
Molecular Insight into Gut Microbiota and Rheumatoid Arthritis

 , ,
, ,
Abstract
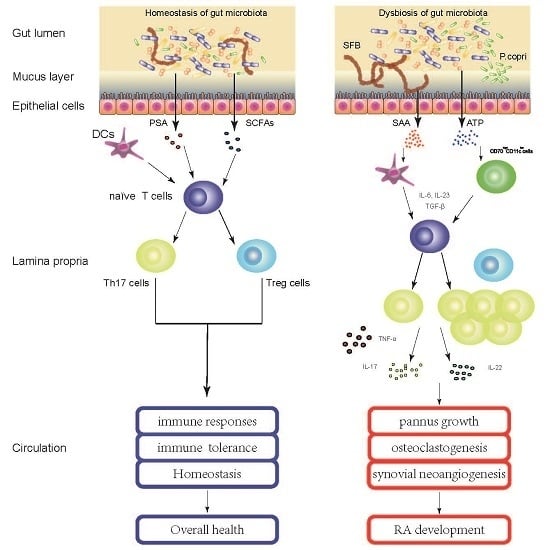
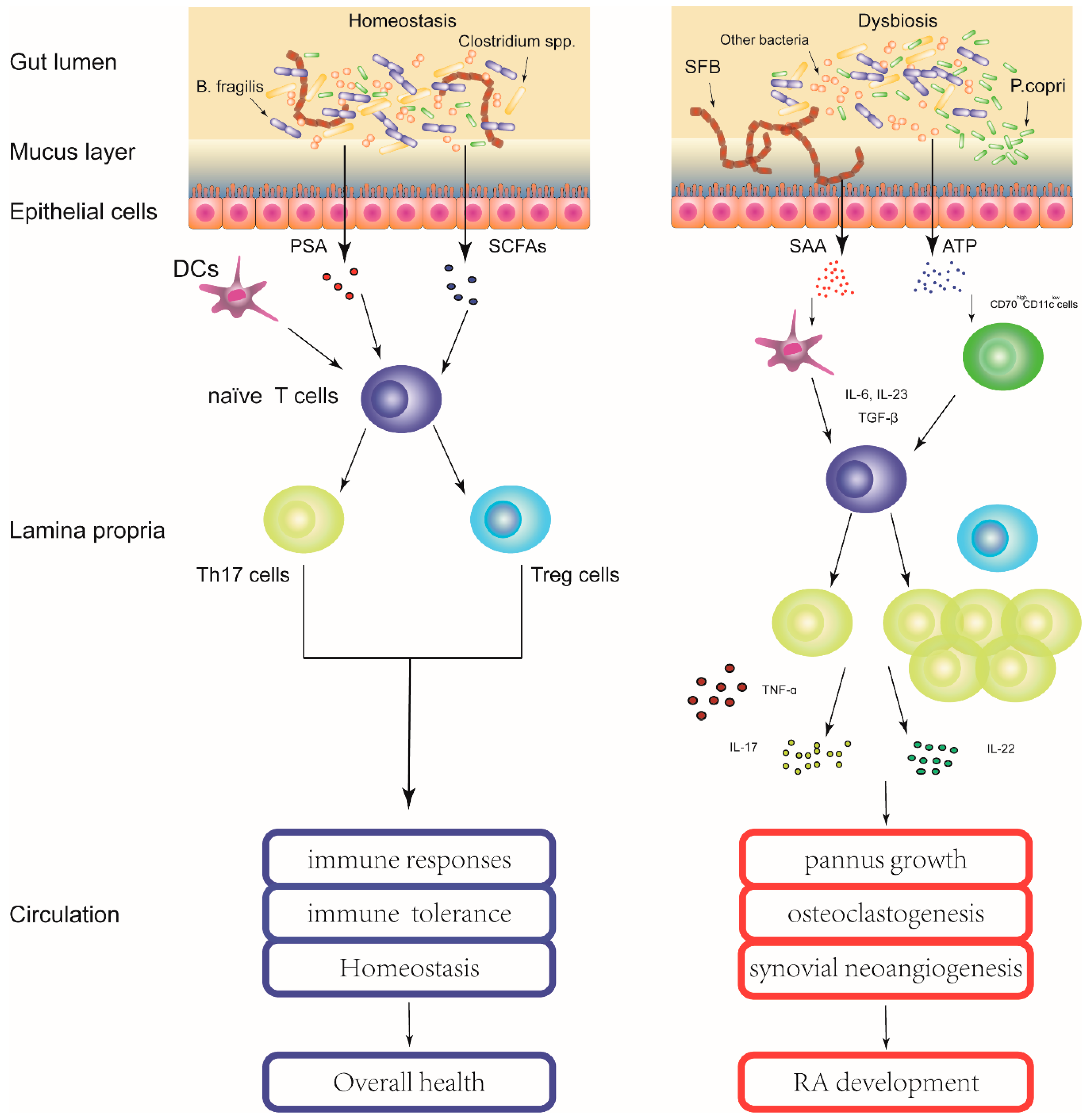
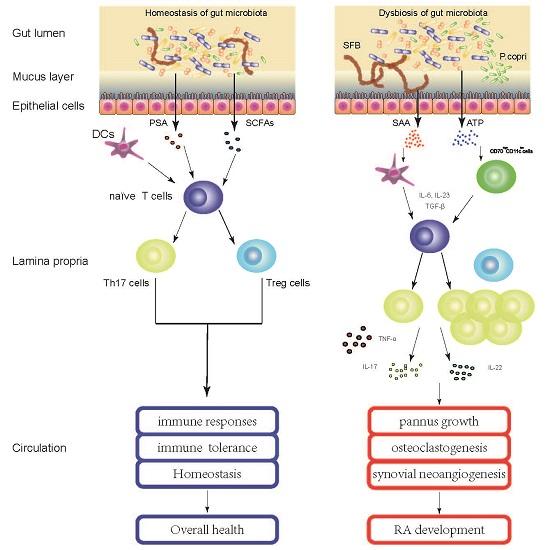
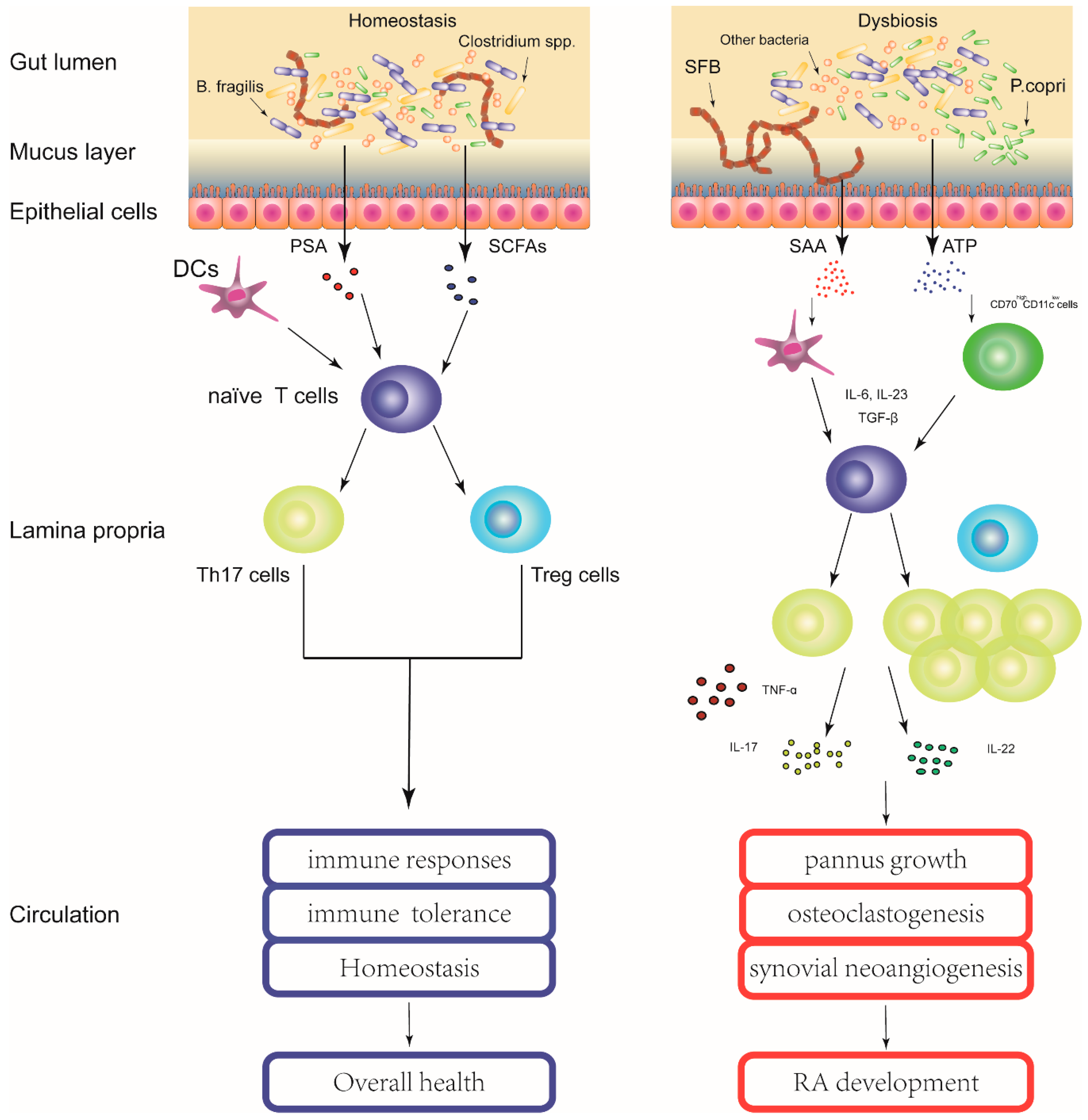
:
1. Introduction
2. The Human Gut Microbiota
3. Alterations of Gut Microbiota in RA
4. Gut Microbiota Contribute to the Pathogenesis of RA
5. Perspective: Gut Microbiota as Potential Drug Targets for RA?
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RA | Rheumatoid Arthritis |
| SCFAs | short-chain fatty acids |
| Treg cell | regulatory T cell |
| Th cell | T helper cell |
| PSA | polysaccharide A |
| SFB | Segmented Filamentous Bacterium |
| IL | Interleukin |
| NORA | new-onset untreated RA |
| anti-CCP | autoantibodies anticyclic citrullinated peptide |
| DMARDs | disease modifying anti-rheumatic drugs |
| ACPAs | anti-citrullinated protein autoantibodies |
| SAA | serum amyloid A |
| ATP | adenosine 5′-triphosphate |
| GM-CSF | granulocyte–macrophage colonystimulating factor |
| TNF-α | tumor necrosis factor ɑ |
| VEGF | vascular endothelial growth factor |
| FLS | fibroblast like synoviocytes |
| MMP | matrix metalloproteinase |
| NSAIDs | non-steroidal anti-inflammatory drugs |
References
- Tobón, G.J.; Youinou, P.; Saraux, A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J. Autoimmun. 2010, 35, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Deane, K.D.; Scher, J.U. Gene, environment, microbiome and mucosal immune tolerance in rheumatoid arthritis. Rheumatology 2016, 55, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; MacGregor, A.J.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W.E. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Br. J. Rheumatol. 1993, 32, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; Newman, J.; MacGregor, A.J. Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheum. 1996, 39, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Salgado, E.; Bes-Rastrollo, M.; de Irala, J.; Carmona, L.; Gómez-Reino, J.J. High sodium intake is associated with self-reported rheumatoid arthritis: A cross sectional and case control analysis within the SUN cohort. Medicine 2015, 94, 924. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Littman, D.R.; Abramson, S.B. Review: Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol. 2016, 68, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Telesford, K.M.; Yan, W.; Ochoa-Reparaz, J.; Pant, A.; Kircher, C.; Christy, M.A.; Begum-Haque, S.; Kasper, D.L.; Kasper, L.H. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+) Foxp3(+) T cells and Treg function. Gut Microbes 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kasper, D.L. Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Curr. Opin. Immunol. 2010, 22, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Lunney, P.C.; Leong, R.W. Review article: Ulcerative colitis, smoking and nicotine therapy. Aliment. Pharmacol. Ther. 2012, 36, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Zhou, L.; Zhang, J.; Wang, B. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int. J. Clin. Exp. Pathol. 2015, 8, 5153–5160. [Google Scholar] [PubMed]
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes 2015, 64, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS ONE 2015, 10, 0137429. [Google Scholar]
- Gill, T.; Asquith, M.; Rosenbaum, J.T.; Colbert, R.A. The intestinal microbiome in spondyloarthritis. Curr. Opin. Rheumatol. 2015, 27, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Vaahtovuo, J.; Munukka, E.; Korkeamäki, M.; Luukkainen, R.; Toivanen, P. Fecal microbiota in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1500–1505. [Google Scholar] [PubMed]
- Liu, X.; Zou, Q.; Zeng, B.; Fang, Y.; Wei, H. Analysis of fecal Lactobacillus community structure in patients with early rheumatoid arthritis. Curr. Microbiol. 2013, 67, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Gul’neva, M.I.U.; Noskov, S.M. Colonic microbial biocenosis in rheumatoid arthritis. Klin. Med. 2011, 89, 45–48. [Google Scholar]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011, 17, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Weisman, M.H.; Simonian, P.L.; Lynch, D.A.; Sachs, P.B.; Pedraza, I.F.; Harrington, A.R.; Kolfenbach, J.R.; Striebich, C.C.; Pham, Q.N.; et al. Brief report: Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arthritis Rheum. 2012, 64, 1756261. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Joshua, V.; Klareskog, L.; Malmström, V. Mechanisms involved in triggering rheumatoid arthritis. Immunol. Rev. 2016, 269, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Koenders, M.I.; Devesa, I.; Roelofs, M.F.; Radstake, T.R.; Heuvelmans-Jacobs, M.; Akira, S.; Nicklin, M.J.; Ribeiro-Dias, F.; et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Investig. 2008, 118, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Lv, T.T.; Yin, Z.J.; Wang, X.B. Elevated circulating Th17 and follicular helper CD4+ T cells in patients with rheumatoid arthritis. APMIS 2015, 123, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; van den Bersselaar, L.; Oppers-Walgreen, B.; Schwarzenberger, P.; Coenen-de Roo, C.J.; Kolls, J.K.; Joosten, L.A.; van den Berg, W.B. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-κB ligand/osteoprotegerin balance. J. Immunol. 2003, 17, 2655–2662. [Google Scholar] [CrossRef]
- Kim, K.W.; Kim, H.R.; Park, J.Y.; Park, J.S.; Oh, H.J.; Woo, Y.J.; Park, M.K.; Cho, M.L.; Lee, S.H. Interleukin-22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis Rheum. 2012, 64, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Lee, J.H.; Kim, S.I. IL-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clin. Rheumatol. 2006, 25, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.M.; Mullan, R.; McCormick, J.; Connolly, M.; Sullivan, O.; Fitzgerald, O.; Bresnihan, B.; Veale, D.J.; Fearon, U. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: Synergy with tumour necrosis factor-α, Oncostatin M and response to biologic therapies. Arthritis Res. Ther. 2009, 11, R113. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-β and TNF-α, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Maurice, M.M.; Nakamura, H.; Gringhuis, S.; Okamoto, T.; Yoshida, S.; Kullmann, F.; Lechner, S.; van der Voort, E.A.; Leow, A.; Versendaal, J.; et al. Expression of the thioredoxin-thioredoxin reductase system in the inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42, 2430–2439. [Google Scholar] [CrossRef]
- Toivanen, P. Normal intestinal microbiota in the aetiopathogenesis of rheumatoid arthritis. Ann. Rheum Dis. 2003, 62, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.; Luckey, D.; Yeoman, C.J.; Marietta, E.V.; Berg Miller, M.E.; Murray, J.A.; White, B.A.; Taneja, V. Loss of sex and age driven differences in the gut microbiome characterize arthritis-susceptible 0401 mice but not arthritis-resistant 0402 mice. PLoS ONE 2012, 7, e36095. [Google Scholar]
- Jia, W.; Li, H.; Zhao, L.; Nicholson, J.K. Gut microbiota: A potential new territory for drug targeting. Nat. Rev. Drug Discov. 2008, 7, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lian, F.; Zhao, L.; Zhao, Y.; Chen, X.; Zhang, X.; Guo, Y.; Zhang, C.; Zhou, Q.; Xue, Z.; et al. Structural modulation of gut microbiota during alleviation of type 2 diabetes with a Chinese herbal formula. ISME J. 2015, 9, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Lin, C.S.; Lu, C.C.; Martel, J.; Ko, Y.F.; Ojcius, D.M.; Tseng, S.F.; Wu, T.R.; Chen, Y.Y.; Young, J.D.; et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2015, 6, 7489. [Google Scholar] [CrossRef] [PubMed]
- Pullar, T.; Hunter, J.A.; Capell, H.A. Which component of sulphasalazine is active in rheumatoid arthritis? Br. Med. J. 1985, 290, 1535–1538. [Google Scholar] [CrossRef]
- Ogrendik, M. Effects of clarithromycin in patients with active rheumatoid arthritis. Curr. Med. Res. Opin. 2007, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Saviola, G.; Abdi Ali, L.; Rossini, P.; Campostrini, L.; Coppini, A.; Gori, M.; Ianaro, A.; Bucci, M.; de Nucci, G.; Cirino, G. Clarithromycin in rheumatoid arthritis patients not responsive to disease-modifying antirheumatic drugs: An open, uncontrolled pilot study. Clin. Exp. Rheumatol. 2002, 20, 373–378. [Google Scholar] [PubMed]
- Dorożyńska, I.; Majewska-Szczepanik, M.; Marcińska, K.; Szczepanik, M. Partial depletion of natural gut flora by antibiotic aggravates collagen induced arthritis (CIA) in mice. Pharmacol. Rep. 2014, 66, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Syer, S.D.; Blackler, R.W.; Martin, R.; de Palma, G.; Rossi, L.; Verdu, E.; Bercik, P.; Surette, M.G.; Aucouturier, A.; Langella, P.; et al. NSAID enteropathy and bacteria: A complicated relationship. J. Gastroenterol. 2015, 50, 387–393. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Kwon, H.K.; Lee, C.G.; Yi, H.J.; Park, J.A.; Lim, S.Y.; Hwang, K.C.; Jeon, Y.H.; Im, S.H. Lactobacillus casei suppresses experimental arthritis by down-regulating T helper 1 effector functions. Mol. Immunol. 2008, 45, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Studygroups | Sample Type | Technology Employed | Bacterial Taxa (↓low, ↑enriched) | Ref. |
|---|---|---|---|---|
| Early RA (51) vs. Fibromyalgia (50) | Stool | 16S rRNA hybridization, and DNA-staining | ↓Bifidobacteria, ↓Bacteroides-Porphyromonas-Prevotella, ↓Bacteroides fragile, ↓Clostridium coccoides | [24] |
| Early RA (15) vs. Healthy (15) | Stool | Quantitative real-time PCR | ↑Lactobacillus | [25] |
| New-Onset RA (44) vs. Healthy (28) | Stool | 16S rRNA gene and WGS sequencing | ↑Prevotella copri, ↓Bacteroidetes | [26] |
| RA (30) vs.Healthy (30) | Stool | 16S rRNA gene and WGS sequencing | ↑Enterococci, ↑Clostridia, ↑Colibacteria, ↓Lactobacteria | [27] |
| Treatment-naïve RA (94) vs. Healthy (97) | Stool, Dental, Saliva | Metagenomic shotgun sequencing | ↑Lactobacillus salivarius, ↑Gordonibacter pamelaeae, ↑Clostridium asparagiforme, …, ↓Haemophilus spp. | [28] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; He, B.; Liu, J.; Feng, H.; Ma, Y.; Li, D.; Guo, B.; Liang, C.; Dang, L.; Wang, L.; et al. Molecular Insight into Gut Microbiota and Rheumatoid Arthritis. Int. J. Mol. Sci. 2016, 17, 431. https://doi.org/10.3390/ijms17030431
Wu X, He B, Liu J, Feng H, Ma Y, Li D, Guo B, Liang C, Dang L, Wang L, et al. Molecular Insight into Gut Microbiota and Rheumatoid Arthritis. International Journal of Molecular Sciences. 2016; 17(3):431. https://doi.org/10.3390/ijms17030431
Chicago/Turabian StyleWu, Xiaohao, Bing He, Jin Liu, Hui Feng, Yinghui Ma, Defang Li, Baosheng Guo, Chao Liang, Lei Dang, Luyao Wang, and et al. 2016. "Molecular Insight into Gut Microbiota and Rheumatoid Arthritis" International Journal of Molecular Sciences 17, no. 3: 431. https://doi.org/10.3390/ijms17030431