
Crystal Structure of Cytochrome P450 (CYP105P2) from Streptomyces peucetius and Its Conformational Changes in Response to Substrate Binding

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Enzymatic Role of CYP105P2 and Bioinformatics Analysis
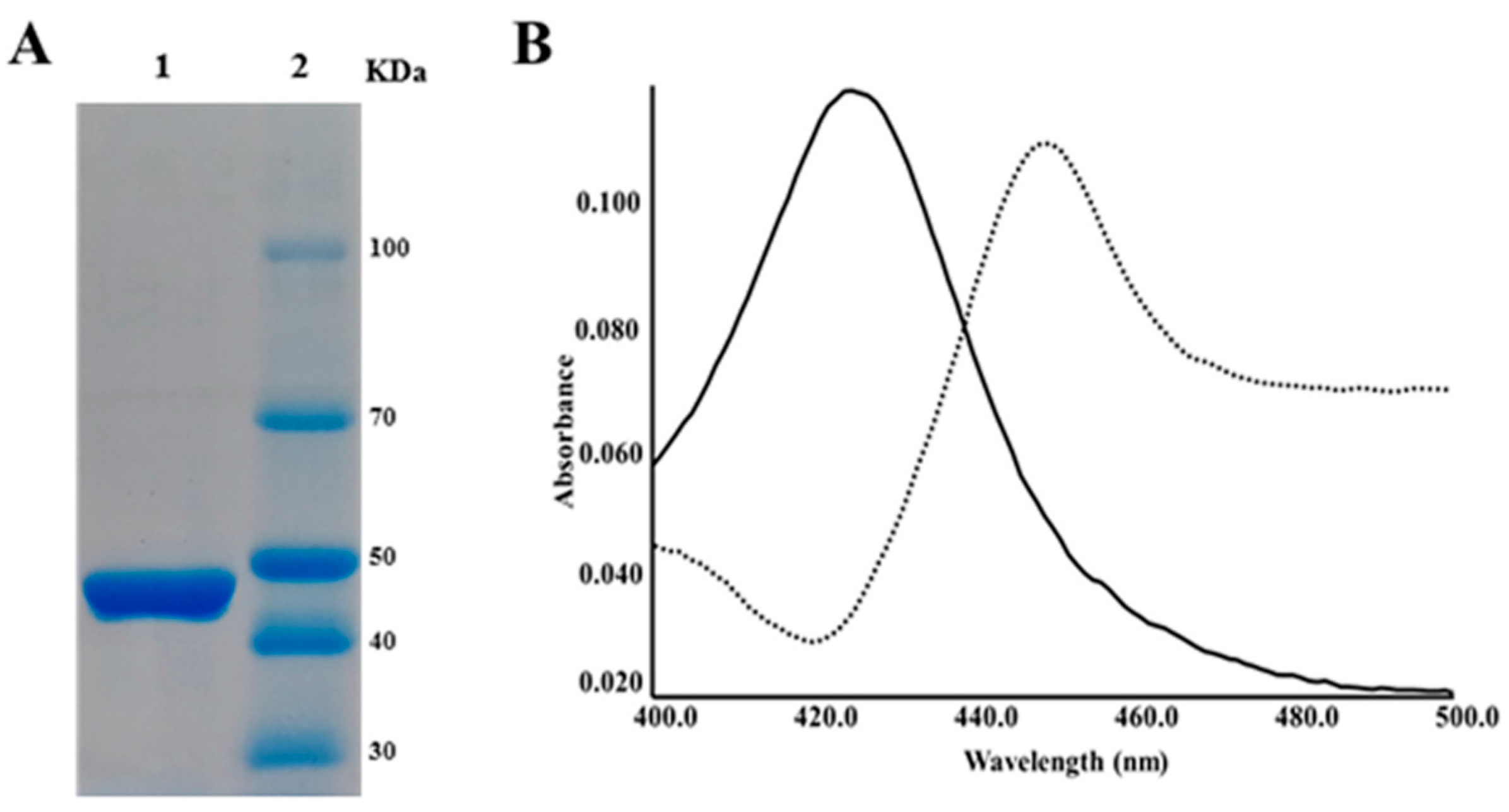
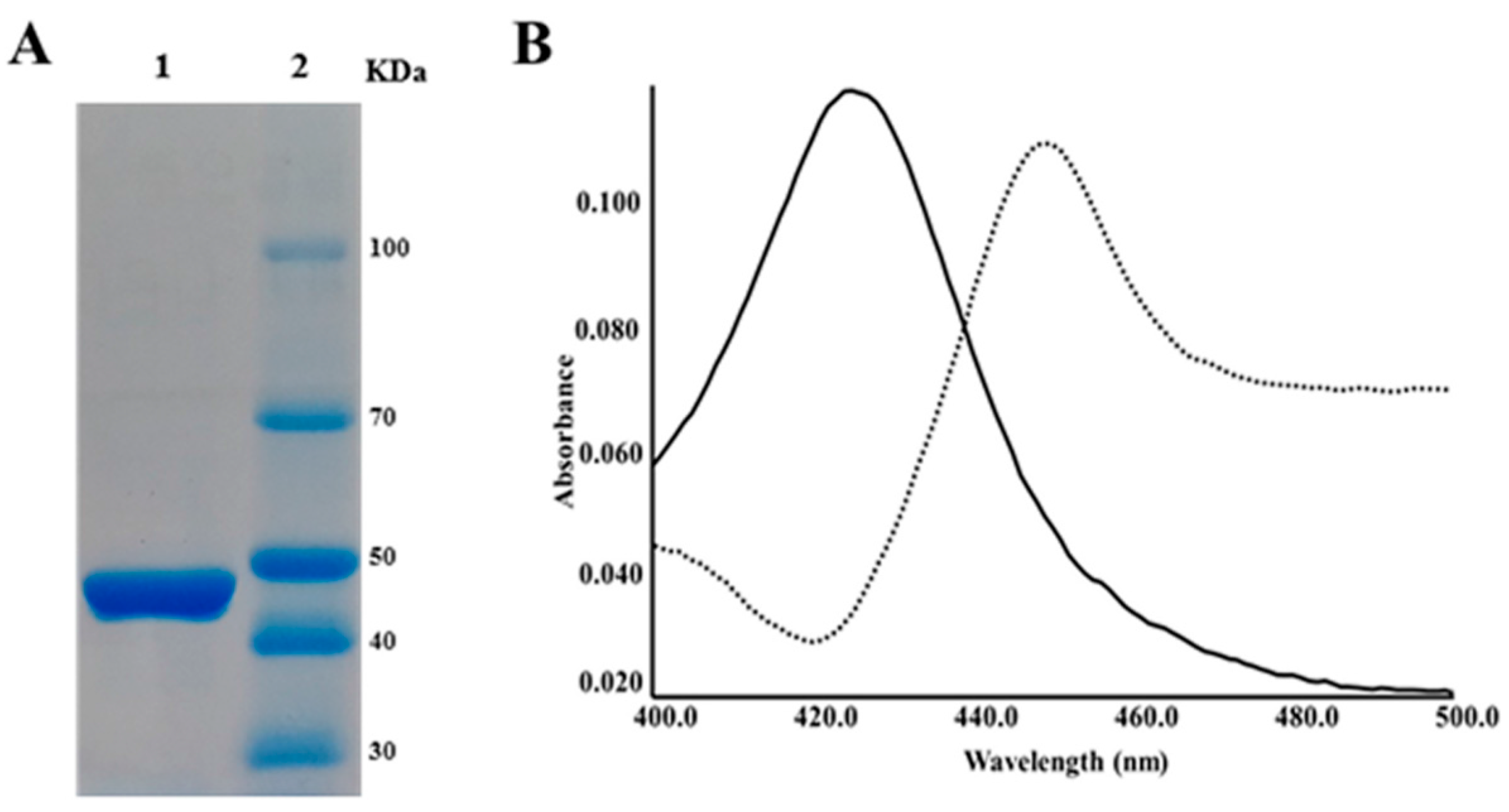
2.2. CYP105P2 over-Expression, Purification, and Spectral Analysis
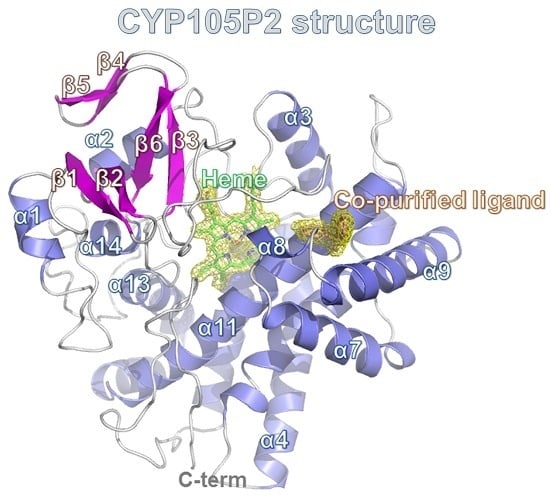
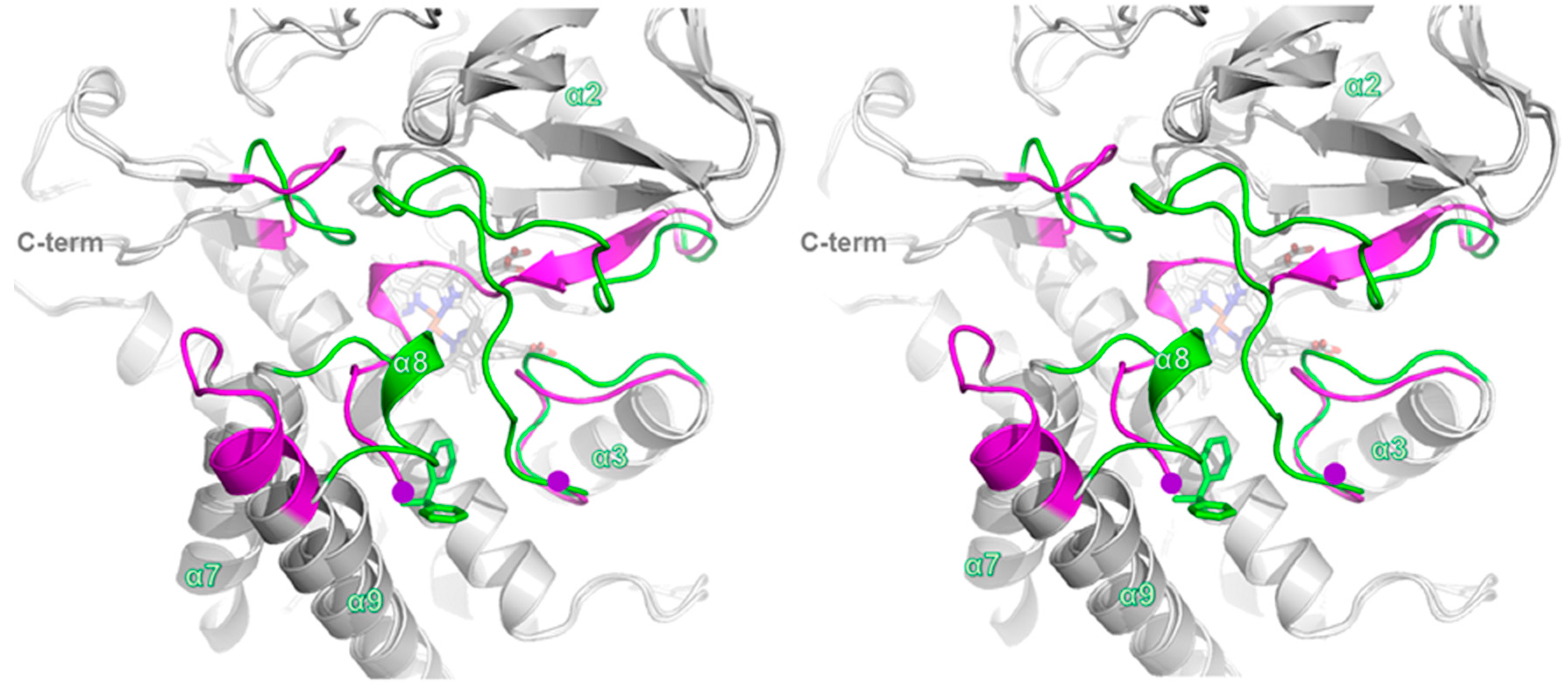
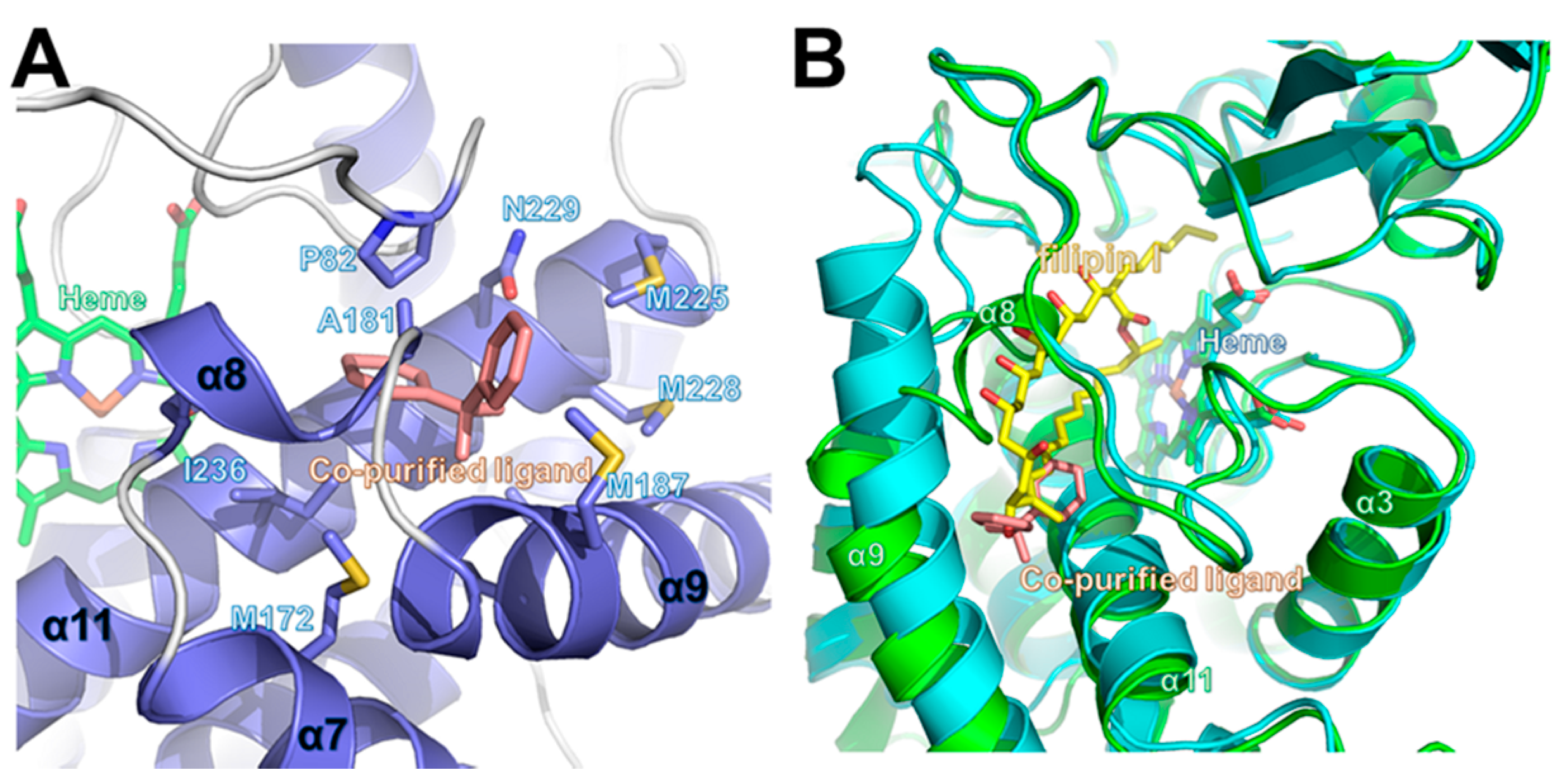
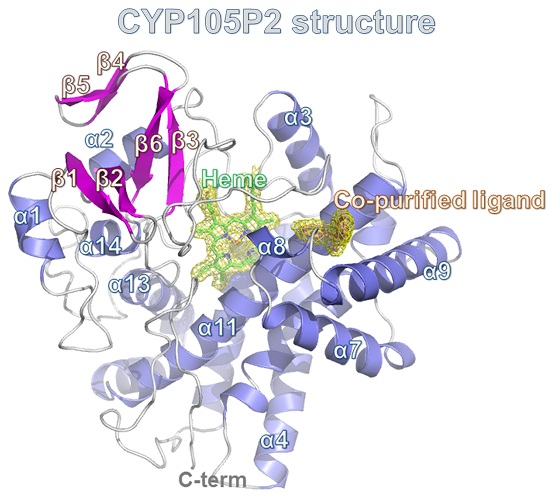
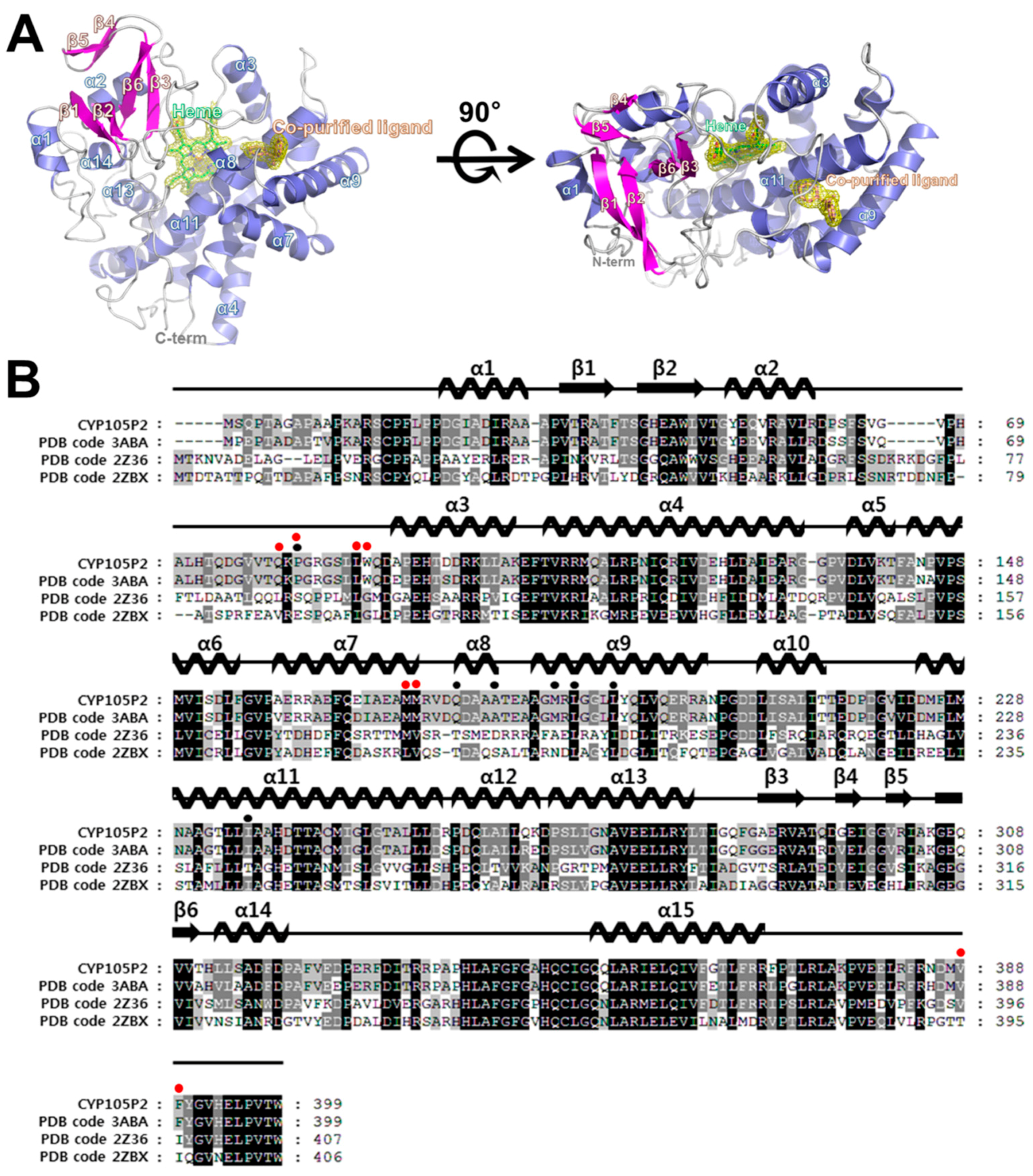
2.3. Structure of CYP105P2
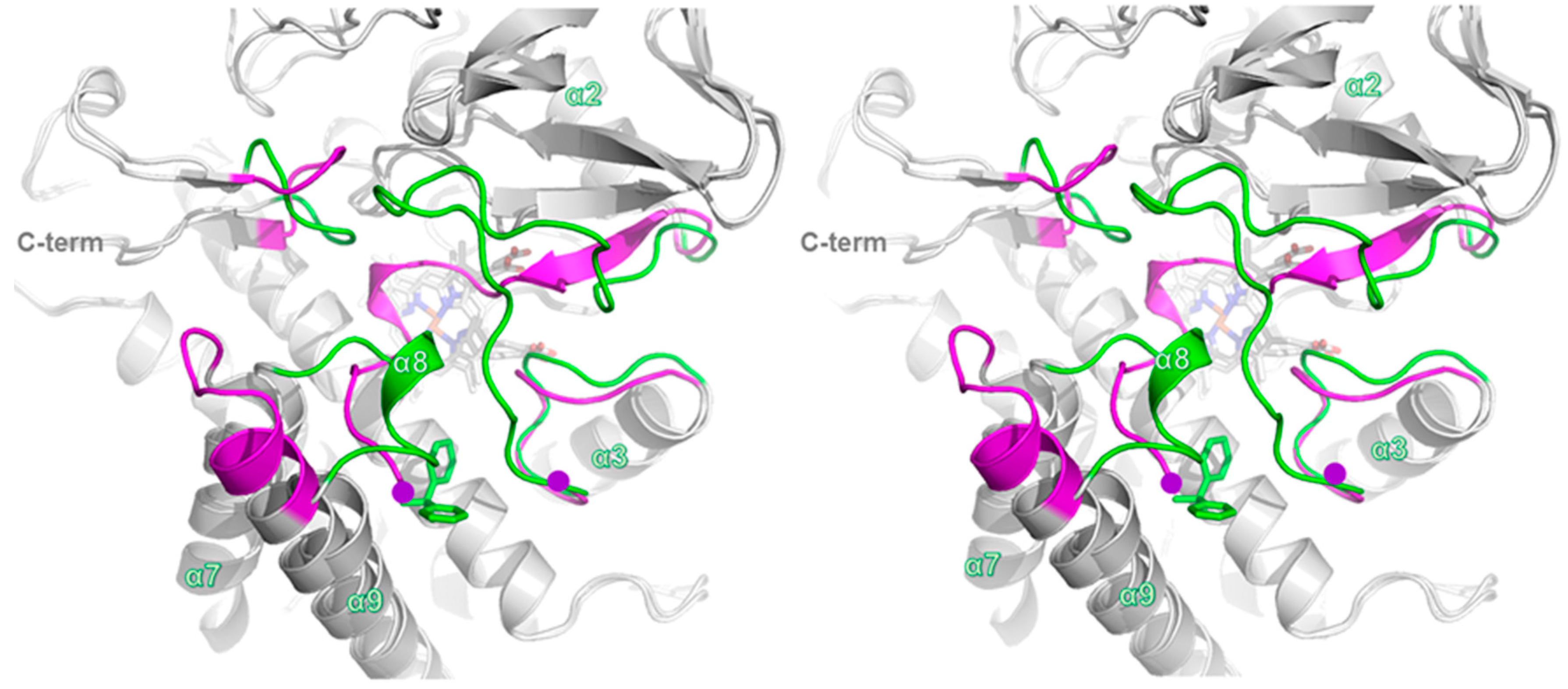
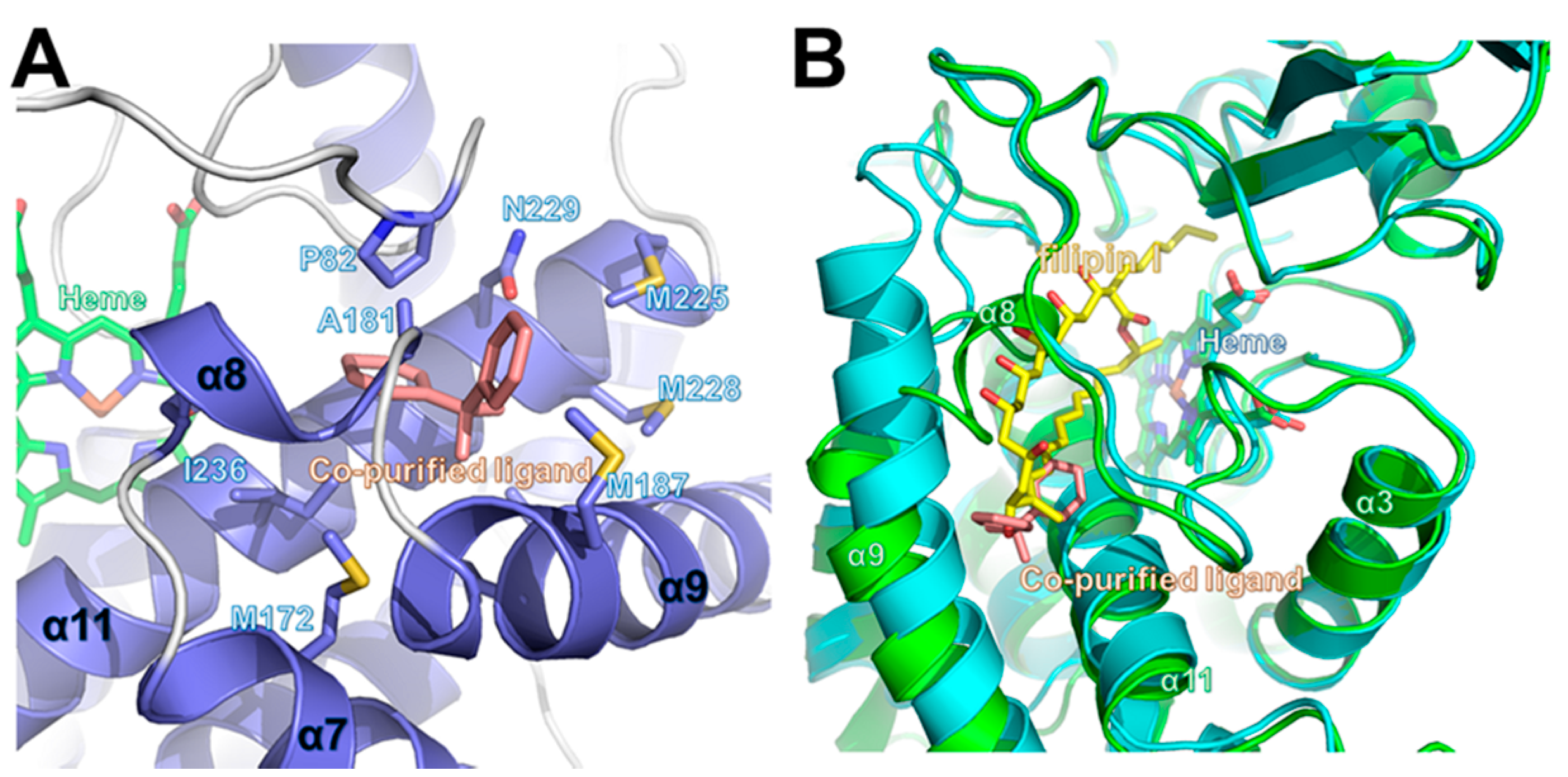
2.4. Comparison of Ligand-Bound CYP105P2 Structure with Other CYP Structures
3. Experimental Section
3.1. Purification of Streptomyces peucetius CYP105P2
3.2. Crystallization and Data Collection
3.3. Structure Determination and Refinement of CYP105P2
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CYP | Cytochrome P450 |
| ALA | 5-α-aminolevulinic acid |
| MCS | multiple-cloning site |
References
- Guengerich, F.P. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 2002, 1, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Lamb, D.C.; Waterman, M.R.; Kelly, S.L.; Guengerich, F.P. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol. 2007, 18, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Omura, T. Structural diversity of cytochrome P450 enzyme system. J. Biochem. 2010, 147, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R. Cytochrome P450: Structure, function, and generation of reactive oxygen species. Rev. Physiol. Biochem. Pharmacol. 1996, 127, 137–221. [Google Scholar] [PubMed]
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Structure 1995, 3, 41–62. [Google Scholar] [CrossRef]
- Zhang, Z.; Sibbesen, O.; Johnson, R.A.; Ortiz de Montellano, P.R. The substrate specificity of cytochrome P450cam. Bioorg. Med. Chem. 1998, 6, 1501–1508. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Guengerich, F.P.; Kelly, S.L.; Waterman, M.R. Exploiting Streptomyces coelicolor A3(2) P450s as a model for application in drug discovery. Expert Opin. Drug Metab. Toxicol. 2006, 2, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P. Streptomyces: Molecular Biology and Biotechnology; Caister Academic Press: Norfolk, UK, 2011. [Google Scholar]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends Biotechnol. 2012, 30, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Skaug, T.; Song, H.L.; Jackson, C.J.; Podust, L.M.; Waterman, M.R.; Kell, D.B.; Kelly, D.E.; Kelly, S.L. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem. 2002, 277, 24000–24005. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Shinose, M.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M.; Omura, S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003, 21, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Ikeda, H.; Nelson, D.R.; Ishikawa, J.; Skaug, T.; Jackson, C.; Omura, S.; Waterman, M.R.; Kelly, S.L. Cytochrome p450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2). Biochem. Biophys. Res. Commun. 2003, 307, 610–619. [Google Scholar] [CrossRef]
- McLean, K.J.; Clift, D.; Lewis, D.G.; Sabri, M.; Balding, P.R.; Sutcliffe, M.J.; Leys, D.; Munro, A.W. The preponderance of P450s in the Mycobacterium tuberculosis genome. Trends Microbiol. 2006, 14, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Waterman, M.R.; Zhao, B. Streptomyces cytochromes P450: Applications in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R.; Urlacher, V.B. Cytochromes P450 as promising catalysts for biotechnological application: Chances and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Moody, S.C.; Hider, R.C.; Lei, L.; Kelly, S.L.; Waterman, M.R.; Lamb, D.C. Structural analysis of cytochrome P450 105N1 involved in the biosynthesis of the zincophore, coelibactin. Int. J. Mol. Sci. 2012, 13, 8500–8513. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Xu, L.H.; Fushinobu, S.; Shoun, H.; Komatsu, M.; Cane, D.E.; Ikeda, H. Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: Hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis. J. Antibiot. 2011, 64, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.C.; Loveridge, E.J. CYP105-diverse structures, functions and roles in an intriguing family of enzymes in Streptomyces. J. Appl. Microbiol. 2014, 117, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Fushinobu, S.; Takamatsu, S.; Wakagi, T.; Ikeda, H.; Shoun, H. Regio- and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J. Biol. Chem. 2010, 285, 16844–16853. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Ikeda, H.; Liu, L.; Arakawa, T.; Wakagi, T.; Shoun, H.; Fushinobu, S. Structural basis for the 4’-hydroxylation of diclofenac by a microbial cytochrome P450 monooxygenase. Appl. Microbiol. Biotechnol. 2015, 99, 3081–3091. [Google Scholar] [CrossRef] [PubMed]
- Niraula, N.P.; Bhattarai, S.; Lee, N.R.; Sohng, J.K.; Oh, T.J. Biotransformation of flavones by CYP105P2 from Streptomyces peucetius. J. Microbiol. Biotechnol. 2012, 22, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Tsuchido, T.; Matsumura, Y. Molecular cloning and characterization of cytochrome P450 and ferredoxin genes involved in bisphenol A degradation in Sphingomonas bisphenolicum strain AO1. J. Appl. Microbiol. 2008, 105, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- Kanth, B.K.; Liou, K.; Sohng, J.K. Homology modeling, binding site identification and docking in flavones hydroxylase CYP105P2 in Streptomyces peucetius ATCC27952. Comput. Biol. Chem. 2010, 34, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, Y.; Imoto, N.; Fujii, Y.; Fujii, T.; Arisawa, A.; Tamura, T. Crystal structure of cytochrome P450 MoxA from Nonomuraea recticatena (CYP105). Biochem. Biophys. Res. Commun. 2007, 361, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Shinkyo, R.; Hayashi, K.; Yoneda, S.; Yamada, M.; Kamakura, M.; Ikushiro, S.; Shiro, Y.; Sakaki, T. Crystal structure of CYP105A1 (P450SU-1) in complex with 1α,25-dihydroxyvitamin D3. Biochemistry 2008, 47, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Fushinobu, S.; Ikeda, H.; Wakagi, T.; Shoun, H. Crystal structures of cytochrome P450 105P1 from Streptomyces avermitilis: Conformational flexibility and histidine ligation state. J. Bacteriol. 2009, 191, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Rimal, H.; Lee, S.W.; Lee, J.H.; Oh, T.J. Understanding of real alternative redox partner of Streptomyces peucetius DoxA: Prediction and validation using in silico and in vitro analyses. Arch. Biochem. Biophys. 2015, 585, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- GeneDoc. Available online: www.psc.edu/index.php/user-resources/software/genedoc (accessed on 18 March 2015).
- Holm, L.; Sander, C. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 1995, 20, 478–480. [Google Scholar] [CrossRef]
- Li, S.; Tietz, D.R.; Rutaganira, F.U.; Kells, P.M.; Anzai, Y.; Kato, F.; Pochapsky, T.C.; Sherman, D.H.; Podust, L.M. Substrate recognition by the multifunctional cytochrome P450 MycG in mycinamicin hydroxylation and epoxidation reactions. J. Biol. Chem. 2012, 287, 37880–37890. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.J.; Hans, M.; Meijrink, B.; van Scheppingen, W.B.; Vollebregt, A.; Tee, K.L.; van der Laan, J.M.; Leys, D.; Munro, A.W.; van den Berg, M.A. Single-step fermentative production of the cholesterol-lowering drug pravastatin via reprogramming of Penicillium chrysogenum. Proc. Natl. Acad. Sci. USA 2015, 112, 2847–2852. [Google Scholar] [CrossRef] [PubMed]
- Montemiglio, L.C.; Parisi, G.; Scaglione, A.; Sciara, G.; Savino, C.; Vallone, B. Functional analysis and crystallographic structure of clotrimazole bound OleP, a cytochrome P450 epoxidase from Streptomyces antibioticus involved in oleandomycin biosynthesis. Biochim. Biophys. Acta 2016, 1860, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Sugimoto, H.; Shinkyo, R.; Yamada, M.; Ikeda, S.; Ikushiro, S.; Kamakura, M.; Shiro, Y.; Sakaki, T. Structure-based design of a highly active vitamin D hydroxylase from Streptomyces griseolus CYP105A1. Biochemistry 2008, 47, 11964–11972. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.2r3pre; Schrödinger LLC: New York, NY, USA, 2002. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PDB Code | DALI Score | Sequence % ID (Aligned Residue Number/Total Residue Number) | Reference |
|---|---|---|---|---|
| CYP105P1 | 3ABA | 60.0 | 92% (385/397) | [21] |
| MycG | 3ZSN | 50.1 | 37% (380/393) | [33] |
| MoxA | 2Z36 | 49.9 | 37% (380/393) | [26] |
| CYP105AS1 | 4OQS | 49.7 | 44% (368/384) | [34] |
| OleP | 4XE3 | 48.7 | 37% (378/394) | [35] |
| CYP107W1 | 4WQ0 | 48.4 | 38% (375/397) | It has not yet been published |
| CYP105N1 | 3TYW | 47.3 | 41% (379/399) | It has not yet been published |
| CYP105A1 | 3CV8 | 46.8 | 42% (379/402) | [36] |
| CYP105D6 | 3ABB | 46.7 | 39% (371/383) | [21] |
| Data Set | CYP105P2 + Biphenyl Molecule |
|---|---|
| X-ray source | PAL 7A beam line |
| Space group | C2 |
| Wavelength (Å) | 0.97934 |
| Resolution (Å) | 50.00–2.10 (2.14–2.10) |
| Total reflections | 1146063 |
| Unique reflections | 165620 (8315) |
| Average I/σ (I) | 39.3 (4.6) |
| Rmerge a | 0.104 (0.823) |
| Redundancy | 6.9 (7.1) |
| Completeness (%) b | 99.5 (100.0) |
| Refinement | |
| Resolution range (Å) | 50.0–2.10 (2.16–2.10) |
| No. of reflections in working set | 157,003 (11,120) |
| No. of reflections in test set | 8315 (564) |
| No. of amino acid residues | 1580 |
| No. of water molecules | 657 |
| Rcryst b | 0.200 (0.317) |
| Rfree c | 0.243 (0.352) |
| RMS bond length (Å) | 0.019 |
| RMS bond angle (°) | 1.927 |
| Average B value (Å2) (protein) | 48.510 |
| Average B value (Å2) (solvent) | 51.306 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.W.; Lee, J.-H.; Rimal, H.; Park, H.; Lee, J.H.; Oh, T.-J. Crystal Structure of Cytochrome P450 (CYP105P2) from Streptomyces peucetius and Its Conformational Changes in Response to Substrate Binding. Int. J. Mol. Sci. 2016, 17, 813. https://doi.org/10.3390/ijms17060813
Lee CW, Lee J-H, Rimal H, Park H, Lee JH, Oh T-J. Crystal Structure of Cytochrome P450 (CYP105P2) from Streptomyces peucetius and Its Conformational Changes in Response to Substrate Binding. International Journal of Molecular Sciences. 2016; 17(6):813. https://doi.org/10.3390/ijms17060813
Chicago/Turabian StyleLee, Chang Woo, Joo-Ho Lee, Hemraj Rimal, Hyun Park, Jun Hyuck Lee, and Tae-Jin Oh. 2016. "Crystal Structure of Cytochrome P450 (CYP105P2) from Streptomyces peucetius and Its Conformational Changes in Response to Substrate Binding" International Journal of Molecular Sciences 17, no. 6: 813. https://doi.org/10.3390/ijms17060813