
Finding a Potential Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for Type-2 Diabetes Treatment Based on Molecular Docking, Pharmacophore Generation, and Molecular Dynamics Simulation


Abstract
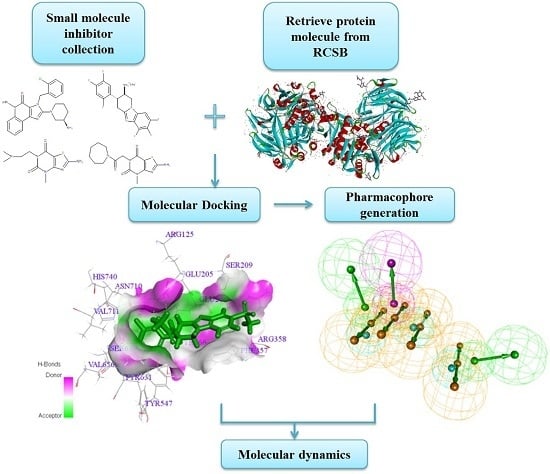
:
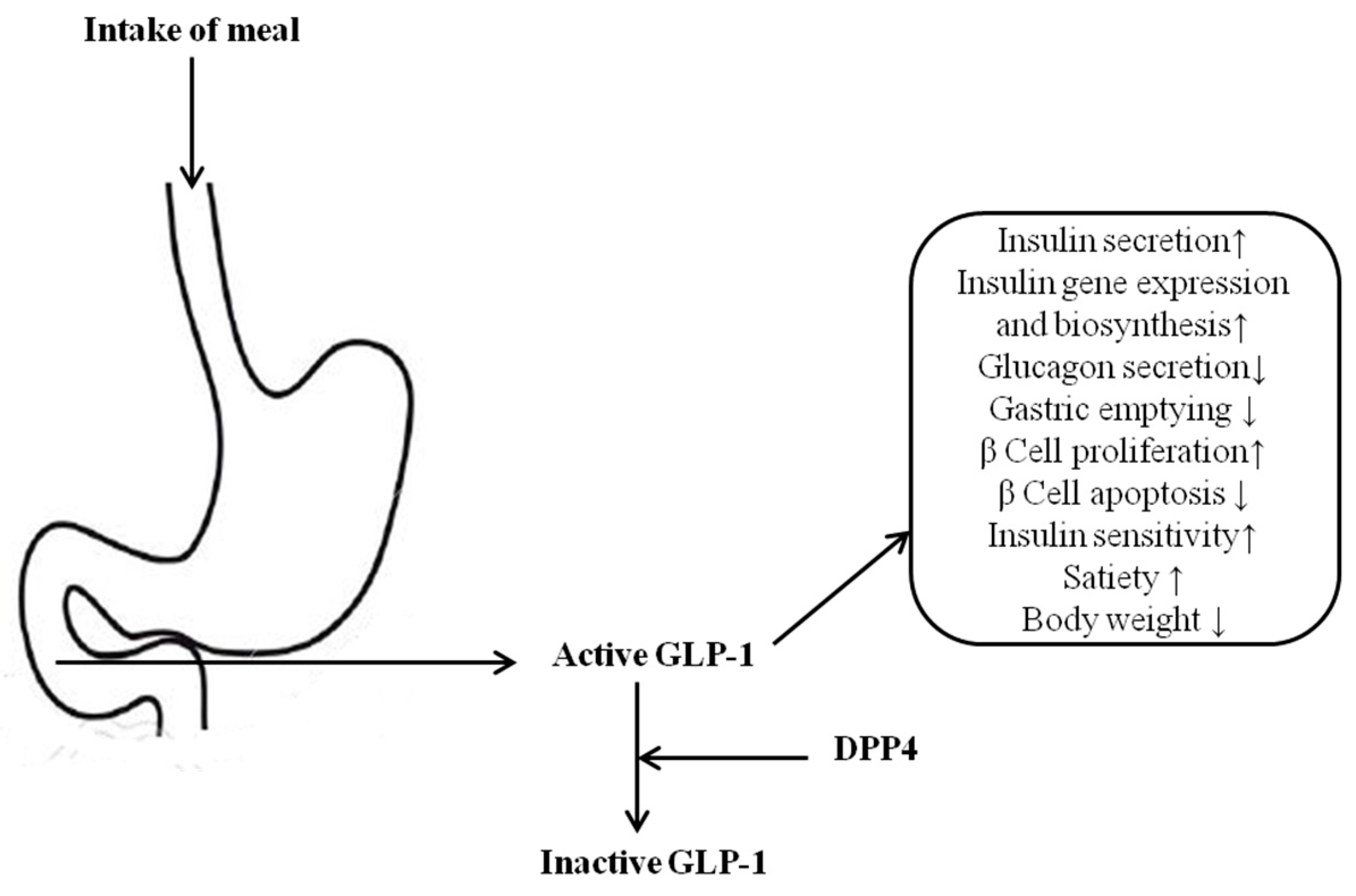
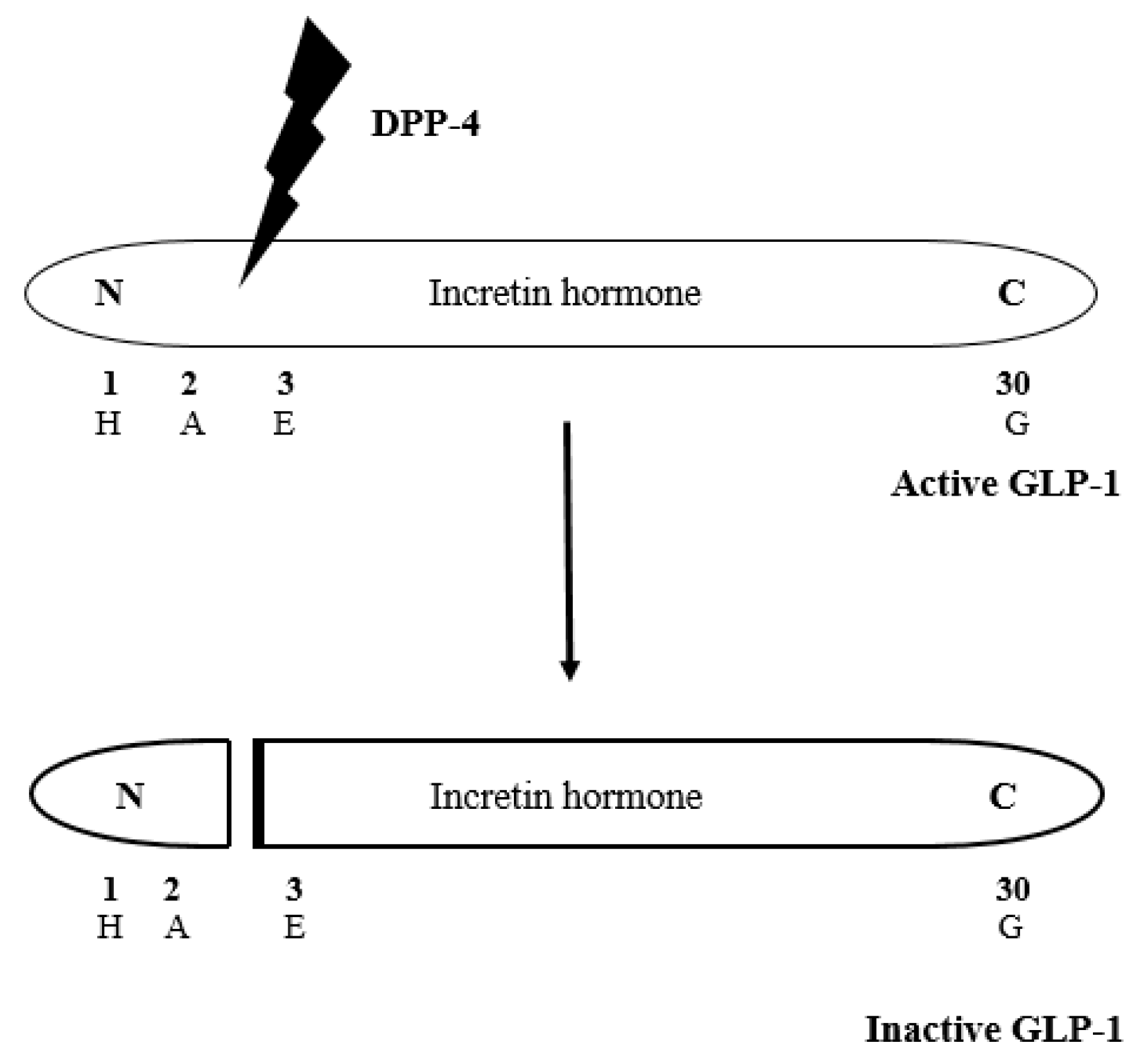
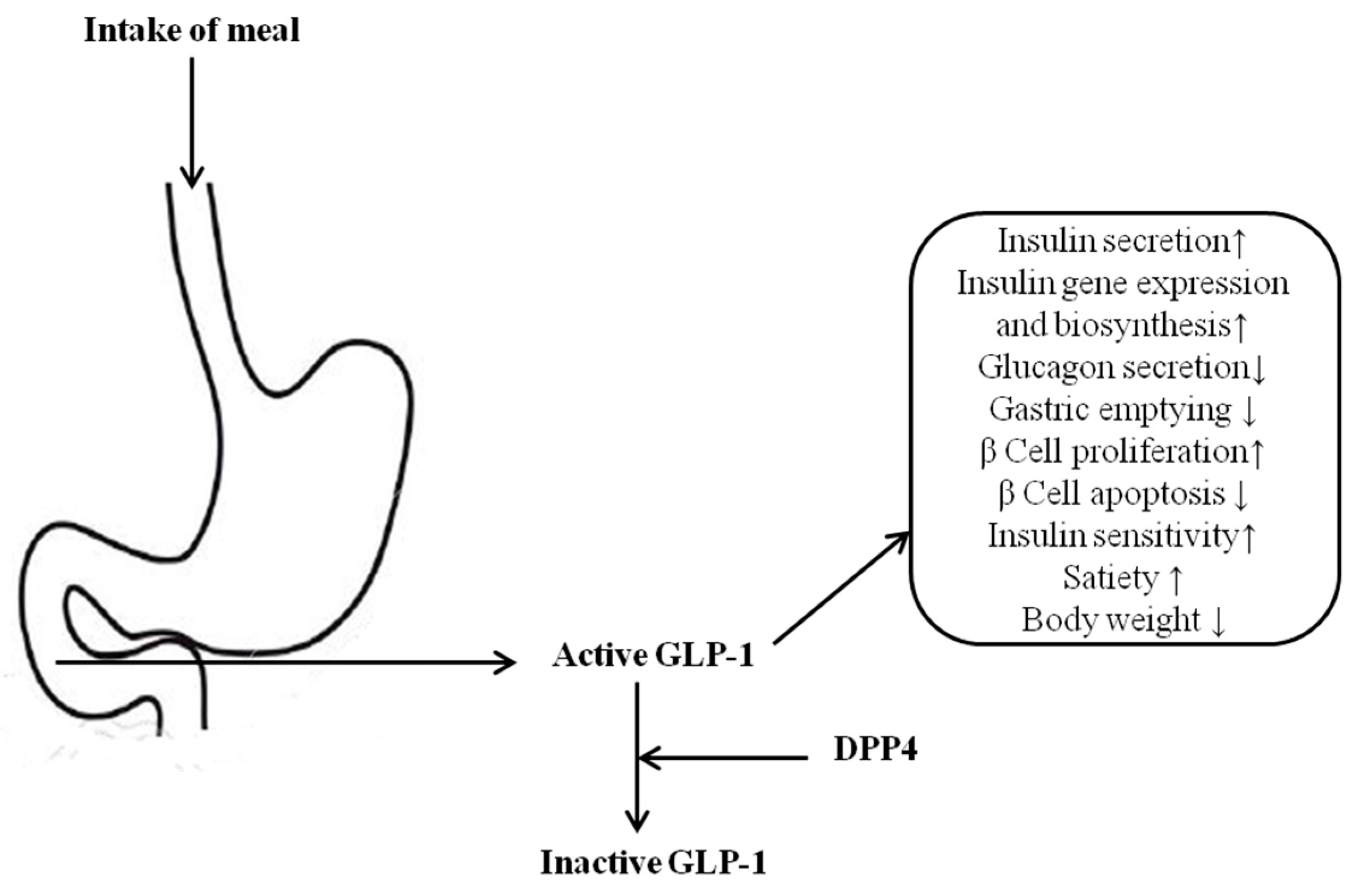
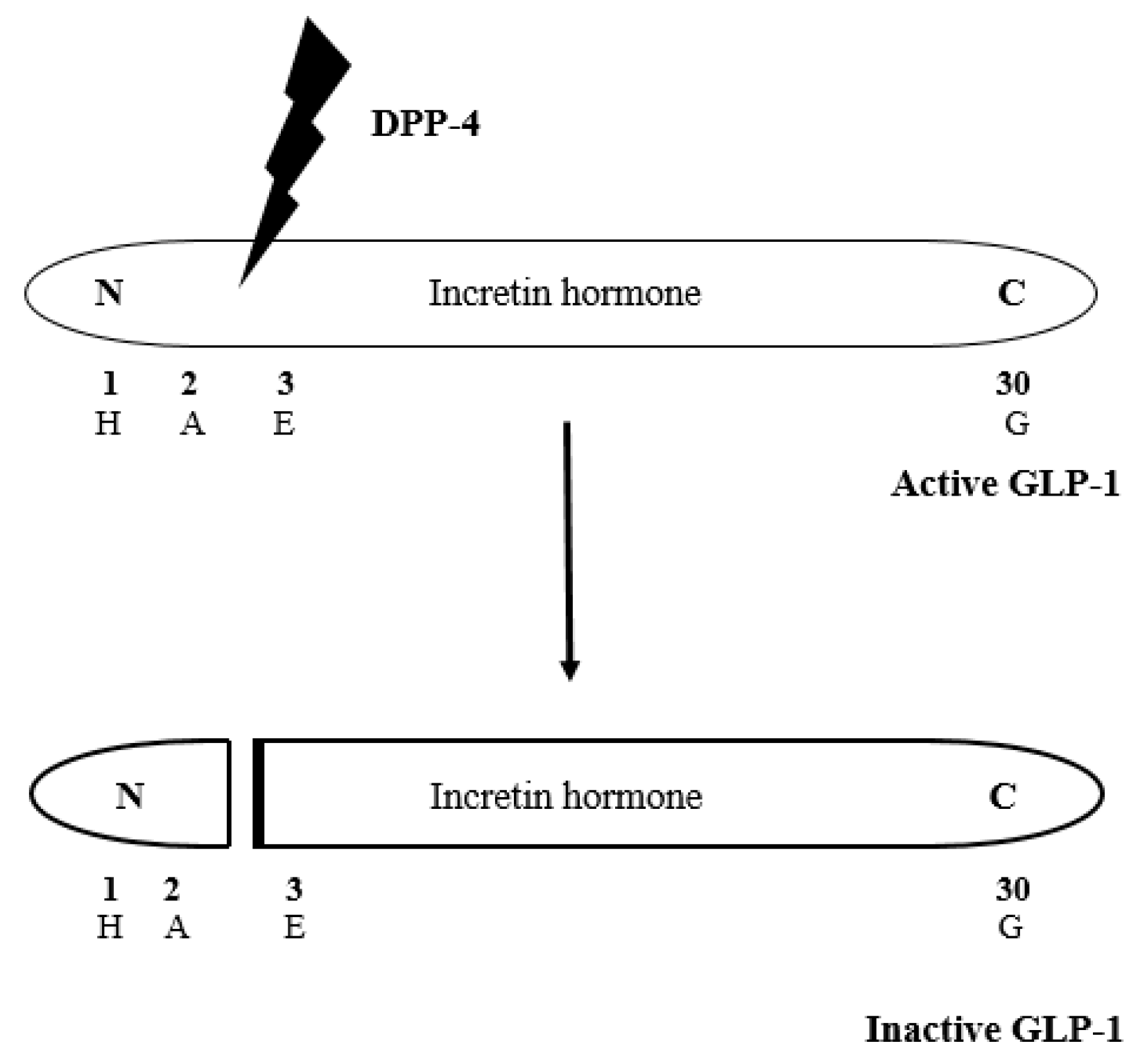
1. Introduction
2. Results and Discussion
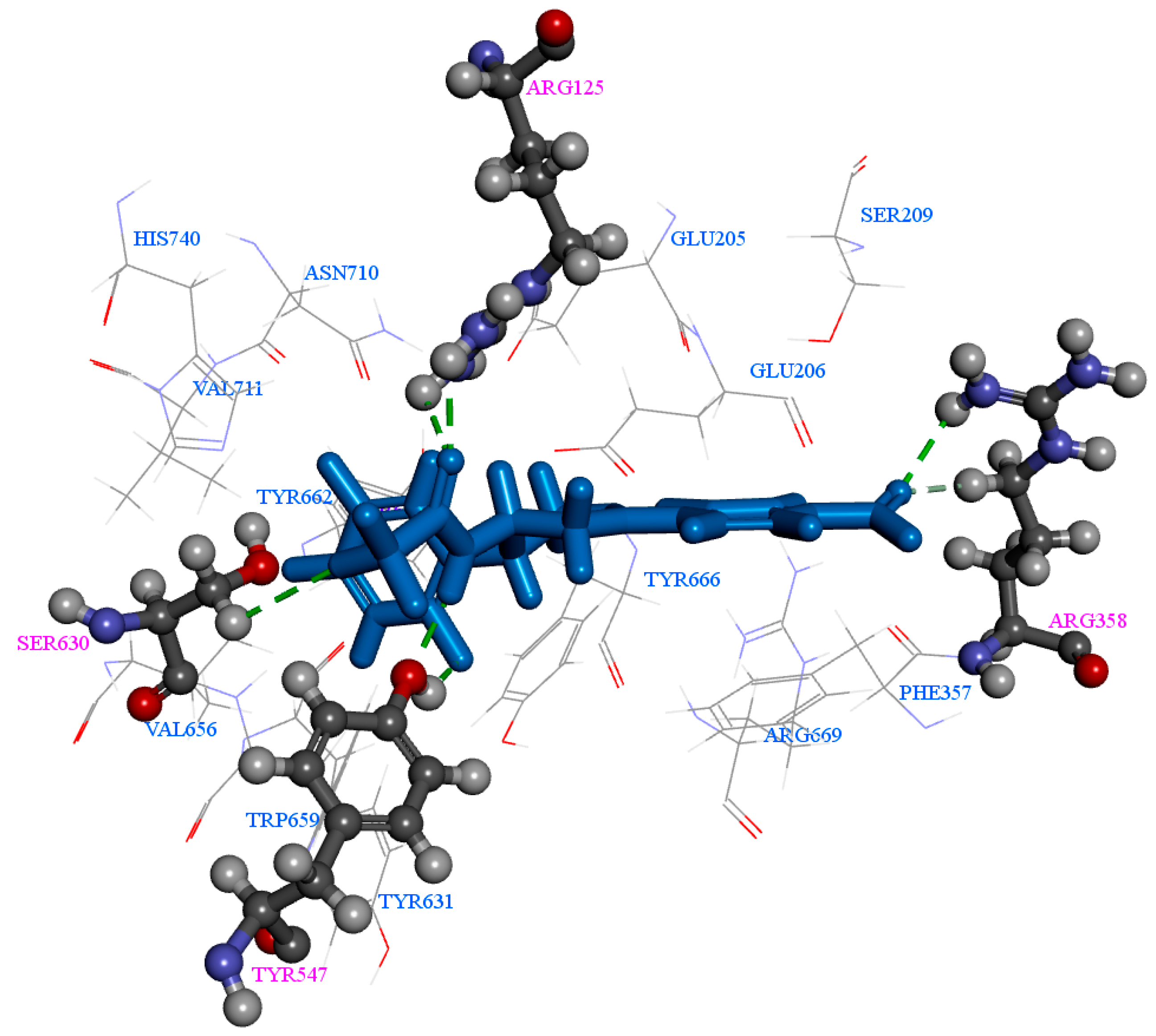
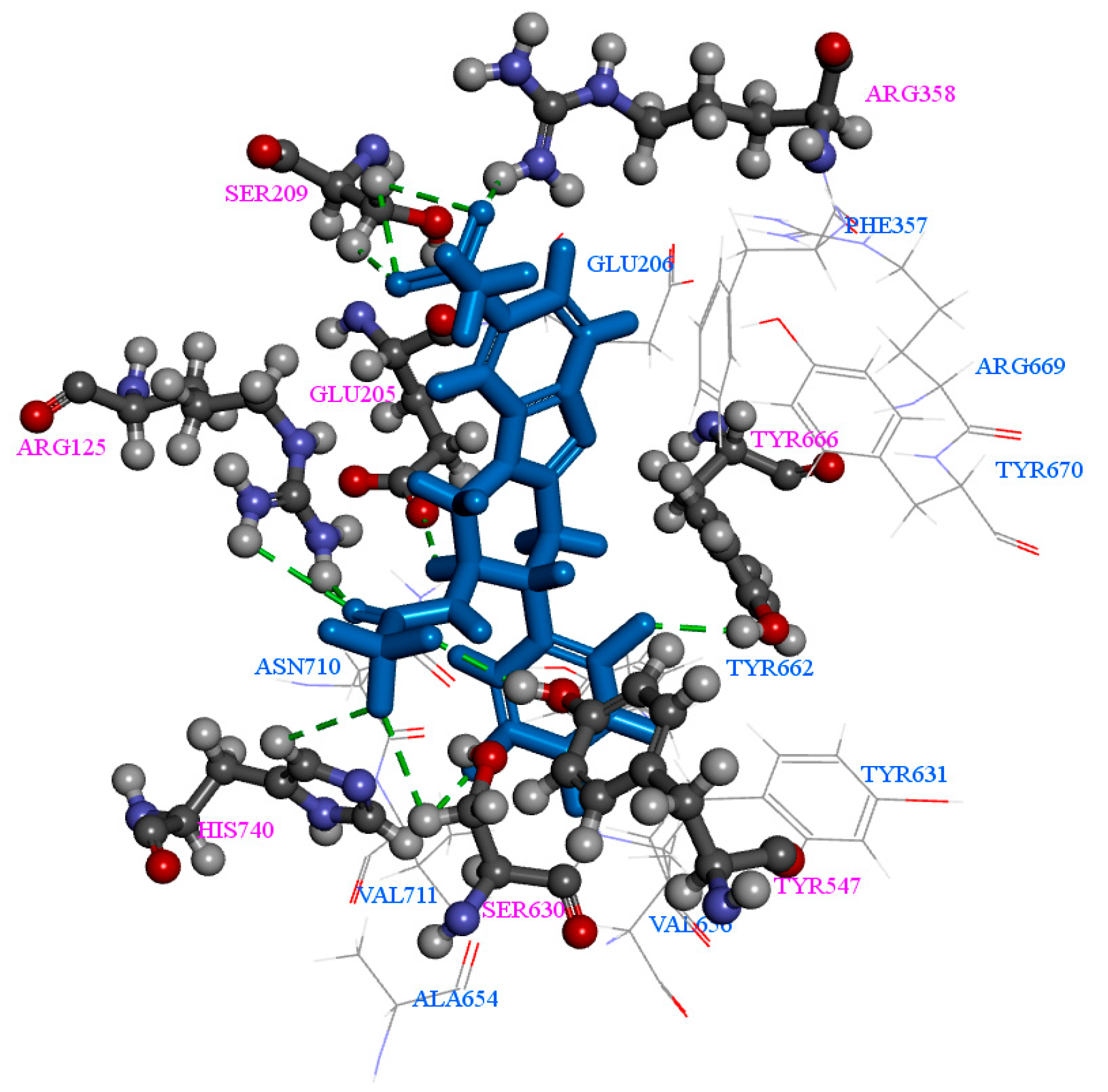
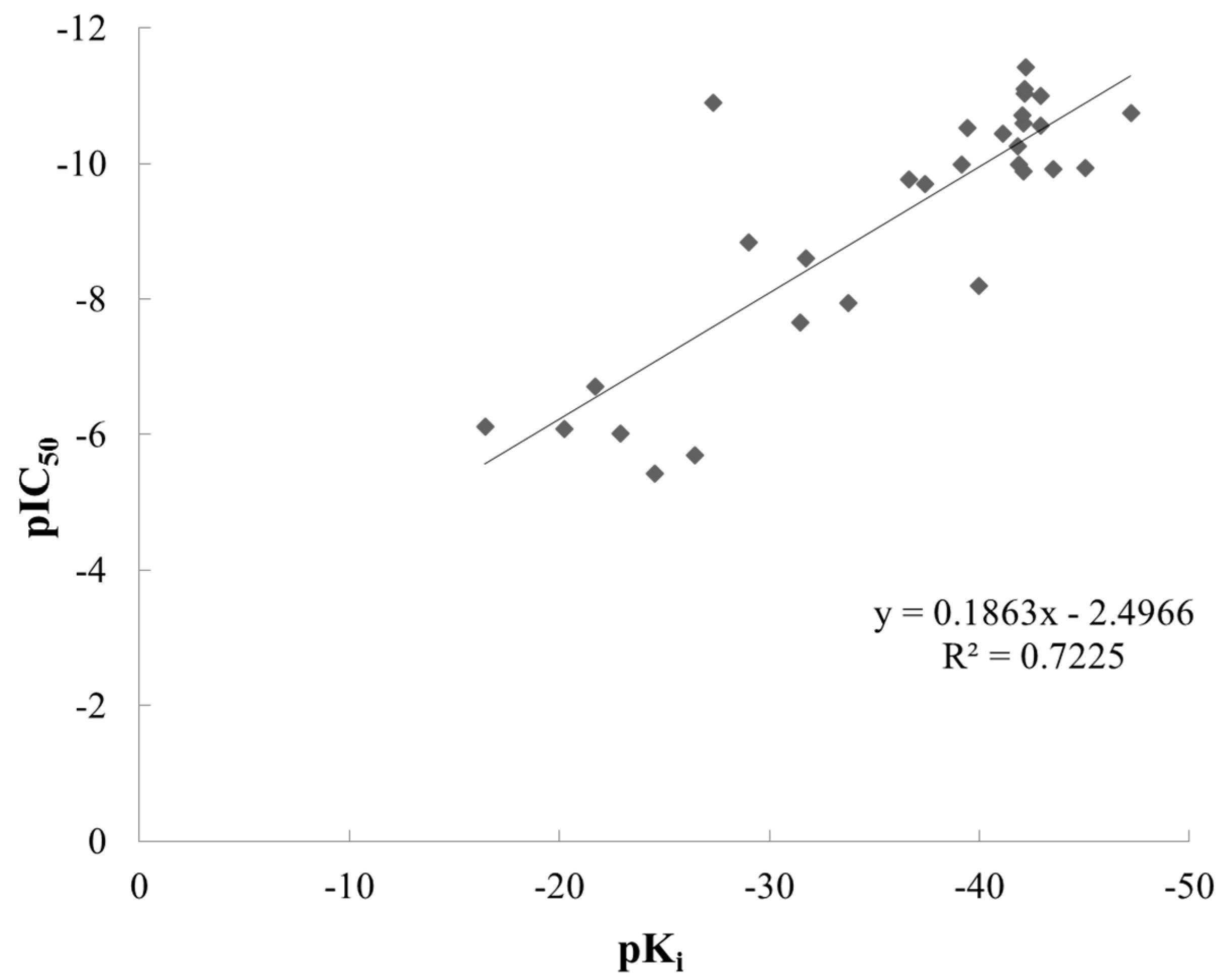
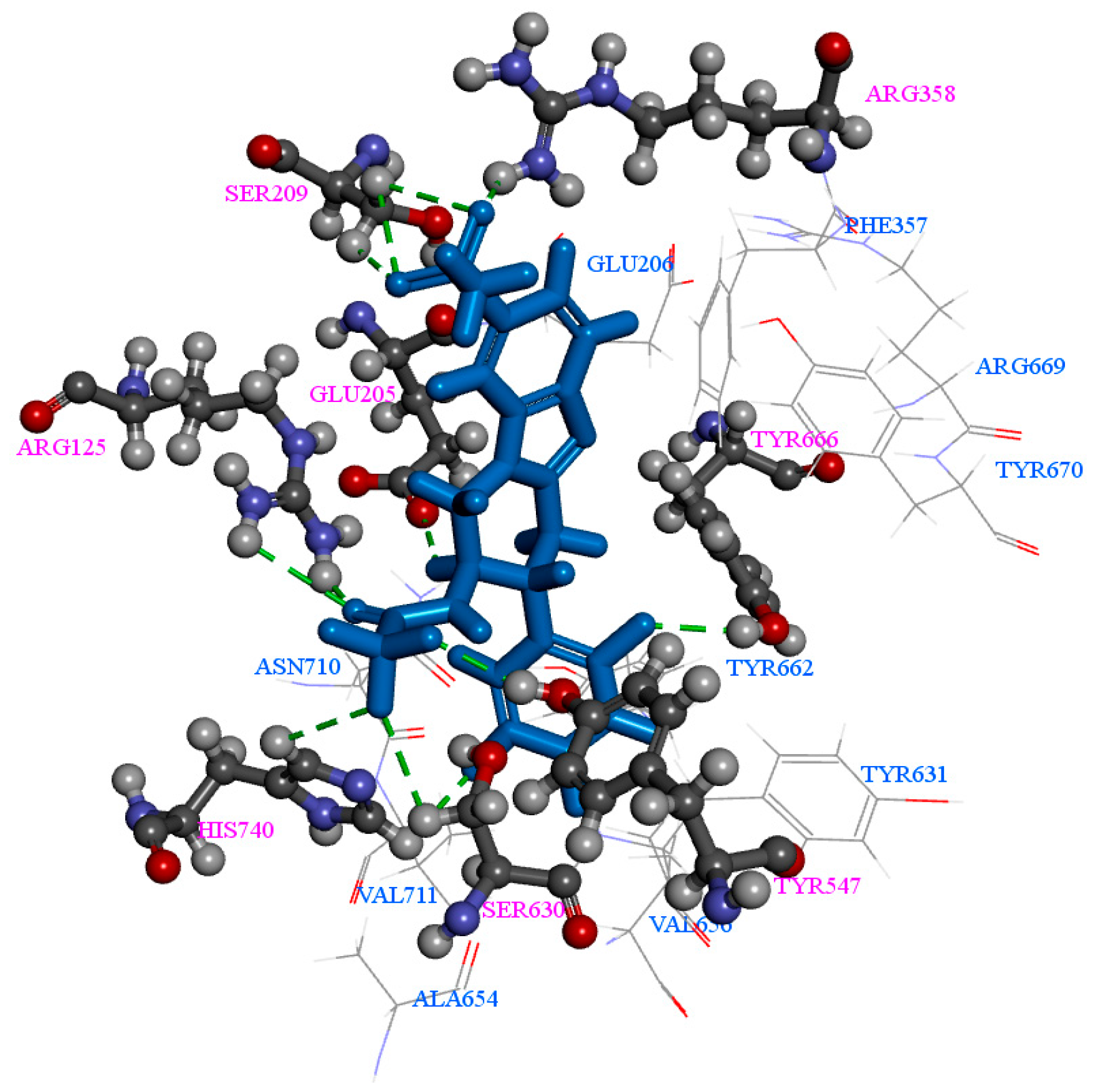
2.1. Molecular Docking
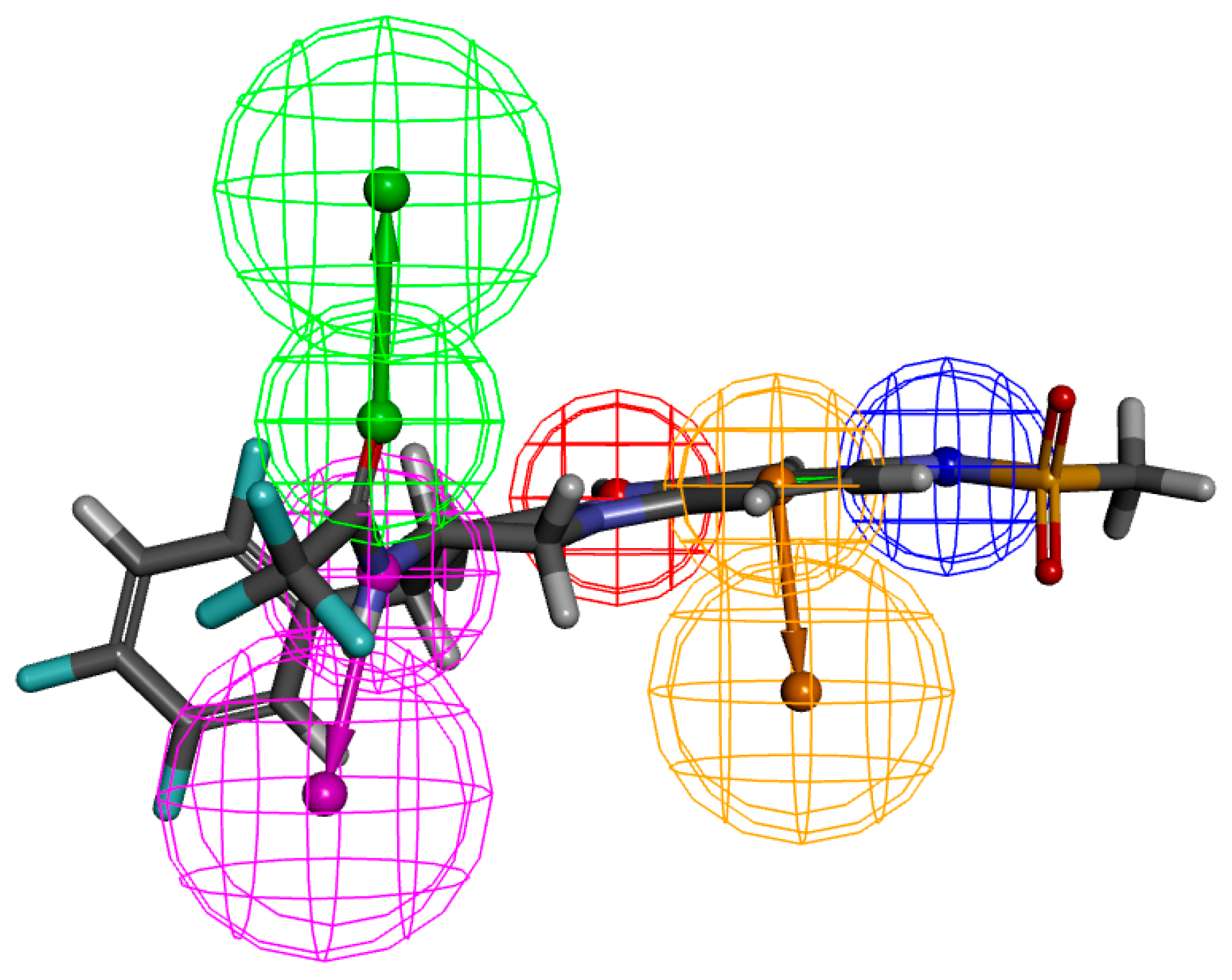
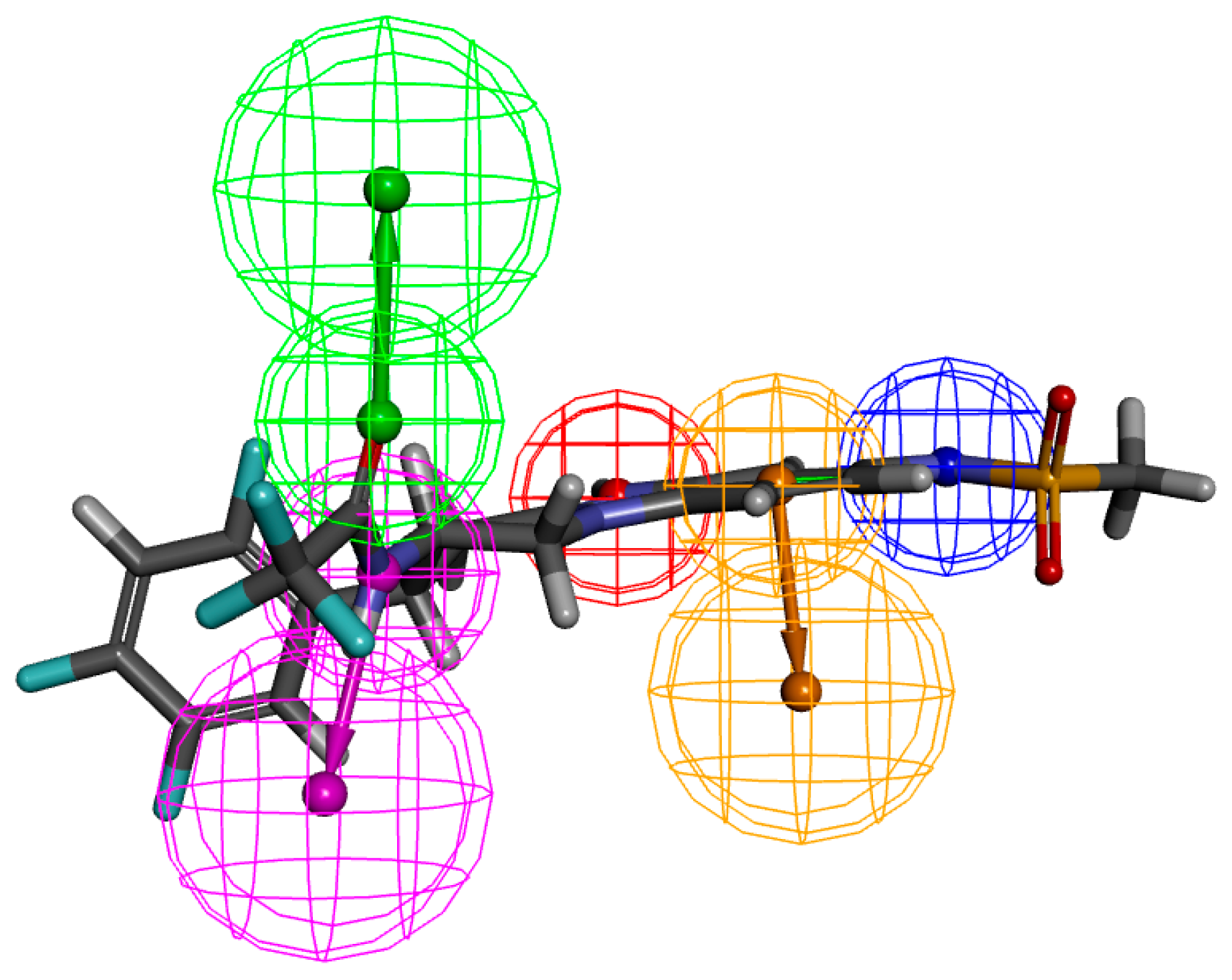
2.2. Pharmacophore Generation
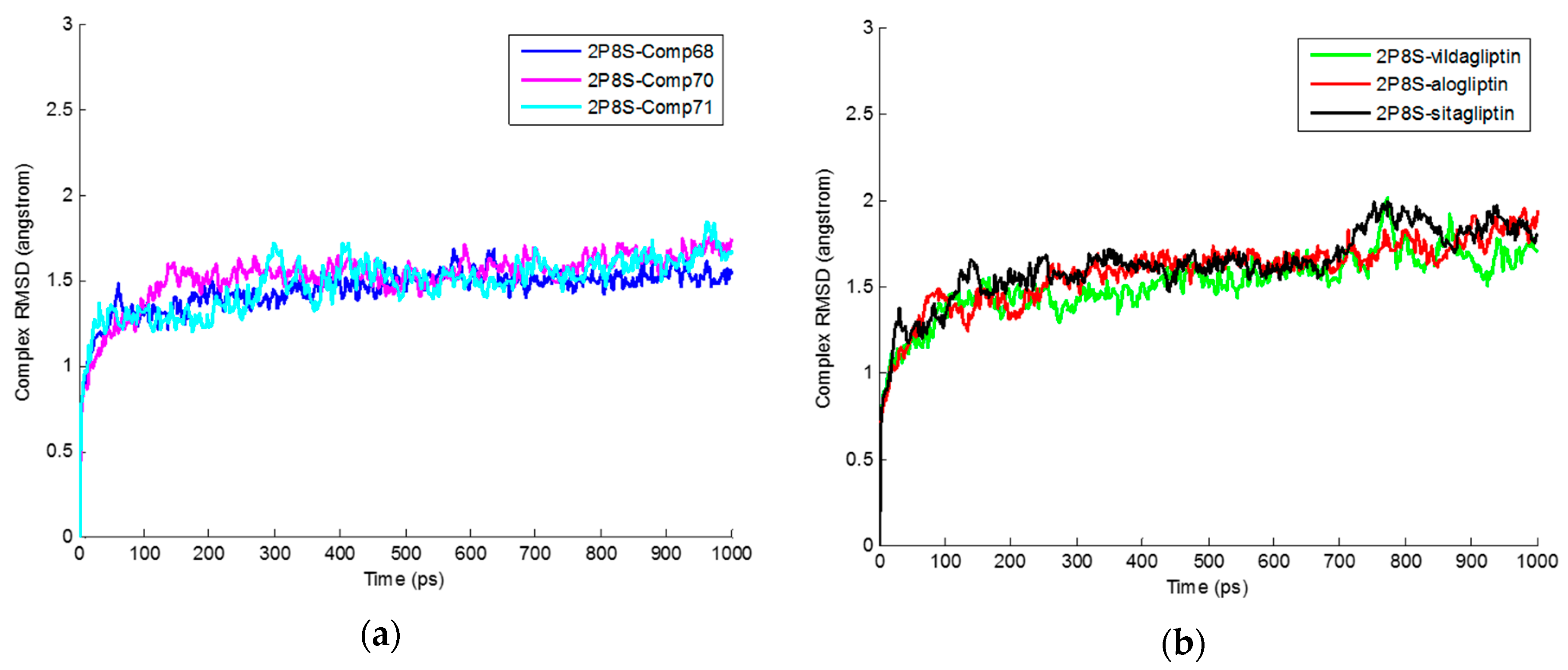
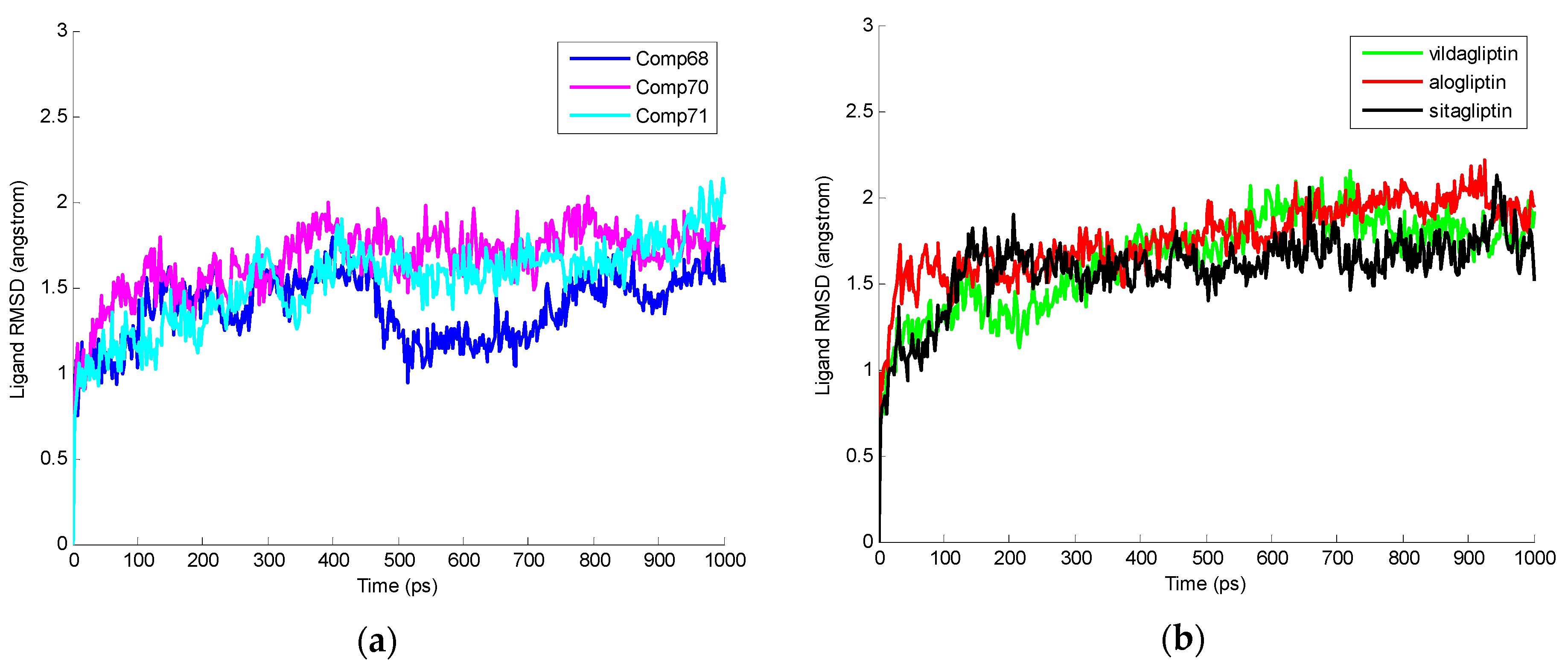
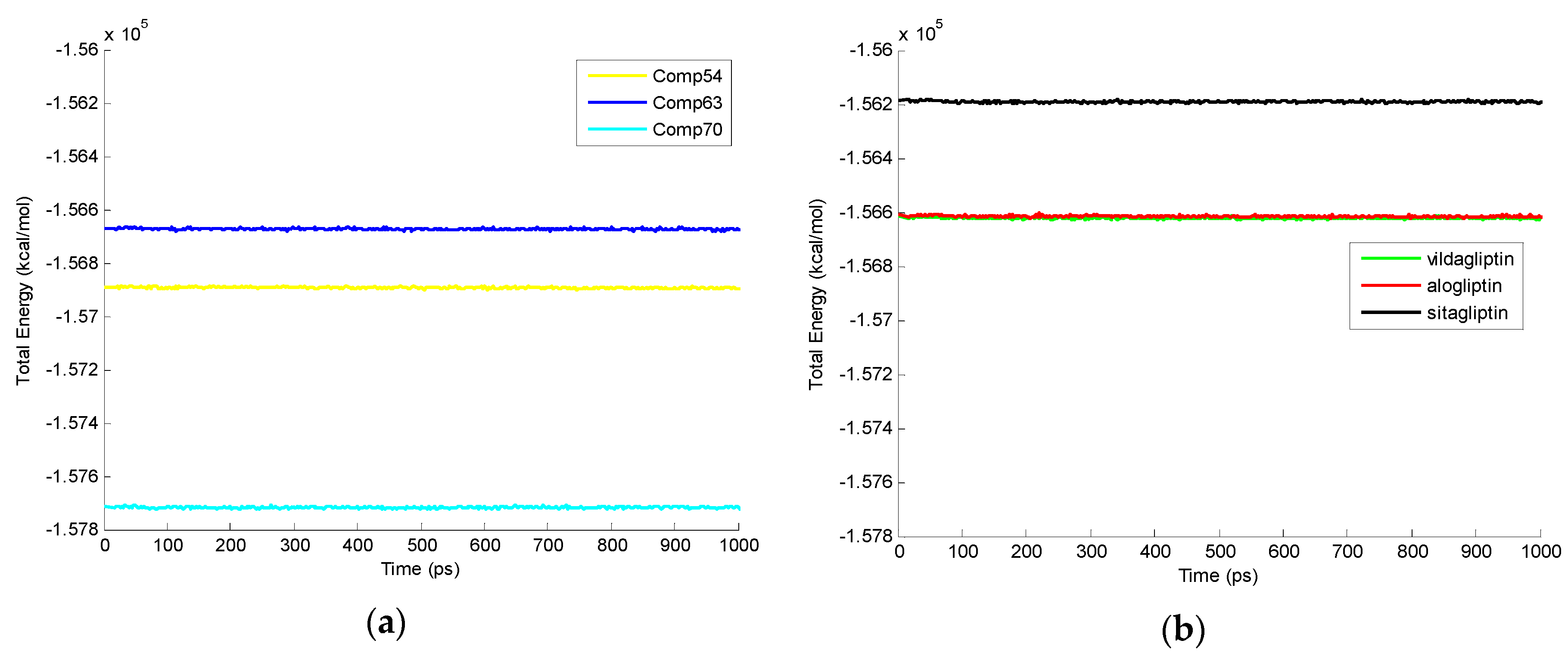
2.3. Molecular Dynamics Simulation
3. Methods and Materials
3.1. Small-Molecules Preparation
3.2. DPP-4 Model Preparation
3.3. Molecular Docking
3.4. Pharmacophore Generation
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE 2012, 7, e38546. [Google Scholar] [CrossRef] [PubMed]
- Anselmino, M. Cardiovascular prevention in type 2 diabetes mellitus patients: The role of oral glucose-lowering agents. J. Diabetes Complicat. 2009, 23, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; deFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Zander, M.; Madsbad, S.; Madsen, J.L.; Holst, J.J. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes—A parallel-group study. Lancet 2002, 359, 824–830. [Google Scholar] [CrossRef]
- Lim, G.E.; Brubaker, P.L. Glucagon-like peptide 1 secretion by the l-cell: The view from within. Diabetes 2006, 55, S70–S77. [Google Scholar] [CrossRef]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Vetere, A.; Choudhary, A.; Burns, S.M.; Wagner, B.K. Targeting the pancreatic beta-cell to treat diabetes. Nat. Rev. Drug Discov. 2014, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Iacomelli, I.; Marchionni, N.; Mannucci, E. Dipeptydil peptidase-4 inhibitors in type 2 diabetes: A meta-analysis of randomized clinical trials. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V. Glucagon-like peptide-1 analogues: An overview. Indian J. Endocrinol. Metab. 2013, 17, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, P.; Chepurny, O.G.; Holz, G.G. Regulation of glucose homeostasis by GLP-1. Prog. Mol. Biol. Transl. Sci. 2014, 121, 23–65. [Google Scholar] [PubMed]
- Fadini, G.P.; Avogaro, A. Cardiovascular effects of DPP-4 inhibition: Beyond GLP-1. Vascul. Pharmacol. 2011, 55, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Cheng, J.H.; Suen, C.S.; Huang, C.H.; Tsai, C.H.; Huang, L.H.; Chen, Y.R.; Wang, A.H.; Jiaang, W.T.; Hwang, M.J.; et al. The dimeric transmembrane domain of prolyl dipeptidase dpp-iv contributes to its quaternary structure and enzymatic activities. Protein Sci. 2010, 19, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Valenzuela, A.N.; Herrera, C.; Lluís, C.; Hovanessian, A.G.; Franco, R. The HIV-1 gp120 inhibits the binding of adenosine deaminase to CD26 by a mechanism modulated by CD4 and cxcr4 expression. FEBS Lett. 2000, 477, 123–128. [Google Scholar] [CrossRef]
- Green, B.D.; Flatt, P.R.; Bailey, C.J. Dipeptidyl peptidase IV (DPP IV) inhibitors-a newly emerging drug class for the treatement of type2 diabetes. Diabetes Vasc. Dis. Res. 2006, 3, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Idris, I.; Donnelly, R. Dipeptidyl peptidase-IV inhibitors: A major new class of oral antidiabetic drug. Diabetes Obes. Metab. 2007, 9, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Fossa, P. Docking-based 3D-QSAR analyses of pyrazole derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Mol. Model. 2012, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Espinoza, S.; Franchini, S.; Guariento, S.; Brasili, L.; Gainetdinov, R.R.; Fossa, P. Further insights into the pharmacology of the human trace amine-associated receptors: Discovery of novel ligands for TAAR1 by a virtual screening approach. Chem. Biol. Drug Des. 2014, 84, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; D’Ursi, P.; Moscatelli, M.; Bruno, O.; Orro, A.; Rotolo, C.; Milanesi, L.; Fossa, P. Homology modeling, docking studies and molecular dynamic simulations using graphical processing unit architecture to probe the type-11 phosphodiesterase catalytic site: A computational approach for the rational design of selective inhibitors. Chem. Biol. Drug Des. 2013, 82, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Rocca, R.; Costa, G.; Artese, A.; Parrotta, L.; Ortuso, F.; Maccioni, E.; Pinato, O.; Greco, M.L.; Sissi, C.; Alcaro, S.; et al. Hit identification of a novel dual binder for H-TELO/C-MYC G-Quadruplex by a combination of pharmacophore structure-based virtual screening and docking refinement. ChemMedChem 2016, 11, 1–14. [Google Scholar]
- Chan, D.S.; Yang, H.; Kwan, M.H.; Cheng, Z.; Lee, P.; Bai, L.P.; Jiang, Z.H.; Wong, C.Y.; Fong, W.F.; Leung, C.H.; et al. Structure-based optimization of FDA-approved drug methylene blue as a c-myc G-quadruplex DNA stabilizer. Biochimie 2011, 93, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Lee, H.M.; Yang, F.; Che, C.M.; Wong, C.C.; Abagyan, R.; Leung, C.H.; Ma, D.L. Structure-based discovery of natural-product-like TNF-α inhibitors. Angew. Chem. Int. Ed. Engl. 2010, 49, 2860–2864. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Chan, D.S.; Leung, C.H. Molecular docking for virtual screening of natural product databases. Chem. Sci. 2011, 2, 1656–1665. [Google Scholar] [CrossRef]
- Eckhardt, M.; Langkopf, E.; Mark, M.; Tadayyon, M.; Thomas, L.; Nar, H.; Pfrengle, W.; Guth, B.; Lotz, R.; Sieger, P.; et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. [Google Scholar] [CrossRef] [PubMed]
- Biftu, T.; Sinha-Roy, R.; Chen, P.; Qian, X.; Feng, D.; Kuethe, J.T.; Scapin, G.; Gao, Y.D.; Yan, Y.; Krueger, D.; et al. Omarigliptin (MK-3102): A novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes. J. Med. Chem. 2014, 57, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Gupta, M.; Singh, D.; Kumar, M.; Kaur, P. Synthesis, evaluation and molecular docking of thiazolopyrimidine derivatives as dipeptidyl peptidase iv inhibitors. Chem. Biol. Drug Des. 2012, 80, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Ikuma, Y.; Hochigai, H.; Kimura, H.; Nunami, N.; Kobayashi, T.; Uchiyama, K.; Furuta, Y.; Sakai, M.; Horiguchi, M.; Masui, Y.; et al. Discovery of 3h-imidazo[4,5-c]quinolin-4(5h)-ones as potent and selective dipeptidyl peptidase IV (DPP-4) inhibitors. Bioorg. Med. Chem. 2012, 20, 5864–5883. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.D.; Mastracchio, A.; Cox, J.M.; Eiermann, G.J.; He, H.; Lyons, K.A.; Patel, R.A.; Patel, S.B.; Petrov, A.; Scapin, G.; et al. Aminopiperidine-fused imidazoles as dipeptidyl peptidase-IV inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4097–4101. [Google Scholar] [CrossRef] [PubMed]
- Biftu, T.; Scapin, G.; Singh, S.; Feng, D.; Becker, J.W.; Eiermann, G.; He, H.; Lyons, K.; Patel, S.; Petrov, A.; et al. Rational design of a novel, potent, and orally bioavailable cyclohexylamine DPP-4 inhibitor by application of molecular modeling and x-ray crystallography of sitagliptin. Bioorg. Med. Chem. Lett. 2007, 17, 3384–3387. [Google Scholar] [CrossRef] [PubMed]
- Comunity, B. Releasenotes_accelrysdraw_4.1. 2015. Available online: http://download.accelrys.com/freeware/accelrys_draw/ReleaseNotes_AccelrysDraw_4.1.pdf (accessed on 15 April 2016).
- Singla, D.; Dhanda, S.K.; Chauhan, J.S.; Bhardwaj, A.; Brahmachari, S.K.; Open Source Drug Discovery Consortium; Raghava, G.P. Open source software and web services for designing therapeutic molecules. Curr. Top. Med. Chem. 2013, 13, 1172–1191. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking a case study of cdocker—A charmm-based md. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (iupac recommendations 1998). Pure Appl. Chem. 2009, 70, 1129–1143. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. Ligandscout 3-d pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Meslamani, J.; Li, J.; Sutter, J.; Stevens, A.; Bertrand, H.O.; Rognan, D. Protein-ligand-based pharmacophores: Generation and utility assessment in computational ligand profiling. J. Chem. Inf. Model. 2012, 52, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.R.; Wang, P.; Smith, C.L.; Zhu, B.T. Synthesis of novel estrogen receptor antagonists using metal-catalyzed coupling reactions and characterization of their biological activity. J. Med. Chem. 2013, 56, 2779–2790. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | CDOCKER Energy (kcal/mol) | Number of H-Bonds |
|---|---|---|
| 71 | −47.22 | 7 |
| 72 | −45.05 | 7 |
| 64 | −43.55 | 6 |
| 65 | −42.93 | 7 |
| 74 | −42.93 | 7 |
| 68 | −42.20 | 7 |
| 69 | −42.16 | 7 |
| 73 | −42.16 | 7 |
| 61 | −42.09 | 7 |
| 70 | −42.07 | 6 |
| Sitagliptin | −39.43 | 6 |
| Alogliptin | −25.64 | 10 |
| Vildagliptin | −5.64 | 6 |
| Compounds | Number of Features | Feature Set | Selectivity Score |
|---|---|---|---|
| Comp73 | 5 | A 1D 2N 3P 4R 5 | 11.72 |
| Comp74 | 5 | ADDH 6P | 10.89 |
| Comp55 | 6 | AAAAHP | 10.58 |
| Comp71 | 6 | AADHNR | 10.46 |
| Comp63 | 7 | AADHHPR | 10.05 |
| Comp57 | 5 | AADHP | 9.64 |
| Comp56 | 4 | AADP | 8.33 |
| Comp54 | 4 | AAAP | 7.42 |
| Comp72 | 4 | ADHH | 7.01 |
| Comp65 | 5 | ADHHR | 6.94 |
| Alogliptin | 4 | AAPP | 8.19 |
| Vildagliptin | 4 | AAHP | 7.07 |
| Sitagliptin | 3 | AHP | 5.63 |
| Compounds | Total Energy at 1000 ps (kcal/mol) | Different Time Points (ps) | ||||
|---|---|---|---|---|---|---|
| 200 | 400 | 600 | 800 | 1,000 | ||
| Comp70 | −157,714.28 | 11 | 9 | 7 | 11 | 12 |
| Comp54 | −156,891.72 | 8 | 6 | 5 | 5 | 6 |
| Comp63 | −156,672.61 | 12 | 7 | 8 | 5 | 7 |
| Comp55 | −156,558.42 | 9 | 14 | 8 | 11 | 10 |
| Comp64 | −156,449.51 | 6 | 5 | 5 | 5 | 5 |
| Comp68 | −156,440.91 | 6 | 5 | 8 | 4 | 5 |
| Comp61 | −156,331.64 | 8 | 9 | 10 | 10 | 6 |
| Comp57 | −156,297.55 | 5 | 10 | 8 | 9 | 7 |
| Comp56 | −155,533.69 | 5 | 6 | 10 | 5 | 6 |
| Comp72 | −155,408.55 | 6 | 7 | 4 | 8 | 5 |
| Comp71 | −155,240.66 | 3 | 8 | 6 | 6 | 6 |
| Comp69 | −155,187.79 | 5 | 5 | 10 | 9 | 9 |
| Comp73 | −155,187.79 | 5 | 5 | 10 | 9 | 9 |
| Comp65 | −154,805.59 | 8 | 7 | 7 | 6 | 9 |
| Comp74 | −154,805.59 | 8 | 7 | 7 | 6 | 9 |
| Vildagliptin | −156,620.70 | 7 | 7 | 8 | 10 | 7 |
| Alogliptin | −156,613.52 | 14 | 12 | 18 | 11 | 12 |
| Sitagliptin | −156,192.52 | 14 | 17 | 15 | 14 | 12 |
| Average RMSD Values for Protein-Ligand Complexes | Average RMSD Values for Ligands |
|---|---|
| 2P8S_Comp68 (1.44 ± 0.13 Å) | Comp68 (1.37 ± 0.20 Å) |
| 2P8S_Comp70 (1.52 ± 0.16 Å) | Comp70 (1.66 ± 0.20 Å) |
| 2P8S_Comp71 (1.47 ± 0.16 Å) | Comp71 (1.53 ± 0.24 Å) |
| 2P8S_Sitagliptin (1.62 ± 0.21 Å) | Sitagliptin (1.58 ± 0.21 Å) |
| 2P8S_Vildagliptin (1.51 ± 0.19 Å) | Vildagliptin (1.64 ± 0.26 Å) |
| 2P8S_Alogliptin (1.58 ± 0.20 Å) | Alogliptin (1.76 ± 0.21 Å) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meduru, H.; Wang, Y.-T.; Tsai, J.J.P.; Chen, Y.-C. Finding a Potential Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for Type-2 Diabetes Treatment Based on Molecular Docking, Pharmacophore Generation, and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2016, 17, 920. https://doi.org/10.3390/ijms17060920
Meduru H, Wang Y-T, Tsai JJP, Chen Y-C. Finding a Potential Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for Type-2 Diabetes Treatment Based on Molecular Docking, Pharmacophore Generation, and Molecular Dynamics Simulation. International Journal of Molecular Sciences. 2016; 17(6):920. https://doi.org/10.3390/ijms17060920
Chicago/Turabian StyleMeduru, Harika, Yeng-Tseng Wang, Jeffrey J. P. Tsai, and Yu-Ching Chen. 2016. "Finding a Potential Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for Type-2 Diabetes Treatment Based on Molecular Docking, Pharmacophore Generation, and Molecular Dynamics Simulation" International Journal of Molecular Sciences 17, no. 6: 920. https://doi.org/10.3390/ijms17060920