
The Obesity-Breast Cancer Conundrum: An Analysis of the Issues


Abstract
:
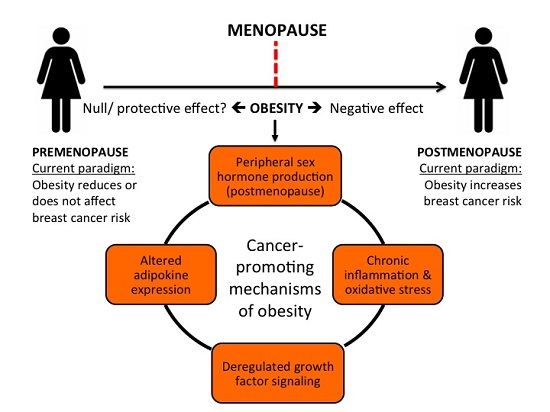{kind=link}
1. Overview
2. Breast Cancer and Obesity: What Is the Connection?
3. Menopausal Status, Obesity, and Breast Cancer
3.1. Premenopausal Breast Cancer
3.2. Postmenopausal Breast Cancer
4. Obesity Negatively Influences Breast Cancer Outcomes
5. Other Mechanisms Underlying the Effects of Obesity on Breast Carcinogenesis
6. Deregulated Insulin Signaling in Obesity and Cancer
- (1)
- The difficulty in measuring circulating levels of bioavailable IGF-1, as no validated assays are currently available;
- (2)
- Uncertainty relative to the usefulness of reported levels of serum insulin, which are highly dependent on the state and duration of fasting, assay characteristics and genetic factors;
- (3)
- Possible reporting bias for studies describing significant associations between either insulin or IGF-1 and cancer risk [54];
- (4)
- (5)
- Concern that the extensive preclinical literature on supraphysiological levels of insulin on tumor development are not applicable to humans [57]; and
- (6)
7. Chronic Inflammation as a Permissive Environment for Breast Carcinogenesis
8. Altered Adipose Tissue Endocrine Function in Obesity Has Implications on Breast Cancer
8.1. Leptin and Breast Cancer
8.2. Adiponectin and Breast Cancer
8.3. Other Considerations
9. The Multiple-Hits Hypothesis for Carcinogenesis
10. Effects of Obesity on Other Mechanisms of Carcinogenesis: Relevance of the Cancer Hallmarks
11. Preclinical Models: What Tools Are Available?
12. Rodent Models of Obesity and Breast Cancer
12.1. Monogenic Models of Obesity
12.2. Polygenic Mouse Models of Obesity
12.3. Polygenic Rat Models of Obesity
12.4. Mammary Carcinogenesis
13. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures 2012; American Cancer Society: Boston, MA, USA, 2012. [Google Scholar]
- Economopoulou, P.; Dimitriadis, G.; Psyrri, A. Beyond BRCA: New hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015, 41, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Wiseman, M. The second World Cancer Research Fund/American Institute for Cancer Research expert report. Food, nutrition, physical activity, and the prevention of cancer: A global perspective. Proc. Nutr. Soc. 2008, 67, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Overweight and Obesity Statistics. NIH Publication No. 04-4158. Available online: http://win.niddk.nih.gov/publications/PDFs/stat904z.pdf (accessed on 15 August 2013).
- Ford, E.S.; Li, C.; Zhao, G.; Tsai, J. Trends in obesity and abdominal obesity among adults in the United States from 1999–2008. Int. J. Obes. 2011, 35, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Anderson, A.S.; Clarke, R.B.; Duffy, S.W.; Evans, D.G.; Garcia-Closas, M.; Gescher, A.J.; Key, T.J.; Saxton, J.M.; Harvie, M.N. Risk determination and prevention of breast cancer. Breast Cancer Res. 2014, 16, 446. [Google Scholar] [CrossRef] [PubMed]
- Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective. Available online: http://www.dietandcancerreport.org/expert_report/report_contents/index.php (accessed on 15 August 2013).
- Berstad, P.; Coates, R.J.; Bernstein, L.; Folger, S.G.; Malone, K.E.; Marchbanks, P.A.; Weiss, L.K.; Liff, J.M.; McDonald, J.A.; Strom, B.L.; et al. A case-control study of body mass index and breast cancer risk in white and African-American women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Cheraghi, Z.; Poorolajal, J.; Hashem, T.; Esmailnasab, N.; Irani, A.D. Effect of Body Mass Index on Breast Cancer during Premenopausal and Postmenopausal Periods: A Meta-Analysis. PLoS ONE 2012, 7, e51446. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.L.; Neuhouser, M.L. Obesity and the Risk for Premenopausal and Postmenopausal Breast Cancer. Cancer Prev. Res. 2012, 5, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.S.; Costantino, J.P.; Cauley, J.A.; Cronin, W.M.; Wickerham, D.L.; Land, S.R.; Weissfeld, J.L.; Wolmark, N. Body Mass Index and the Risk for Developing Invasive Breast Cancer among High-Risk Women in NSABP P-1 and STAR Breast Cancer Prevention Trials. Cancer Prev. Res. 2012, 5, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Whiteman, M.K.; Flaws, J.A.; Langenberg, P.; Tkaczuk, K.H.; Bush, T.L. Body mass and stage of breast cancer at diagnosis. Int. J. Cancer 2002, 98, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G. Obesity and cancer in Asia-Pacific populations. Lancet Oncol. 2010, 11, 704–705. [Google Scholar] [CrossRef]
- Huang, Z.; Hankinson, S.E.; Colditz, G.A.; Stampfer, M.J.; Hunter, D.J.; Manson, J.E.; Hennekens, C.H.; Rosner, B.; Speizer, F.E.; Willett, W.C. Dual effects of weight and weight gain on breast cancer risk. JAMA 1997, 278, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; van Noord, P.A.H.; den Tonkelaar, I.; Peeters, P.H.M.; Riboli, E.; Grobbee, D.E. Breast-cancer incidence in relation to height, weight and body-fat distribution in the Dutch “DOM” cohort. Int. J. Cancer 1998, 76, 647–651. [Google Scholar] [CrossRef]
- Lahmann, P.H.; Hoffmann, K.; Allen, N.; van Gils, C.H.; Khaw, K.T.; Tehard, B.; Berrino, F.; Tjonneland, A.; Bigaard, J.; Olsen, A.; et al. Body size and breast cancer risk: Findings from the European Prospective Investigation into Cancer And Nutrition (EPIC). Int. J. Cancer 2004, 111, 762–771. [Google Scholar] [PubMed]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Spiegelman, D.; Barbieri, R.L.; Dowsett, M.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 2006, 98, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Rinaldi, S.; Key, T.J.; Berrino, F.; Peeters, P.H.M.; Biessy, C.; Dossus, L.; Lukanova, A.; Binghan, S.; Khaw, K.T.G.; et al. Postmenopausal serum androgens, oestrogens and breast cancer risk: The European prospective investigation into cancer and nutrition. Endocr. Relat. Cancer 2005, 12, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Orsini, N.; Saji, S.; Key, T.J.; Wolk, A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status-A meta-analysis. Int. J. Cancer 2009, 124, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Schatzkin, A.; Lacey, J.V.; Albanes, D.; Ballard-Barbash, R.; Adams, K.F.; Kipnis, V.; Mouw, T.; Hollenbeck, A.R.; Leitzmann, M.F. Adiposity, adult weight change, and postmenopausal breast cancer risk. Arch. Intern. Med. 2007, 167, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Feigelson, H.S.; Patel, A.V.; Teras, L.R.; Gansler, T.; Thun, M.J.; Calle, E.E. Adult weight gain and histopathologic characteristics of breast cancer among postmenopausal women. Cancer 2006, 107, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.R.; Adams-Campbell, L.L.; Boggs, D.A.; Wise, L.A.; Rosenberg, L. A prospective study of body size and breast cancer in black women. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.D.; Cerhan, J.R.; Sellers, T.A.; Mcgovern, P.G.; Drinkard, C.; Kushi, L.R.; Folsom, A.R. Progesterone and Estrogen-Receptors and Mammary Neoplasia in the Iowa Womens Health Study—How Many Kinds of Breast-Cancer Are There. Cancer Epidemiol. Biomark. Prev. 1995, 4, 319–326. [Google Scholar]
- Suzuki, R.; Rylander-Rudqvist, T.; Ye, W.M.; Saji, S.; Wolk, A. Body weight and postmenopausal breast cancer risk defined by estrogen and progesterone receptor status among Swedish women: A prospective cohort study. Int. J. Cancer 2006, 119, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; Mccann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II Endometrial Cancers: Have They Different Risk Factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Ritte, R.; Lukanova, A.; Berrino, F.; Dossus, L.; Tjonneland, A.; Olsen, A.; Overvad, T.F.; Overvad, K.; Clavel-Chapelon, F.; Fournier, A.; et al. Adiposity, hormone replacement therapy use and breast cancer risk by age and hormone receptor status: A large prospective cohort study. Breast Cancer Res. 2012, 14, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, A.I.; Chlebowski, R.T.; Prentice, R.; McTiernan, A.; Stefanick, M.L.; Wactawski-Wende, J.; Kuller, L.H.; Adams-Campbell, L.L.; Lane, D.; Vitolins, M.; et al. Body Size, Physical Activity, and Risk of Triple-Negative and Estrogen Receptor-Positive Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.; Dorgan, J.F.; Longcope, C.; Stanczyk, F.Z.; Stephenson, H.E.; Falk, R.T.; Miller, R.; et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl. Cancer Inst. 2003, 95, 1218–1226. [Google Scholar] [PubMed]
- Biglia, N.; Peano, E.; Sgandurra, P.; Moggio, G.; Pecchio, S.; Maggiorotto, F.; Sismondi, P. Body mass index (BMI) and breast cancer: Impact on tumor histopathologic features, cancer subtypes and recurrence rate in pre and postmenopausal women. Gynecol. Endocrinol. 2013, 29, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Milne, R.L.; Friedlander, M.L.; McCredie, M.R.E.; Giles, G.G.; Hopper, J.L.; Phillips, K.A. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, A.R. Obesity and prognosis of breast cancer. Obes. Rev. 2006, 7, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewertz, M.; Jensen, M.; Gunnarsdottir, K.A.; Hojris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of obesity on prognosis after early-stage breast cancer. J. Clin. Oncol. 2011, 29, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.T.; Ibrahim, J.G.; Stevens, J.; Cleveland, R.; Abrahamson, P.E.; Satia, J.A.; Teitelbaum, S.L.; Neugut, A.I.; Gammon, M.D. Postdiagnosis change in bodyweight and survival after breast cancer diagnosis. Epidemiology 2012, 23, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Finer, N. Redefining type 2 diabetes: ‘Diabesity’ or ‘obesity dependent diabetes mellitus’? Obes. Rev. 2000, 1, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, M.E.; Fantus, I.G.; Ezzat, S.; McKeown-Eyssen, G.; Page, D.; Goodwin, P.J. Insulin and related factors in premenopausal breast cancer risk. Breast Cancer Res. Treat. 1998, 47, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. Insulin-like growth factor-I and binding protein-3 and risk of cancer. Horm. Res. 1999, 51 (Suppl. 3), 34–41. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin Resistance and Cancer Risk: An Overview of the Pathogenetic Mechanisms. Exp. Diabetes Res. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Leitner, J.W.; Solomon, S.; Golovchenko, I.; Goalstone, M.L.; Draznin, B. Effect of insulin on cell cycle progression in MCF-7 breast cancer cells—Direct and potentiating influence. J. Biol. Chem. 2001, 276, 38023–38028. [Google Scholar] [PubMed]
- Creighton, C.J.; Casa, A.; Lazard, Z.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Lee, A.V. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J. Clin. Oncol. 2008, 26, 4078–4085. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Krishnaswamy, G.; Karnad, A.; Peiris, A.N. Insulin: A novel factor in carcinogenesis. Am. J. Med. Sci. 2002, 323, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: Pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010, 11, 530–542. [Google Scholar] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Vona-Davis, L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr. Relat. Cancer 2012, 19, R225–R241. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Calori, G.; Lattuada, G.; Piemonti, L.; Garancini, M.P.; Ragogna, F.; Villa, M.; Mannino, S.; Crosignani, P.; Bosi, E.; Luzi, L.; et al. Prevalence, metabolic features, and prognosis of metabolically healthy obese Italian individuals: The Cremona Study. Diabetes Care 2011, 34, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Papatheodorou, S.I.; Evangelou, E.; Ioannidis, J.P.A. Evaluation of Excess Statistical Significance in Meta-analyses of 98 Biomarker Associations with Cancer Risk. J. Natl. Cancer Inst. 2012, 104, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Badrick, E.; Renehan, A.G. Diabetes and cancer: 5 years into the recent controversy. Eur. J. Cancer 2014, 50, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, L.; Yakubovich, N.; Dagenais, G.R.; Rosenstock, J.; Probstfield, J.; Yu, P.C.; Ryden, L.E.; Pirags, V.; Spinas, G.A.; Birkeland, K.I.; et al. The Association of Basal Insulin Glargine and/or n-3 Fatty Acids with Incident Cancers in Patients with Dysglycemia. Diabetes Care 2014, 37, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Leroith, D. Can endogenous hyperinsulinaemia explain the increased risk of cancer development and mortality in type 2 diabetes: evidence from mouse models. Diabetes Metab. Res. Rev. 2010, 26, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Harvie, M.; Howell, A. Insulin-like growth factor (IGF)-I, IGF binding protein-3 and breast cancer risk: Eight years on. Endocr. Relat. Cancer 2006, 13, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Cottam, D.R.; Mattar, S.G.; Barinas-Mitchell, E.; Eid, G.; Kuller, L.; Kelley, D.E.; Schauer, P.R. The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: Implications and effects of weight loss. Obes. Surg. 2004, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar] [PubMed]
- Allin, K.H.; Nordestgaard, B.G. Elevated C-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit. Rev. Clin. Lab. Sci. 2011, 48, 155–170. [Google Scholar] [CrossRef]
- Bochud, M.; Marquant, F.; Marques-Vidal, P.M.; Vollenweider, P.; Beckmann, J.S.; Mooser, V.; Paccaud, F.; Rousson, V. Association between C-reactive protein and adiposity in women. J. Clin. Endocrinol. Metab. 2009, 94, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Dossus, L.; Jimenez-Corona, A.; Romieu, I.; Boutron-Ruault, M.C.; Boutten, A.; Dupre, T.; Fagherazzi, G.; Clavel-Chapelon, F.; Mesrine, S. C-reactive protein and postmenopausal breast cancer risk: Results from the E3N cohort study. Cancer Causes Control 2014, 25, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, K.; Harris, R.; Lowe, G.; Rumley, A.; Yarnell, J.; Gallacher, J.; Ben-Shlomo, Y.; Ebrahim, S.; Lawlor, D.A. Associations of circulating C-reactive protein and interleukin-6 with cancer risk: Findings from two prospective cohorts and a meta-analysis. Cancer Causes Control 2009, 20, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Il’yasova, D.; Colbert, L.H.; Harris, T.B.; Newman, A.B.; Bauer, D.C.; Satterfield, S.; Kritchevsky, S.B. Circulating levels of inflammatory markers and cancer risk in the health aging and body composition cohort. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2413–2418. [Google Scholar] [CrossRef] [PubMed]
- Siemes, C.; Visser, L.E.; Coebergh, J.W.; Splinter, T.A.; Witteman, J.C.; Uitterlinden, A.G.; Hofman, A.; Pols, H.A.; Stricker, B.H. C-reactive protein levels, variation in the C-reactive protein gene, and cancer risk: The Rotterdam Study. J. Clin. Oncol. 2006, 24, 5216–5222. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulos, D.; Psaltopoulou, T.; Orfanos, P.; Trichopoulou, A.; Boffetta, P. Plasma C-reactive protein and risk of cancer: A prospective study from Greece. Cancer Epidemiol. Biomark. Prev. 2006, 15, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Lin, J.; Cook, N.R.; Lee, I.M.; Manson, J.E.; Buring, J.E.; Ridker, P.M. C-reactive protein and risk of breast cancer. J. Natl. Cancer Inst. 2007, 99, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Ollberding, N.J.; Kim, Y.; Shvetsov, Y.B.; Wilkens, L.R.; Franke, A.A.; Cooney, R.V.; Maskarinec, G.; Hernandez, B.Y.; Henderson, B.E.; Le, M.L.; et al. Prediagnostic leptin, adiponectin, C-reactive protein, and the risk of postmenopausal breast cancer. Cancer Prev. Res. 2013, 6, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Ballard-Barbash, R.; Bernstein, L.; Baumgartner, R.N.; Neuhouser, M.L.; Wener, M.H.; Baumgartner, K.B.; Gilliland, F.D.; Sorensen, B.E.; McTiernan, A.; et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J. Clin. Oncol. 2009, 27, 3437–3444. [Google Scholar] [CrossRef] [PubMed]
- Sheen-Chen, S.M.; Chen, W.J.; Eng, H.L.; Chou, F.F. Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res. Treat. 1997, 43, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Maimoun, L.; Puech, A.M.; Manetta, J.; Badiou, S.; Paris, F.; Ohanna, F.; Rossi, M.; Sultan, C. Circulating leptin concentrations can be used as a surrogate marker of fat mass in acute spinal cord injury patients. Metabolism 2004, 53, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Silha, J.V.; Weiler, H.A.; Murphy, L.J. Plasma adipokines and body composition in response to modest dietary manipulations in the mouse. Obesity 2006, 14, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Cust, A.E.; Stocks, T.; Lukanova, A.; Lundin, E.; Hallmans, G.; Kaaks, R.; Jonsson, H.; Stattin, P. The influence of overweight and insulin resistance on breast cancer risk and tumour stage at diagnosis: A prospective study. Breast Cancer Res. Treat. 2009, 113, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.M.; Patel, A.V.; Teras, L.R.; Sun, J.; Campbell, P.T.; Stevens, V.L.; Jacobs, E.J.; Gapstur, S.M. Obesity-related markers and breast cancer in CPS-II Nutrition Cohort. Int. J. Mol. Epidemiol. Genet. 2013, 4, 156–166. [Google Scholar] [PubMed]
- Gross, A.L.; Newschaffer, C.J.; Hoffman-Bolton, J.; Rifai, N.; Visvanathan, K. Adipocytokines, inflammation, and breast cancer risk in postmenopausal women: A prospective study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.R.; Tworoger, S.S.; Hankinson, S.E.; Rosner, B.A.; Michels, K.B. Plasma Leptin Levels and Risk of Breast Cancer in Premenopausal Women. Cancer Prev. Res. 2011, 4, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Chou, Y.C.; Chou, W.Y.; Hsu, G.C.; Chu, C.H.; Yu, C.P.; Yu, J.C.; Sun, C.A. Circulating levels of leptin, adiposity and breast cancer risk. Br. J. Cancer 2009, 100, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Jarde, T.; Caldefie-Chezet, F.; Damez, M.; Mishellany, F.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on human primary breast carcinoma. Oncol. Rep. 2008, 19, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Caldefie-Chezet, F.; Damez, M.; de, L.M.; Konska, G.; Mishellani, F.; Fusillier, C.; Guerry, M.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin: A proliferative factor for breast cancer? Study on human ductal carcinoma. Biochem. Biophys. Res. Commun. 2005, 334, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Al-Delaimy, W.K.; Flatt, S.W.; Natarajan, L.; Laughlin, G.A.; Rock, C.L.; Gold, E.B.; Caan, B.J.; Parker, B.A.; Pierce, J.P. IGF1 and risk of additional breast cancer in the WHEL study. Endocr. Relat. Cancer 2011, 18, 235–244. [Google Scholar] [PubMed]
- Oh, S.W.; Park, C.Y.; Lee, E.S.; Yoon, Y.S.; Lee, E.S.; Park, S.S.; Kim, Y.; Sung, N.J.; Yun, Y.H.; Lee, K.S.; et al. Adipokines, insulin resistance, metabolic syndrome, and breast cancer recurrence: A cohort study. Breast Cancer Res. 2011, 13, R34. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Taylor, S.K.; Hood, N. Insulin- and obesity-related variables in early-stage breast cancer: Correlations and time course of prognostic associations. J. Clin. Oncol. 2012, 30, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Jarde, T.; Delort, L.; Billard, H.; Bernard-Gallon, D.; Berger, E.; Geloen, A.; Vasson, M.P.; Caldefie-Chezet, F. Leptin induces a proliferative response in breast cancer cells but not in normal breast cells. Nutr. Cancer 2014, 66, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Sun, K.; Yu, K. Leptin promotes the proliferation and migration of human breast cancer through the extracellular-signal regulated kinase pathway. Mol. Med. Rep. 2014, 9, 350–354. [Google Scholar] [PubMed]
- Lautenbach, A.; Budde, A.; Wrann, C.D.; Teichmann, B.; Vieten, G.; Karl, T.; Nave, H. Obesity and the associated mediators leptin, estrogen and IGF-I enhance the cell proliferation and early tumorigenesis of breast cancer cells. Nutr. Cancer 2009, 61, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Palianopoulou, M.; Papanikolaou, V.; Stefanou, N.; Tsezou, A. The activation of leptin-mediated survivin is limited by the inducible suppressor SOCS-3 in MCF-7 cells. Exp. Biol. Med. 2011, 236, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.B.; Oprea-Ilies, G.M.; Nagalingam, A.; Yang, L.; Cohen, C.; Saxena, N.K.; Sharma, D. Survivin upregulation, dependent on leptin-EGFR-Notch1 axis, is essential for leptin-induced migration of breast carcinoma cells. Endocr. Relat. Cancer 2011, 18, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Newman, G.; Gonzalez-Perez, R.R. Leptin–cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shukla, S.; Sinha, S.; Meeran, S.M. Role of adipokines and cytokines in obesity-associated breast cancer: therapeutic targets. Cytokine Growth Factor Rev. 2013, 24, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Revillion, F.; Charlier, M.; Lhotellier, V.; Hornez, L.; Giard, S.; Baranzelli, M.C.; Djiane, J.; Peyrat, J.P. Messenger RNA expression of leptin and leptin receptors and their prognostic value in 322 human primary breast cancers. Clin. Cancer Res. 2006, 12, 2088–2094. [Google Scholar] [CrossRef] [PubMed]
- Tworoger, S.S.; Eliassen, A.H.; Kelesidis, T.; Colditz, G.A.; Willett, W.C.; Mantzoros, C.S.; Hankinson, S.E. Plasma adiponectin concentrations and risk of incident breast cancer. J. Clin. Endocrinol. Metab. 2007, 92, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Wang, M.; Ma, Z.B.; Yu, L.X.; Zhang, Q.; Gao, D.Z.; Wang, F.; Yu, Z.G. The role of adiponectin in breast cancer: A meta-analysis. PLoS ONE 2013, 8, e73183. [Google Scholar] [CrossRef] [PubMed]
- Macis, D.; Guerrieri-Gonzaga, A.; Gandini, S. Circulating adiponectin and breast cancer risk: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Jia, J.; Dong, S.; Zhang, C.; Yu, S.; Li, L.; Mao, C.; Wang, D.; Chen, J.; Yuan, G. Circulating adiponectin levels and the risk of breast cancer: A meta-analysis. Eur. J. Cancer Prev. 2014, 23, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Funahashi, T.; Kihara, S.; Taguchi, T.; Tamaki, Y.; Matsuzawa, Y.; Noguchi, S. Association of serum adiponectin levels with breast cancer risk. Clin. Cancer Res. 2003, 9, 5699–5704. [Google Scholar] [PubMed]
- Mantzoros, C.; Petridou, E.; Dessypris, N.; Chavelas, C.; Dalamaga, M.; Alexe, D.M.; Papadiamantis, Y.; Markopoulos, C.; Spanos, E.; Chrousos, G.; et al. Adiponectin and breast cancer risk. J. Clin. Endocrinol. Metab. 2004, 89, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Karaduman, M.; Bilici, A.; Ozet, A.; Sengul, A.; Musabak, U.; Alomeroglu, M. Tissue levels of adiponectin in breast cancer patients. Med. Oncol. 2007, 24, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Cheng, K.K.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Cross-talk between adipose tissue and vasculature: Role of adiponectin. Acta Physiol. 2011, 203, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Hursting, S.D.; Nunez, N.P.; Varticovski, L.; Vinson, C. The obesity-cancer link: Lessons learned from a fatless mouse. Cancer Res. 2007, 67, 2391–2393. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Ray, A.; Rogozina, O.P.; Dogan, S.; Grossmann, M.E. Targeting the adiponectin: Leptin ratio for postmenopausal breast cancer prevention. Front. Biosci. 2009, 1, 329–357. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Cleary, M.R. The balance between leptin and adiponectin in the control of carcinogenesis—Focus on mammary tumorigenesis. Biochimie 2012, 94, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, S.M.; Wolfe, P.; Paul, D.; Lakoski, S.; Playdon, M.; McGinley, J.N.; Matthews, S.B.; Thompson, H.J. Impact of Weight Loss and Dietary Pattern on Plasma Leptin and Adiponectin in Overweight-to-Obese Post Menopausal Breast Cancer Survivors. Nutrients 2015, 7, 5156–5176. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer risk: Role of environment-response. Science 2015, 347, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, C.R.; Zaykin, D. Is bad luck the main cause of cancer? J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; Mukhtar, H.; Rock, C.L.; Cuzick, J.; Kensler, T.W.; Yang, C.S.; Ramsey, S.D.; Lippman, S.M.; Alberts, D.S. Cancer Prevention: Obstacles, Challenges and the Road Ahead. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Wicha, M.S. Mammary stem cell number as a determinate of breast cancer risk. Breast Cancer Res. 2007, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Labat, M.L.; Vaillant, F.; Sheridan, J.M.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010, 465, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Molyneux, G.; Mackay, A.; Magnay, F.A.; Atienza, M.; Kendrick, H.; Nava-Rodrigues, D.; Lopez-Garcia, M.A.; Milanezi, F.; Greenow, K.; et al. Identification of cellular and genetic drivers of breast cancer heterogeneity in genetically engineered mouse tumour models. J. Pathol. 2014, 233, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Folkerd, E. Reduced progesterone levels explain the reduced risk of breast cancer in obese premenopausal women: A new hypothesis. Breast Cancer Res. Treat. 2015, 149, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Junankar, S.; Baker, L.A.; Roden, D.L.; Nair, R.; Elsworth, B.; Gallego-Ortega, D.; Lacaze, P.; Cazet, A.; Nikolic, I.; Teo, W.S.; et al. ID4 controls mammary stem cells and marks breast cancers with a stem cell-like phenotype. Nat. Commun. 2015, 6, 6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axlund, S.D.; Yoo, B.H.; Rosen, R.B.; Schaack, J.; Kabos, P.; Labarbera, D.V.; Sartorius, C.A. Progesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties. Horm. Cancer 2013, 4, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Kabos, P.; Haughian, J.M.; Wang, X.; Dye, W.W.; Finlayson, C.; Elias, A.; Horwitz, K.B.; Sartorius, C.A. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res. Treat. 2011, 128, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Savarese, T.M.; Strohsnitter, W.C.; Low, H.P.; Liu, Q.; Baik, I.; Okulicz, W.; Chelmow, D.P.; Lagiou, P.; Quesenberry, P.J.; Noller, K.L.; et al. Correlation of umbilical cord blood hormones and growth factors with stem cell potential: Implications for the prenatal origin of breast cancer hypothesis. Breast Cancer Res. 2007, 9, R29. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.M.; Dame, M.; McClintock, S.; Holt, P.R.; Dannenberg, A.J.; Wicha, M.S.; Brenner, D.E. Leptin and Adiponectin Modulate the Self-renewal of Normal Human Breast Epithelial Stem Cells. Cancer Prev. Res. 2015, 8, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Spira, A.; Garber, J.E.; Szabo, E.; Lee, J.J.; Dong, Z.; Dannenberg, A.J.; Hait, W.N.; Blackburn, E.; Davidson, N.E.; et al. Transforming Cancer Prevention through Precision Medicine and Immune-oncology. Cancer Prev. Res. 2016, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Hair, B.Y.; Xu, Z.; Kirk, E.L.; Harlid, S.; Sandhu, R.; Robinson, W.R.; Wu, M.C.; Olshan, A.F.; Conway, K.; Taylor, J.A.; et al. Body mass index associated with genome-wide methylation in breast tissue. Breast Cancer Res. Treat. 2015, 151, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Hair, B.Y.; Troester, M.A.; Edmiston, S.N.; Parrish, E.A.; Robinson, W.R.; Wu, M.C.; Olshan, A.F.; Swift-Scanlan, T.; Conway, K. Body mass index is associated with gene methylation in estrogen receptor-positive breast tumors. Cancer Epidemiol. Biomark. Prev. 2015, 24, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.S.; Ali, S.; Ahmad, A.; Bao, B.; Philip, P.A.; Sarkar, F.H. Expression of microRNAs: Potential molecular link between obesity, diabetes and cancer. Obes. Rev. 2011, 12, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Davis, C.D. MicroRNA, nutrition, and cancer prevention. Adv. Nutr. 2011, 2, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Mattei, E.; Velazquez-Torres, G.; Phan, L.; Zhang, F.; Chou, P.C.; Shin, J.H.; Choi, H.H.; Chen, J.S.; Zhao, R.; Chen, J.; et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.B.; McGinley, J.N.; Neil, E.S.; Thompson, H.J. Premenopausal Obesity and Breast Cancer Growth Rates in a Rodent Model. Nutrients 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Fu, K.K.; Steel, G.G. Growth-Kinetics of a Rat Mammary-Tumor Transplanted into Immune-Suppressed Mice. Cell Tissue Kinet. 1979, 12, 493–499. [Google Scholar] [PubMed]
- Steel, G.G. Some Holes in Tumor-Growth Kinetics. Cell Tissue Kinet. 1978, 11, 689. [Google Scholar]
- Tannock, I.F.; Steel, G.G. Tumor Growth and Cell Kinetics in Chronically Hypoxic Animals. J. Natl. Cancer Inst. 1970, 45, 123–133. [Google Scholar] [PubMed]
- Thompson, H.J.; Strange, R.; Schedin, P.J. Apoptosis in the genesis and prevention of cancer. Cancer Epidemiol. Biomark. Prev. 1992, 1, 597–602. [Google Scholar]
- Farooqi, I.S.; O’Rahilly, S. Monogenic obesity in humans. Annu. Rev. Med. 2005, 56, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Schwartz, M.W. Genetics and pathophysiology of human obesity. Annu. Rev. Med. 2003, 54, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Grande, J.P.; Juneja, S.C.; Maihle, N.J. Diet-induced obesity and mammary tumor development in MMTV-neu female mice. Nutr. Cancer Int. J. 2004, 50, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Grande, J.P.; Maihle, N.J. Effect of high fat diet on body weight and mammary tumor latency in MMTV-TGF-alpha mice. Int. J. Obes. 2004, 28, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Hu, X.; Zhang, Y.; Maihle, N.J.; Grande, J.P.; Cleary, M.P. Effects of high-fat diet and/or body weight on mammary tumor leptin and apoptosis signaling pathways in MMTV-TGF-alpha mice. Breast Cancer Res. 2007, 9, R91. [Google Scholar] [CrossRef] [PubMed]
- Diet-Induced Obesity (DIO) Diets and Phenotypes. Available online: http://jaxmice.jax.org/diomice/diets.html (accessed on 22 May 2016).
- MacLean, P.S.; Higgins, J.A.; Johnson, G.C.; Fleming-Elder, B.K.; Peters, J.C.; Hill, J.O. Metabolic adjustments with the development, treatment, and recurrence of obesity in obesity-prone rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R288–R297. [Google Scholar] [CrossRef] [PubMed]
- MacLean, P.S.; Higgins, J.A.; Jackman, M.R.; Johnson, G.C.; Fleming-Elder, B.K.; Wyatt, H.R.; Melanson, E.L.; Hill, J.O. Peripheral metabolic responses to prolonged weight reduction that promote rapid, efficient regain in obesity-prone rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1577–R1588. [Google Scholar] [CrossRef] [PubMed]
- MacLean, P.S.; Giles, E.D.; Johnson, G.C.; McDaniel, S.M.; Fleming-Elder, B.K.; Gilman, K.A.; Andrianakos, A.G.; Jackman, M.R.; Shroyer, K.R.; Schedin, P.J. A Surprising Link Between the Energetics of Ovariectomy-induced Weight Gain and Mammary Tumor Progression in Obese Rats. Obesity 2010, 18, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.D.; Wellberg, E.A.; Astling, D.P.; Anderson, S.M.; Thor, A.D.; Jindal, S.; Tan, A.C.; Schedin, P.S.; MacLean, P.S. Obesity and Overfeeding Affecting both Tumor and Systemic Metabolism Activates the Progesterone Receptor to Contribute to Postmenopausal Breast Cancer. Cancer Res. 2012, 72, 6490–6501. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Dunn-Meynell, A.A.; Balkan, B.; Keesey, R.E. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am. J. Physiol. 1997, 273, R725–R730. [Google Scholar] [PubMed]
- Madsen, A.N.; Hansen, G.; Paulsen, S.J.; Lykkegaard, K.; Tang-Christensen, M.; Hansen, H.S.; Levin, B.E.; Larsen, P.J.; Knudsen, L.B.; Fosgerau, K.; et al. Long-term characterization of the diet-induced obese and diet-resistant rat model: A polygenetic rat model mimicking the human obesity syndrome. J. Endocrinol. 2010, 206, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, S.J.; Jelsing, J.; Madsen, A.N.; Hansen, G.; Lykkegaard, K.; Larsen, L.K.; Larsen, P.J.; Levin, B.E.; Vrang, N. Characterization of beta-Cell Mass and Insulin Resistance in Diet-induced Obese and Diet-resistant Rats. Obesity 2010, 18, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Thompson, H.J. A comparison of the salient features of mouse, rat, and human mammary tumorigenesis. In Methods in Mammary Gland Biology and Breast Cancer Research; Ip, M.M., Asch, B.B., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2000; pp. 31–35. [Google Scholar]
- Medina, D. Chemical carcinogenesis of rat and mouse mammary glands. Breast Dis. 2007, 28, 63–68. [Google Scholar] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Ao, X.; Grill, C.; Russo, I.H. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res. Treat. 1999, 53, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.; Clarke, R.B. Steroid receptors and cell cycle in normal mammary epithelium. J. Mammary Gland Biol. Neoplasia 2004, 9, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; McGinley, J.N.; Thompson, H.J. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab. Investig. 2000, 80, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.B.; Zhu, Z.; Jiang, W.; McGinley, J.N.; Neil, E.S.; Thompson, H.J. Excess weight gain accelerates 1-methyl-1-nitrosourea-induced mammary carcinogenesis in a rat model of premenopausal breast cancer. Cancer Prev. Res. 2014, 7, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009, 23, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthews, S.B.; Thompson, H.J. The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. Int. J. Mol. Sci. 2016, 17, 989. https://doi.org/10.3390/ijms17060989
Matthews SB, Thompson HJ. The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. International Journal of Molecular Sciences. 2016; 17(6):989. https://doi.org/10.3390/ijms17060989
Chicago/Turabian StyleMatthews, Shawna B., and Henry J. Thompson. 2016. "The Obesity-Breast Cancer Conundrum: An Analysis of the Issues" International Journal of Molecular Sciences 17, no. 6: 989. https://doi.org/10.3390/ijms17060989