
Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish

 and
and 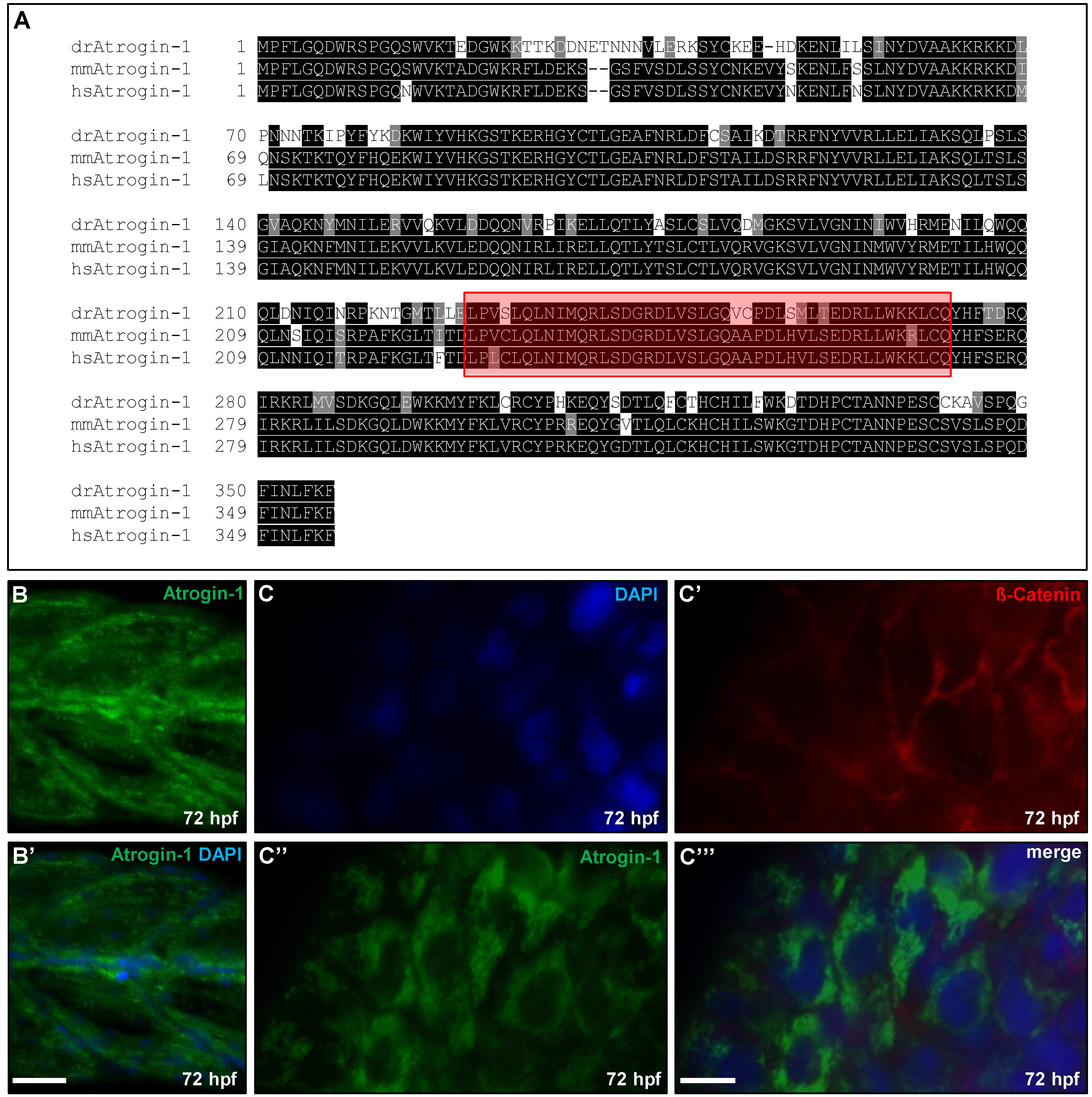{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Zebrafish Atrogin-1 Localizes Cytoplasmatically in Cardiac and Skeletal Muscle Cells
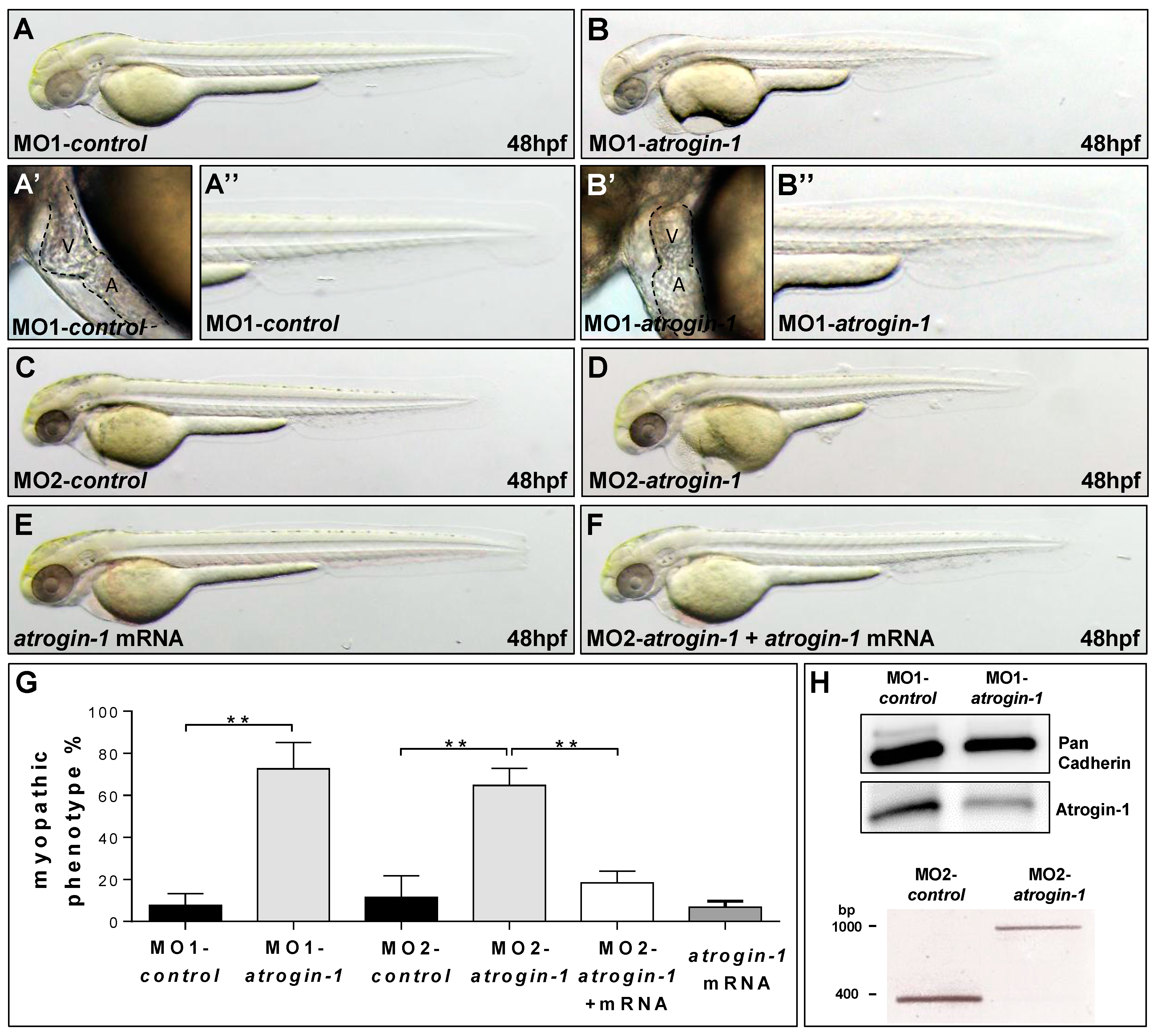
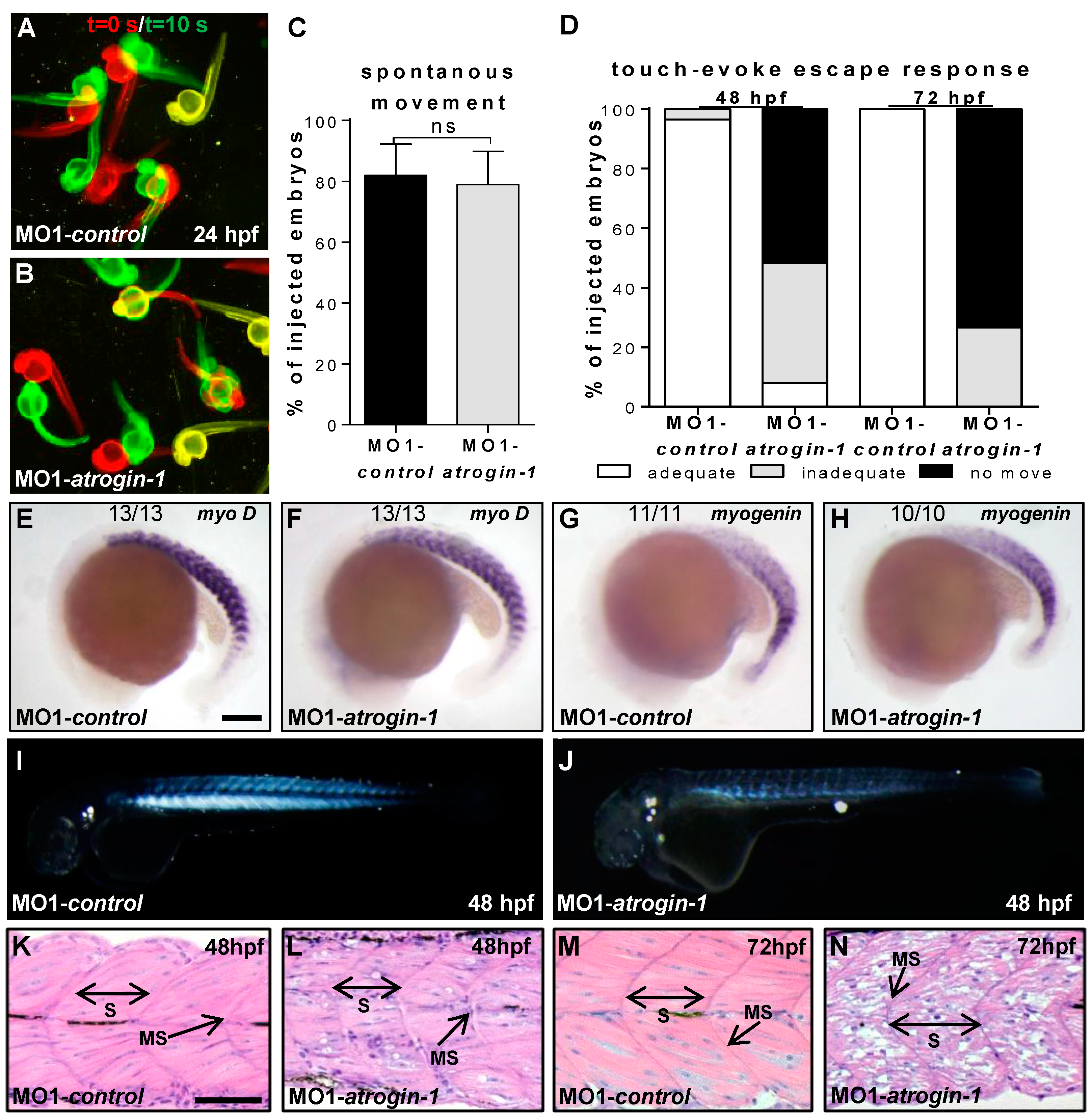
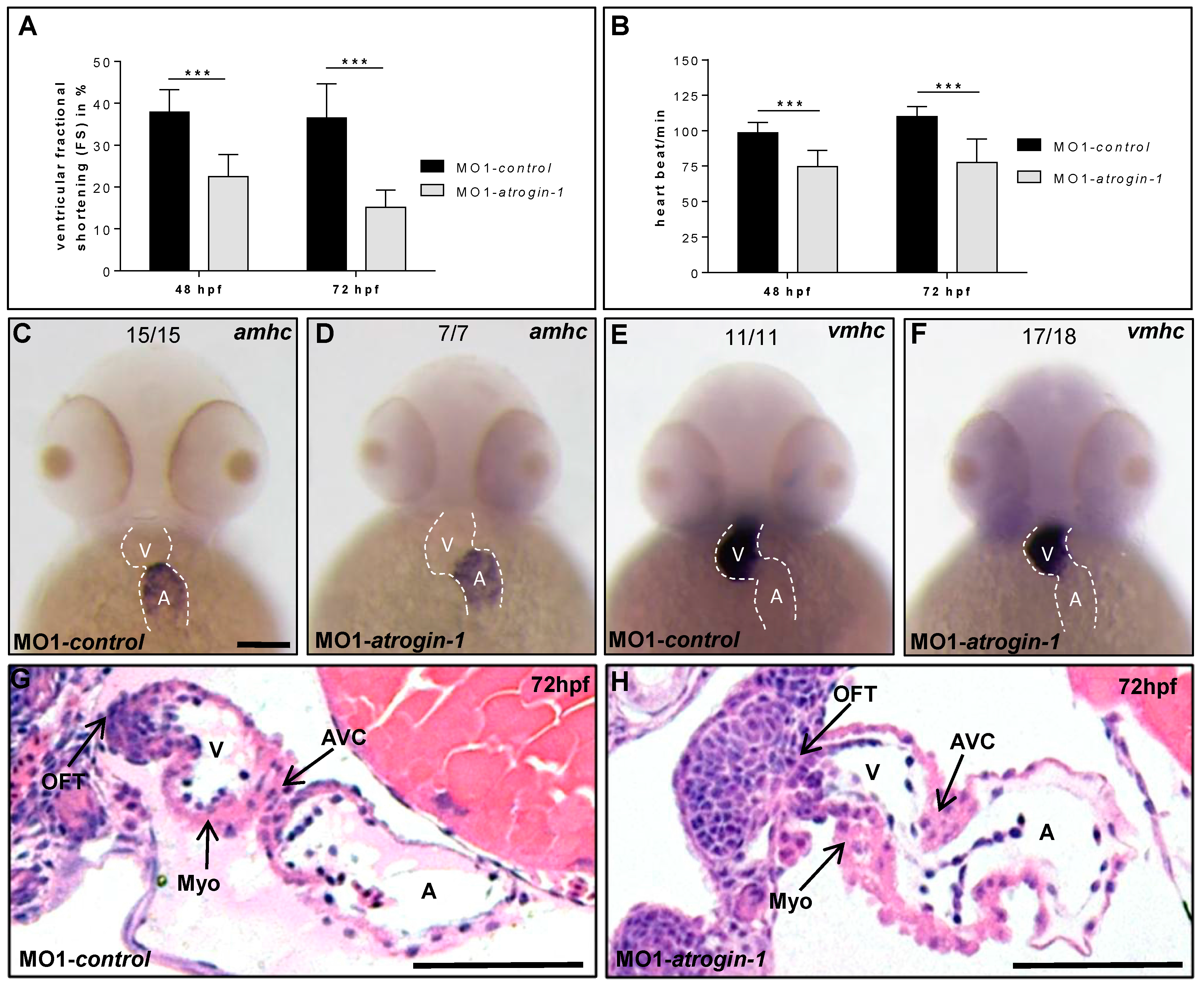
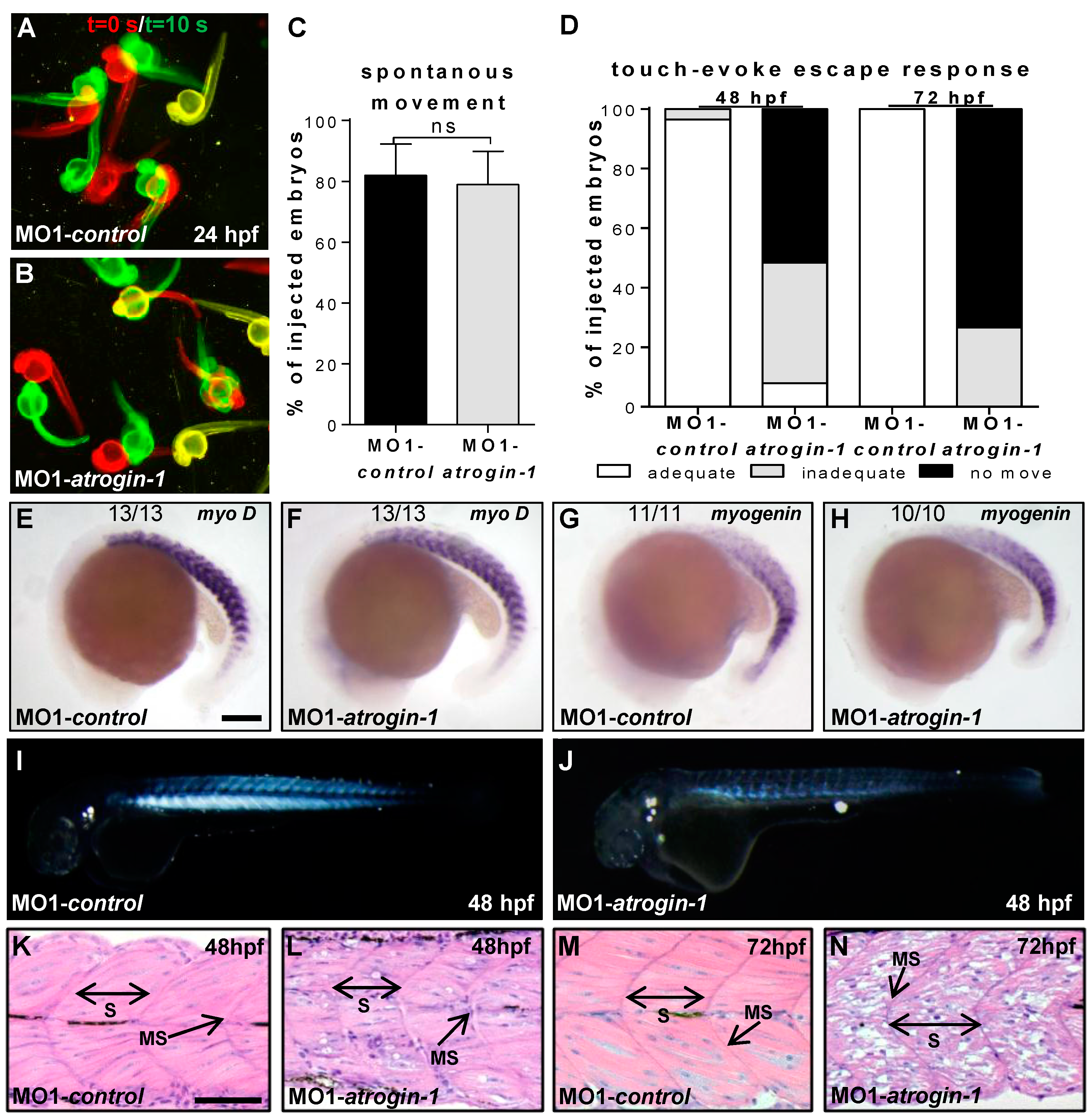
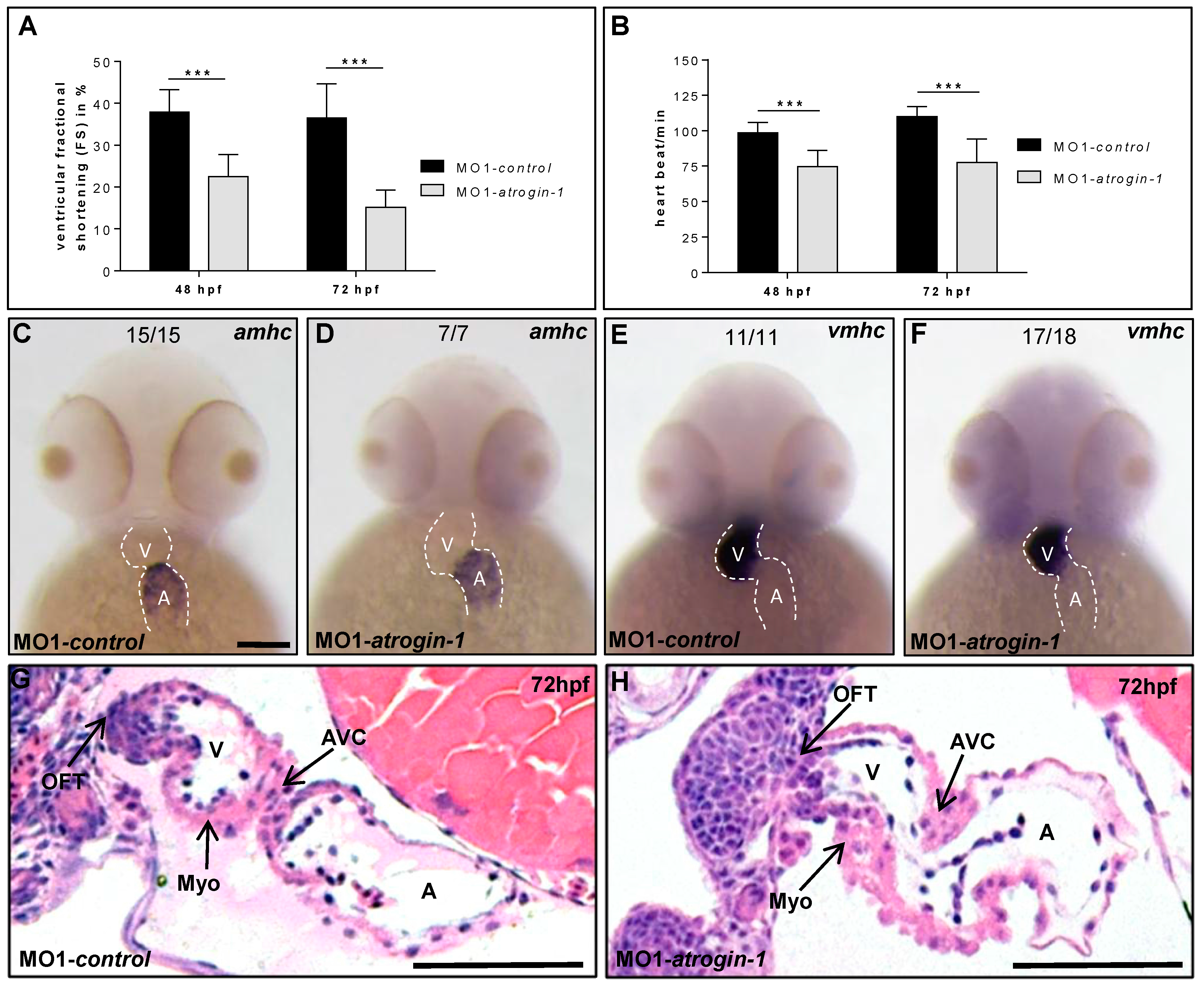
2.2. Inactivation of Atrogin-1 Leads to Myopathy and Heart Failure in Zebrafish Embryos


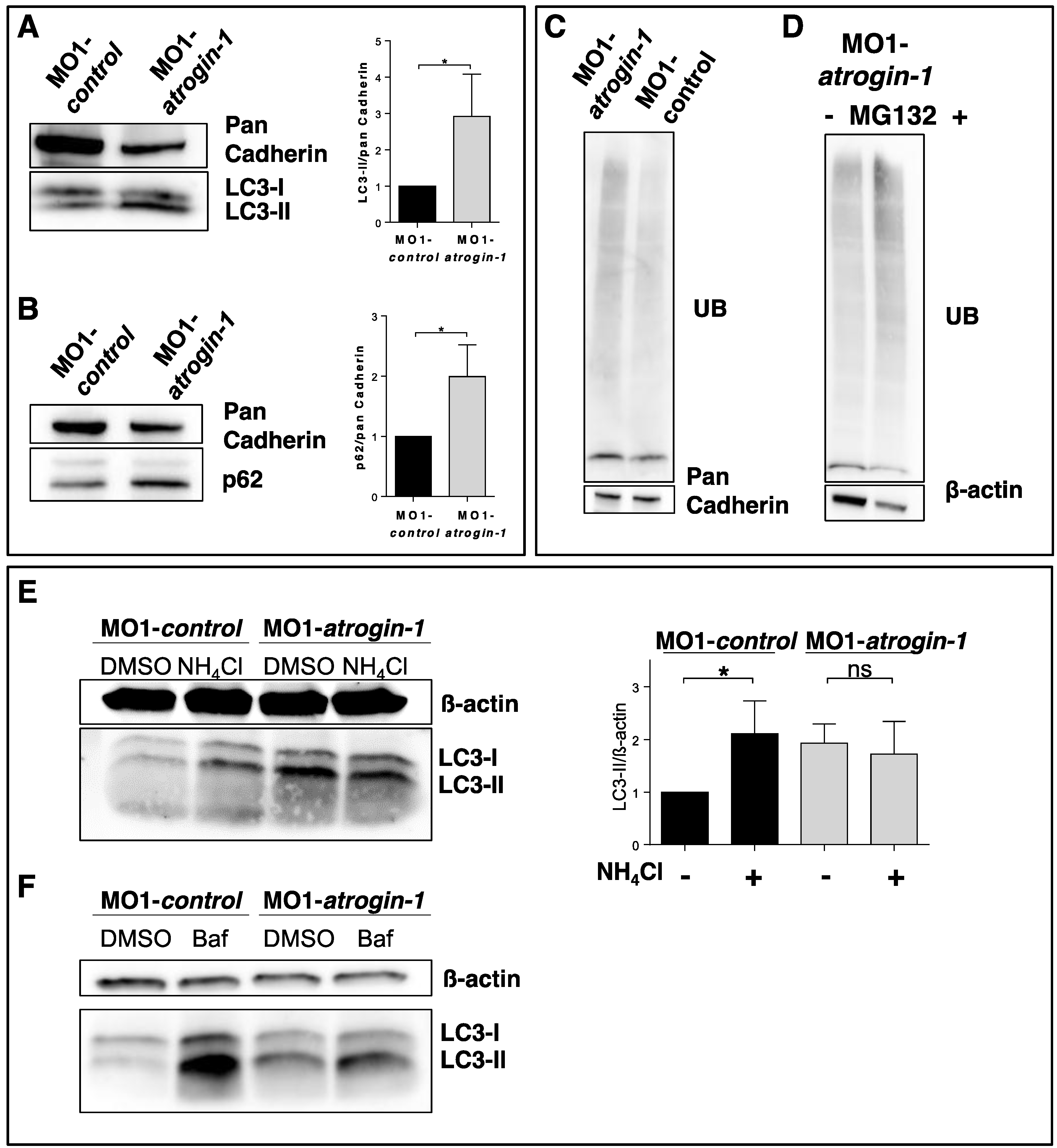
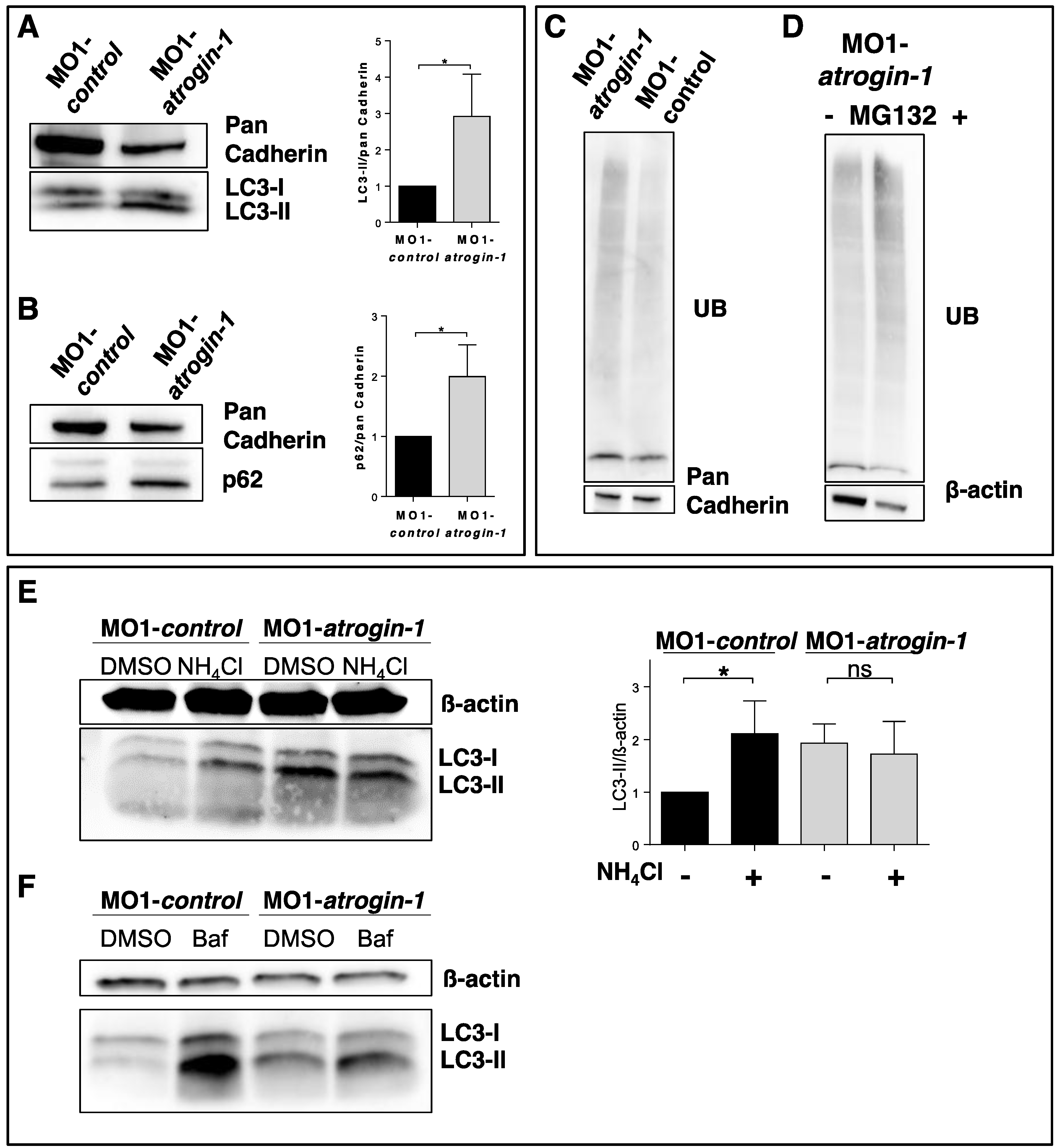
2.3. The Autophagy/Lysosome Machinery Is Impaired in Atrogin-1 Deficient Zebrafish Embryos
2.4. Atrogin-1 Deficiency Leads to an Autophagy-Related Ultrastructural Muscle Pathology

3. Discussion
4. Materials and Methods
4.1. Zebrafish Strains
4.2. Morpholino and mRNA Injection Procedures
4.3. Isolation of Embryonic Zebrafish Hearts and Immunostaining
4.4. Zebrafish Protein Lysate Extraction and Western Blot Analysis
4.5. Ammonium Chloride, Bafilomycin A1, MG132 Treatment
4.6. Reverse Transcriptase (RT)-PCR and RNA in Situ Hybridization
4.7. Birefringence, Spontaneous Movement, and Touch-Evoke Escape Response Assay
4.8. Histology and Transmission Electron Microscopy
4.9. Functional Assessment and Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell. Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Cuervo, A.M. Selective autophagy: Talking with the ups. Cell Biochem. Biophys. 2013, 67, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.; Stoj, J.; Karpowicz, P.; Osmulski, P.A.; Gaczynska, M. The proteasome in health and disease. Curr. Pharm. Des. 2013, 19, 1010–1028. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The function of autophagy in neurodegenerative diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Taneike, M.; Otsu, K. The role of autophagic degradation in the heart. J. Mol. Cell. Cardiol. 2015, 78, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamaguchi, O.; Otsu, K. Degradation systems in heart failure. J. Mol. Cell. Cardiol. 2015, 84, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Mearini, G.; Carrier, L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: Mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2011, 50, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Peter, M. Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Unal, C.; Matthias, J.; Steinert, M.; Eichinger, L. The phenotypes of ATG9, ATG16 and ATG9/16 knock-out mutants imply autophagy-dependent and -independent functions. Open Biol. 2015, 5, 150008. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Marko, M.; Strucksberg, K.H.; Behrens, J.; Wittig, I.; Gartner, L.; Winter, L.; Chevessier, F.; Matthias, J.; Turk, M.; et al. VCP and PSMF1: Antagonistic regulators of proteasome activity. Biochem. Biophys. Res. Commun. 2015, 463, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Zaglia, T.; Milan, G.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Piasentini, E.; Voltarelli, V.A.; Chiavegato, D.; Brum, P.C.; Glass, D.J.; et al. Cardiac sympathetic neurons provide trophic signal to the heart via β2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc. Res. 2013, 97, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific f-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Bührdel, J.B.; Hirth, S.; Kessler, M.; Westphal, S.; Forster, M.; Manta, L.; Wiche, G.; Schoser, B.; Schessl, J.; Schroder, R.; et al. In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2015, 461, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Robbins, J. Proteotoxicity: An underappreciated pathology in cardiac disease. J. Mol. Cell. Cardiol. 2014, 71, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Kedar, V.; Zhang, C.; McDonough, H.; Arya, R.; Wang, D.Z.; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Investig. 2004, 114, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Willis, M.S.; Lockyer, P.; Miller, N.; McDonough, H.; Glass, D.J.; Patterson, C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of forkhead proteins. J. Clin. Investig. 2007, 117, 3211–3223. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: The role of the IGF1-Akt-mTOR-Foxo pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D.; et al. The foxo3a transcription factor regulates cardiac myocyte size downstream of akt signaling. J. Biol. Chem. 2005, 280, 20814–20823. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting Foxo transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Hart, P.D.; Young, M.R. Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: studies of a pathogenic mycobacterium and a nonpathogenic yeast. J. Exp. Med. 1991, 174, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Imarisio, S.; Fleming, A.; Rose, C.; Krishna, G.; Heard, P.; Quick, M.; Korolchuk, V.I.; Renna, M.; Sarkar, S.; et al. Antioxidants.can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 2010, 19, 3413–3429. [Google Scholar] [CrossRef] [PubMed]
- Varga, M.; Fodor, E.; Vellai, T. Autophagy in zebrafish. Methods 2015, 75, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; de Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. 2011, 155, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Maloyan, A.; Robbins, J. Autophagy in desmin-related cardiomyopathy: Thoughts at the halfway point. Autophagy 2010, 6, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Pattison, J.S.; Osinska, H.; Robbins, J. Atg7 induces basal autophagy and rescues autophagic deficiency in cryabr120g cardiomyocytes. Circul. Res. 2011, 109, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Pattison, J.S.; Robbins, J. Autophagy and proteotoxicity in cardiomyocytes. Autophagy 2011, 7, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. Vcp/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef] [PubMed]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef] [PubMed]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Carrier, L. The ubiquitin-proteasome system in cardiomyopathies. Curr. Opin. Cardiol. 2011, 26, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Staudt, D.; Stainier, D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.; Meder, B.; Just, S.; Laufer, C.; Berger, I.; Weber, S.; Katus, H.A.; Rottbauer, W. In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2009, 390, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Rottbauer, W.; Just, S. Recent progress in the use of zebrafish for novel cardiac drug discovery. Exp. Opin. Drug Discov. 2015, 10, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Pott, A.; Rottbauer, W.; Just, S. Functional genomics in zebrafish as a tool to identify novel antiarrhythmic targets. Curr. Med. Chem. 2014, 21, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Pylatiuk, C.; Sanchez, D.; Mikut, R.; Alshut, R.; Reischl, M.; Hirth, S.; Rottbauer, W.; Just, S. Automatic zebrafish heartbeat detection and analysis for zebrafish embryos. Zebrafish 2014, 11, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Spomer, W.; Pfriem, A.; Alshut, R.; Just, S.; Pylatiuk, C. High-throughput screening of zebrafish embryos using automated heart detection and imaging. J. Lab. Autom. 2012, 17, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Berger, I.M.; Just, S.; Rottbauer, W. Loss of dihydrolipoyl succinyltransferase (dlst) leads to reduced resting heart rate in the zebrafish. Basic Res. Cardiol. 2015, 110, 14. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, X. Immunostaining of dissected zebrafish embryonic heart. J. Vis. Exp. 2012, 59, e3510. [Google Scholar] [CrossRef] [PubMed]
- Just, S.; Meder, B.; Berger, I.M.; Etard, C.; Trano, N.; Patzel, E.; Hassel, D.; Marquart, S.; Dahme, T.; Vogel, B.; et al. The myosin-interacting protein smyd1 is essential for sarcomere organization. J. Cell Sci. 2011, 124, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Just, S.; Berger, I.M.; Meder, B.; Backs, J.; Keller, A.; Marquart, S.; Frese, K.; Patzel, E.; Rauch, G.J.; Tubingen Screen, C.; et al. Protein kinase d2 controls cardiac valve formation in zebrafish by regulating histone deacetylase 5 activity. Circulation 2011, 124, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Just, S.; Vogel, B.; Rudloff, J.; Gartner, L.; Dahme, T.; Huttner, I.; Zankl, A.; Katus, H.A.; Rottbauer, W. Junb-cbfbeta signaling is essential to maintain sarcomeric z-disc structure and when defective leads to heart failure. J. Cell Sci. 2010, 123, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bühler, A.; Kustermann, M.; Bummer, T.; Rottbauer, W.; Sandri, M.; Just, S. Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish. Int. J. Mol. Sci. 2016, 17, 187. https://doi.org/10.3390/ijms17020187
Bühler A, Kustermann M, Bummer T, Rottbauer W, Sandri M, Just S. Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish. International Journal of Molecular Sciences. 2016; 17(2):187. https://doi.org/10.3390/ijms17020187
Chicago/Turabian StyleBühler, Anja, Monika Kustermann, Tiziana Bummer, Wolfgang Rottbauer, Marco Sandri, and Steffen Just. 2016. "Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish" International Journal of Molecular Sciences 17, no. 2: 187. https://doi.org/10.3390/ijms17020187