517–6522. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Buffa, F.M.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Phosphorylated ERα, HIF-1α, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J. Clin. Oncol. 2009, 27, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin. Cancer Res. 2006, 12, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ledaki, I.; Turley, H.; Gatter, K.C.; Montero, J.C.; Li, J.L.; Harris, A.L. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y. Acad. Sci. 2009, 1177, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Southey, M.C.; Chang, A.C.; Reddel, R.R.; Willhite, D.; Glynne, R.; Henderson, M.A.; Armes, J.E.; Venter, D.J. Stanniocalcin 2 is an estrogen-responsive gene coexpressed with the estrogen receptor in human breast cancer. Cancer Res. 2002, 62, 1289–1295. [Google Scholar] [PubMed]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.D.; Vigne, J.L.; Minchenko, A.; Lebovic, D.I.; Leitman, D.C.; Taylor, R.N. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors α and β. Proc. Natl. Acad. Sci. USA 2000, 97, 10972–10977. [Google Scholar] [CrossRef] [PubMed]
- Law, A.Y.; Wong, C.K. Stanniocalcin-2 is a HIF-1 target gene that promotes cell proliferation in hypoxia. Exp. Cell Res. 2010, 316, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Hojfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Whetstine, J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 2007, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [PubMed]
- Wellmann, S.; Bettkober, M.; Zelmer, A.; Seeger, K.; Faigle, M.; Eltzschig, H.K.; Buhrer, C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 2008, 372, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; De Los Angeles, A.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Wu, Y.; Guo, L.; et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2012, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.G.; Brown-Kipphut, B.A.; Iqbal, A.; Scott, G.A. Microarray comparative genomic hybridization detection of copy number changes in desmoplastic melanoma and malignant peripheral nerve sheath tumor. Am. J. Dermatopathol. 2011, 33, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, M.; Saso, K.; Tong, K.I.; McQuire, T.; Goto, K.; Son, D.O.; Wakeham, A.; Miyagishi, M.; Mak, T.W.; Okada, H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE 2011, 6, e17830. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Kim, T.D.; Janknecht, R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int. J. Oncol. 2014, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.T.; Fan, Q.; Zhu, Z.; Kwan, S.Y.; Chen, L.; Chen, J.H.; Ying, Z.L.; Zhou, Y.; Gu, W.; Wang, L.H.; et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27kip1. Oncotarget 2015, 6, 31702–31720. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Cho, H.S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Bur, H.; Haapasaari, K.M.; Turpeenniemi-Hujanen, T.; Kuittinen, O.; Auvinen, P.; Marin, K.; Soini, Y.; Karihtala, P. Strong KDM4B and KDM4D Expression associates with radioresistance and aggressive phenotype in classical hodgkin lymphoma. Anticancer Res. 2016, 36, 4677–4683. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Zang, W.; Liu, Z.; Xu, X.; Yu, H.; Yang, Q.; Jia, J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with β-catenin and enhances gastric cancer metastasis. Clin. Cancer Res. 2013, 19, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, L.; Pledger, W.J.; Fang, J.; Chen, J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 2014, 33, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.H.; Wang, L.Y.; Hsu, K.C.; Chen, C.C.; Cheng, H.H.; Wang, S.M.; Wu, C.M.; Chen, T.J.; Li, L.T.; Liu, R.; et al. KDM4B as a target for prostate cancer: Structural analysis and selective inhibition by a novel inhibitor. J. Med. Chem. 2014, 57, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting histone demethylases in MYC-driven neuroblastomas with ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Stepputtis, S.S.; Strietz, J.; Preca, B.T.; Urban, S.; Willmann, D.; Allen, A.; Zenk, F.; Iovino, N.; Bronsert, P.; et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. 2017, 77, 5900–5912. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Bonaldi, T.; Cuomo, A.; del Rosario, J.R.; Hosfield, D.J.; Kanouni, T.; Kao, S.C.; Lai, C.; Lobo, N.A.; Matuszkiewicz, J.; et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med. Chem. Lett. 2017, 8, 869–874. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Estrogen pathway directly drives HIF-1α expression. (A) HIF-1α gene bears a canonical estrogen receptor binding element (ERE), with a FOXA1 binding site downstream of ERE; (B) When ERα is bound by its ligand it drives the expression of HIF-1α. However, ERα antagonists block the expression of HIF-1α; (C) The pathways mediated by hypoxia, estrogen, metabolites, and cancer genes converge on HIF-1α, which drives a plethora of genes that are involved in multiple biological processes, cancer progression, and therapeutic resistance.
Figure 1.
Estrogen pathway directly drives HIF-1α expression. (A) HIF-1α gene bears a canonical estrogen receptor binding element (ERE), with a FOXA1 binding site downstream of ERE; (B) When ERα is bound by its ligand it drives the expression of HIF-1α. However, ERα antagonists block the expression of HIF-1α; (C) The pathways mediated by hypoxia, estrogen, metabolites, and cancer genes converge on HIF-1α, which drives a plethora of genes that are involved in multiple biological processes, cancer progression, and therapeutic resistance.

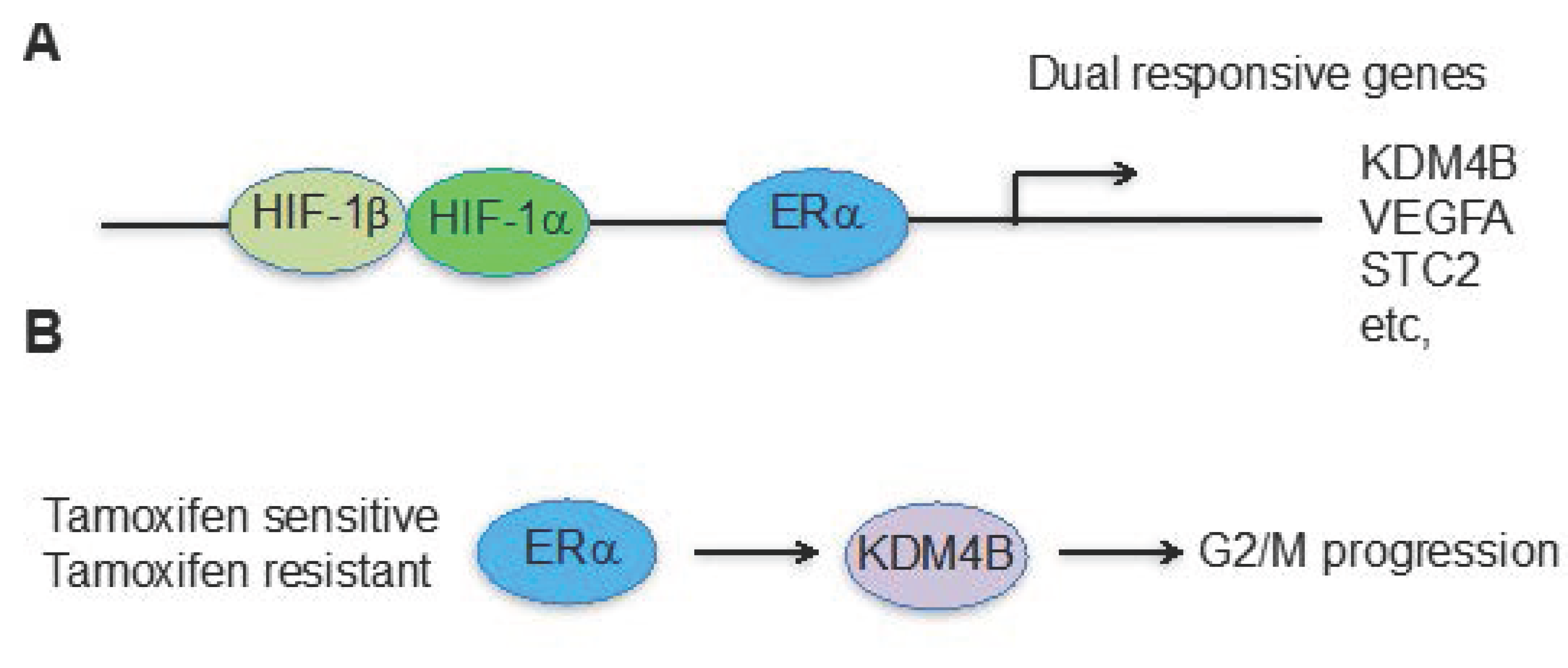
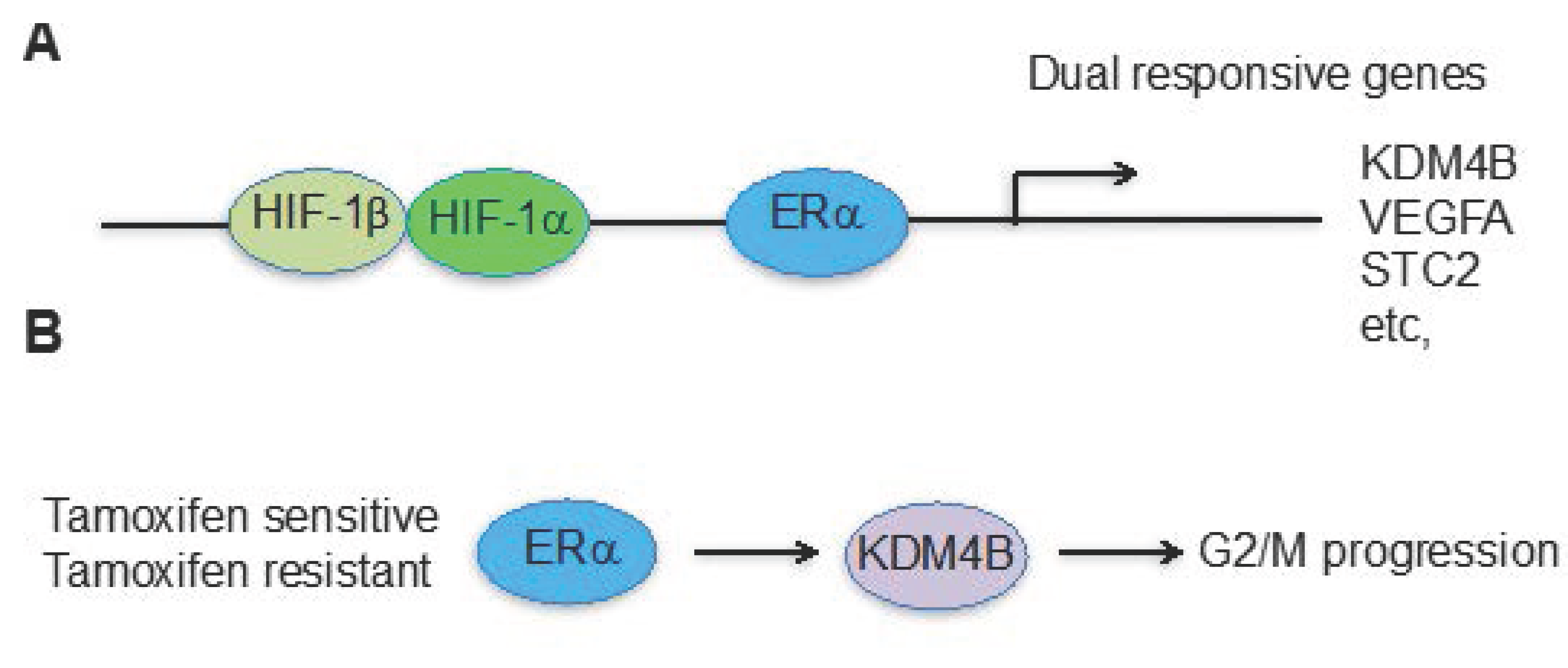
Figure 2.
Hypoxia and estrogen pathways converge at KDM4B for cancer cell proliferation in ERα positive breast cancer. (A) KDM4B is one of the genes responsive to both estrogen and hypoxia-mediated pathways; (B) Regardless of endocrine therapy resistance, ERα drives KDM4B expression, which is required for G2/M phase progression.
Figure 2.
Hypoxia and estrogen pathways converge at KDM4B for cancer cell proliferation in ERα positive breast cancer. (A) KDM4B is one of the genes responsive to both estrogen and hypoxia-mediated pathways; (B) Regardless of endocrine therapy resistance, ERα drives KDM4B expression, which is required for G2/M phase progression.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. https://doi.org/10.3390/ijms19010240
AMA Style
Yang J, Harris AL, Davidoff AM. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. International Journal of Molecular Sciences. 2018; 19(1):240. https://doi.org/10.3390/ijms19010240
Chicago/Turabian StyleYang, Jun, Adrian L. Harris, and Andrew M. Davidoff. 2018. "Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer" International Journal of Molecular Sciences 19, no. 1: 240. https://doi.org/10.3390/ijms19010240
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.