
Henrin A: A New Anti-HIV Ent-Kaurane Diterpene from Pteris henryi

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
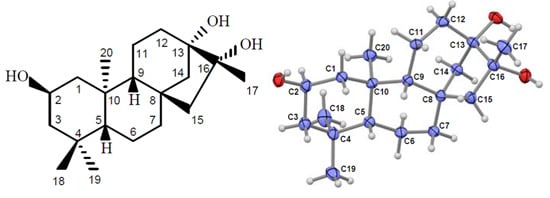
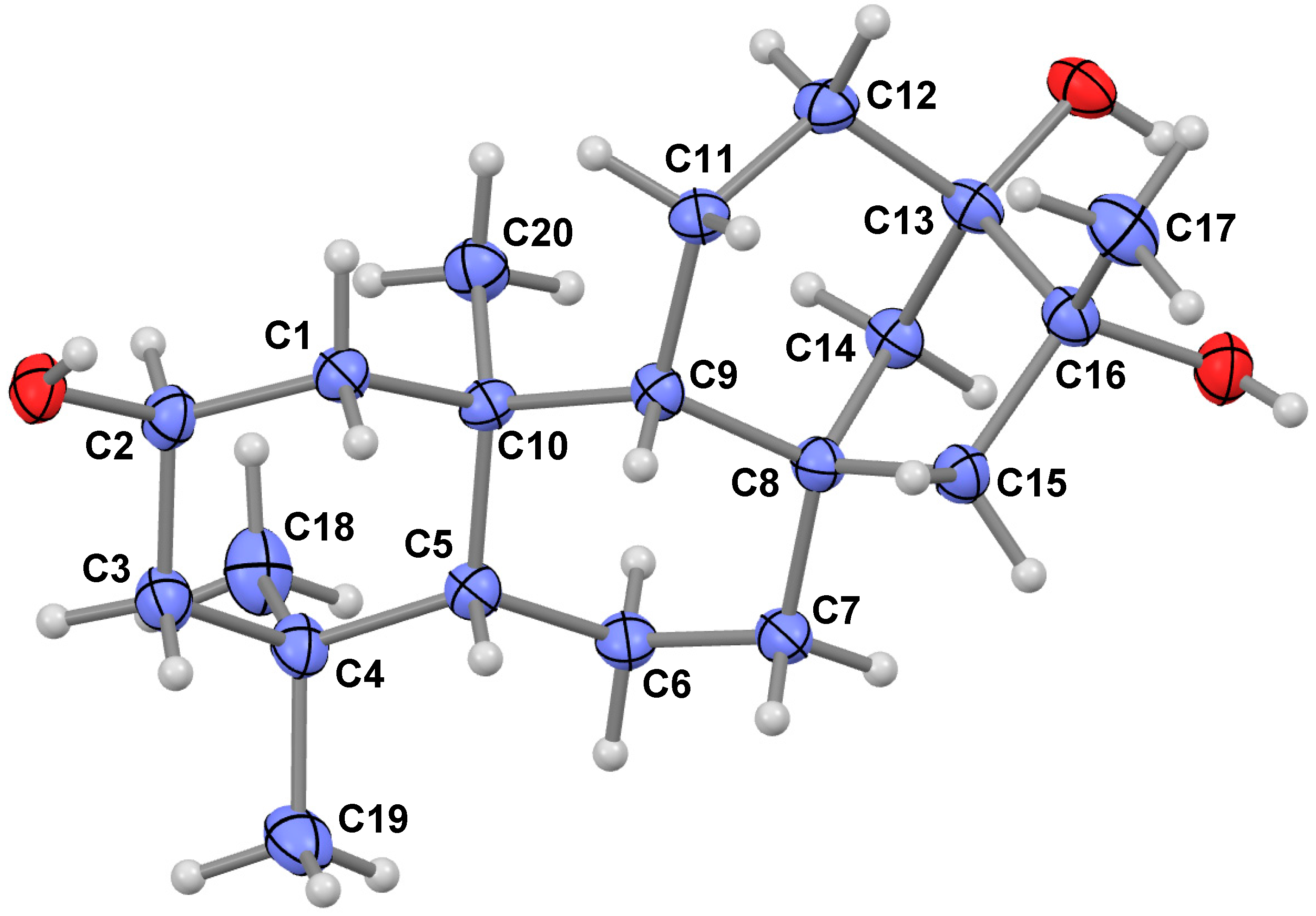
2.1. Compound Identification

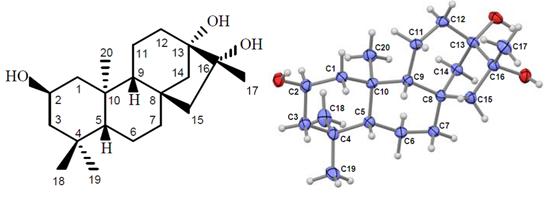{kind=link}
{kind=link}
{kind=link}
{kind=link}
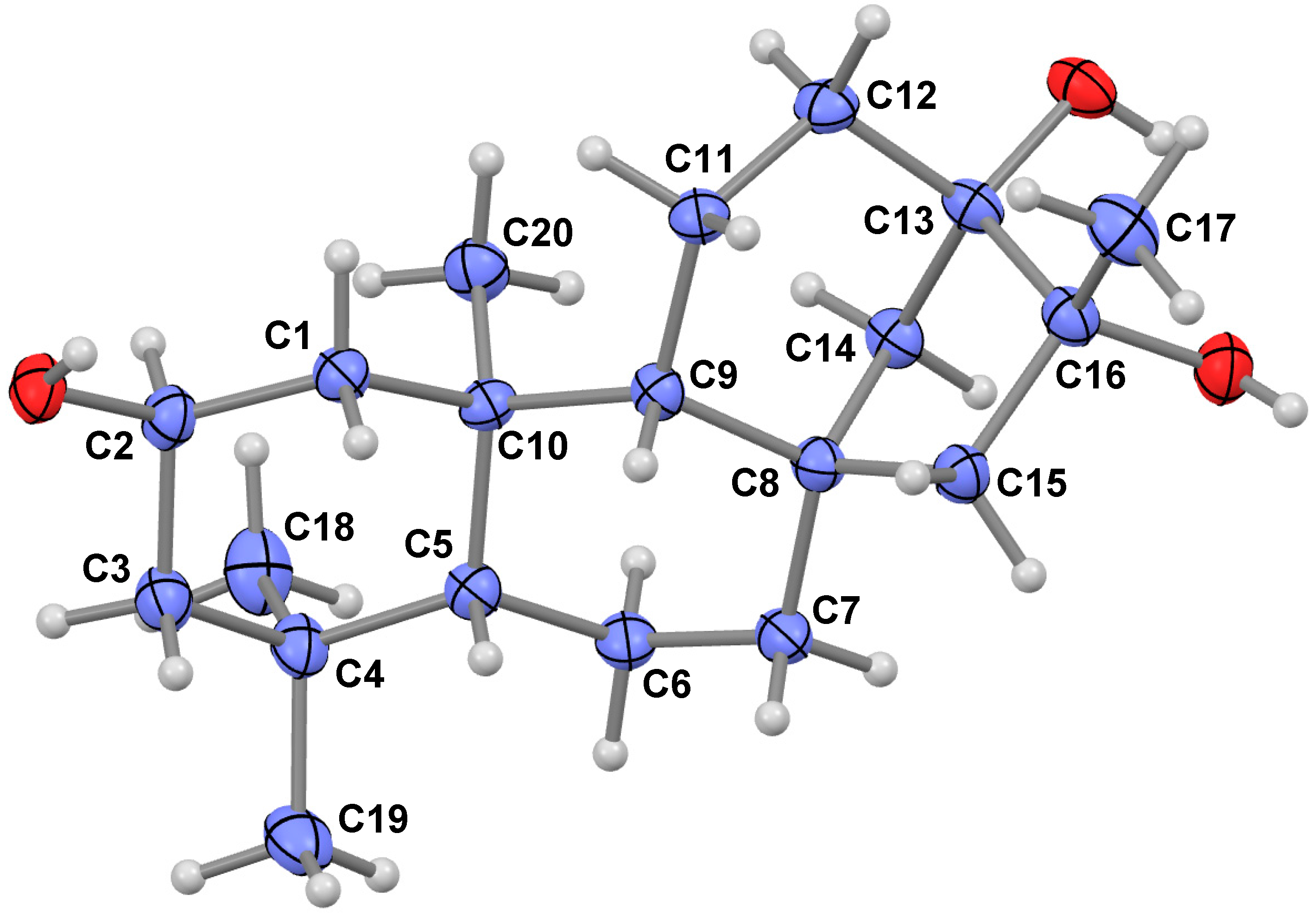{kind=link}
{kind=link}
| No. | δH (mult, J in Hz) b | δC (mult) c | No. | δH (mult, J in Hz) b | δC (mult) c |
|---|---|---|---|---|---|
| 1α | 2.15 (brddd, 12.0, 3.8, 1.9) | 50.3 t | 11α | 1.65 (overlap) | 20.7 t |
| 1β | 0.67 (brt, 11.7) | - | 11β | 1.82 (overlap) | - |
| 2α | 3.87 (brtt, 11.5, 4.3) | 65.4 d | 12α | 1.68 (overlap) | 34.6 t |
| 3α | 1.72 (brddd, 12.5, 4.3, 1.9) | 51.9 t | 12β | 1.64 (overlap) | - |
| 3β | 1.07 (brt, 12.0) | - | 13 | - | 77.4 s |
| 4 | - | 35.7 s | 14α | 1.66 (overlap) | 44.0 t |
| 5β | 0.82 (brd, 11.5) | 57.0 d | 14β | 1.83 (d, 11.9) | - |
| 6α | 1.34 (brqd, 12.1, 1.9) | 21.2 t | 15α | 1.50 (dd, 14.5, 1.3) | 56.7 t |
| 6β | 1.59 (brd, 12.3) | - | 15β | 1.63 (d, 14.7) | - |
| 7α | 1.60 (overlap) | 43.2 t | 16 | - | 81.1 s |
| 7β | 1.44 (brtd, 11.9, 3.4) | - | 17 | 1.18 (s) | 21.3 q |
| 8 | - | 42.2 s | 18 | 0.93 (s) | 34.2 q |
| 9β | 0.96 (brd, 7.2) | 57.1 d | 19 | 0.87 (s) | 22.8 q |
| 10 | - | 41.9 s | 20 | 1.09 (s) | 19.4 q |
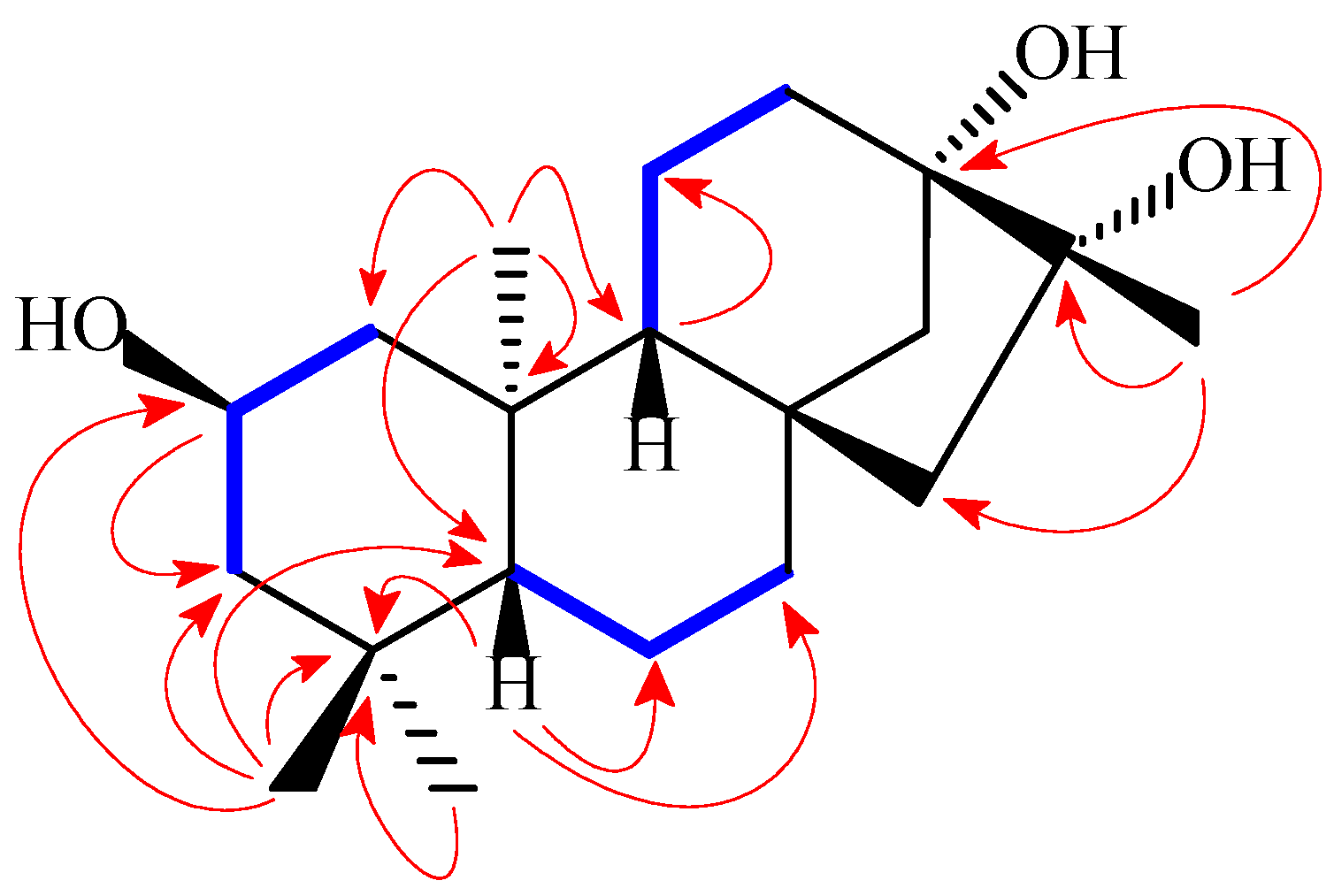
 H)) and HMBC (the red arrows (H
H)) and HMBC (the red arrows (H  C)) correlations for compound 1.
H)) and HMBC (the red arrows (H C)) correlations for compound 1.
C)) correlations for compound 1.
H)) and HMBC (the red arrows (H C)) correlations for compound 1.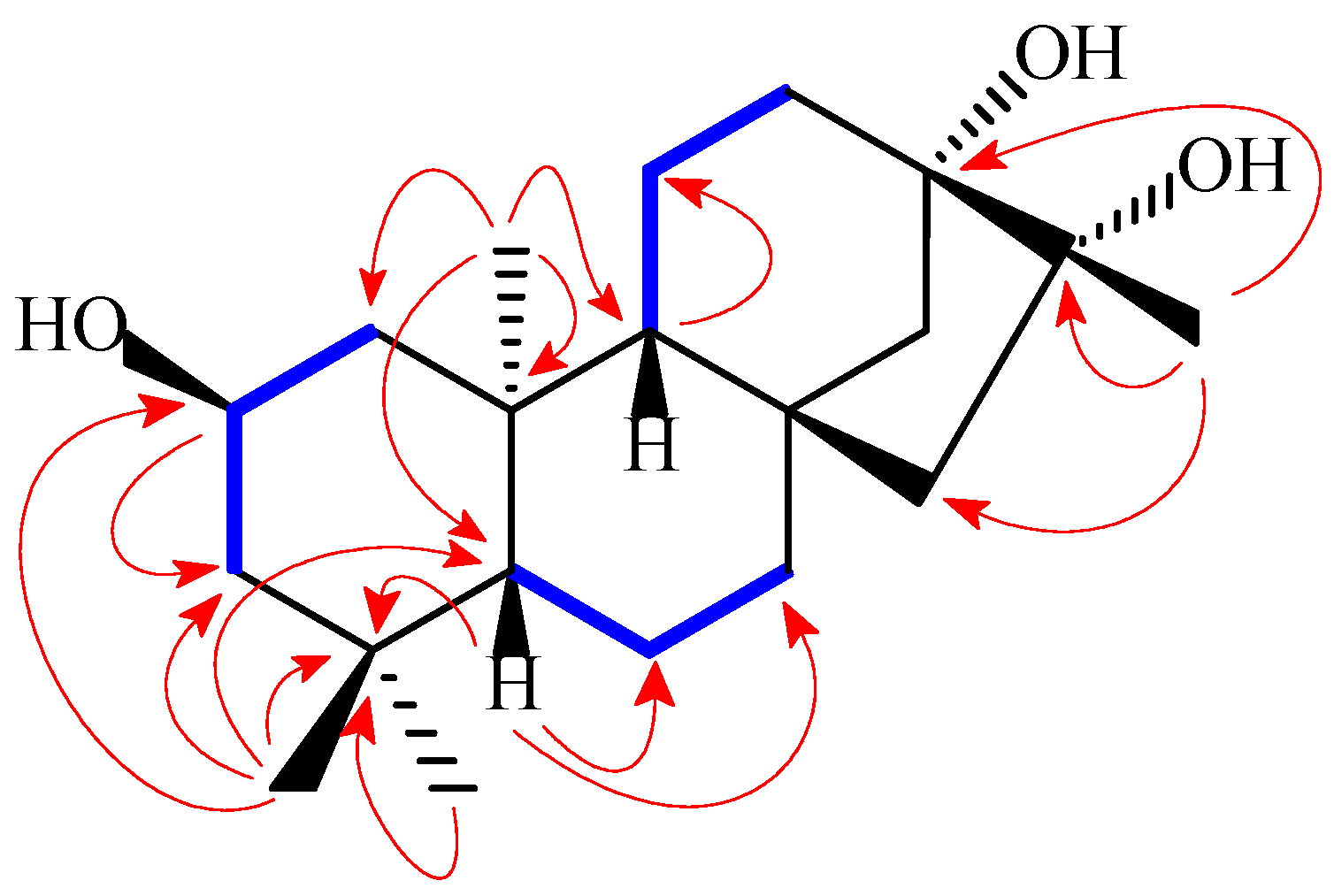
 H)) for compound 1.
H)) for compound 1.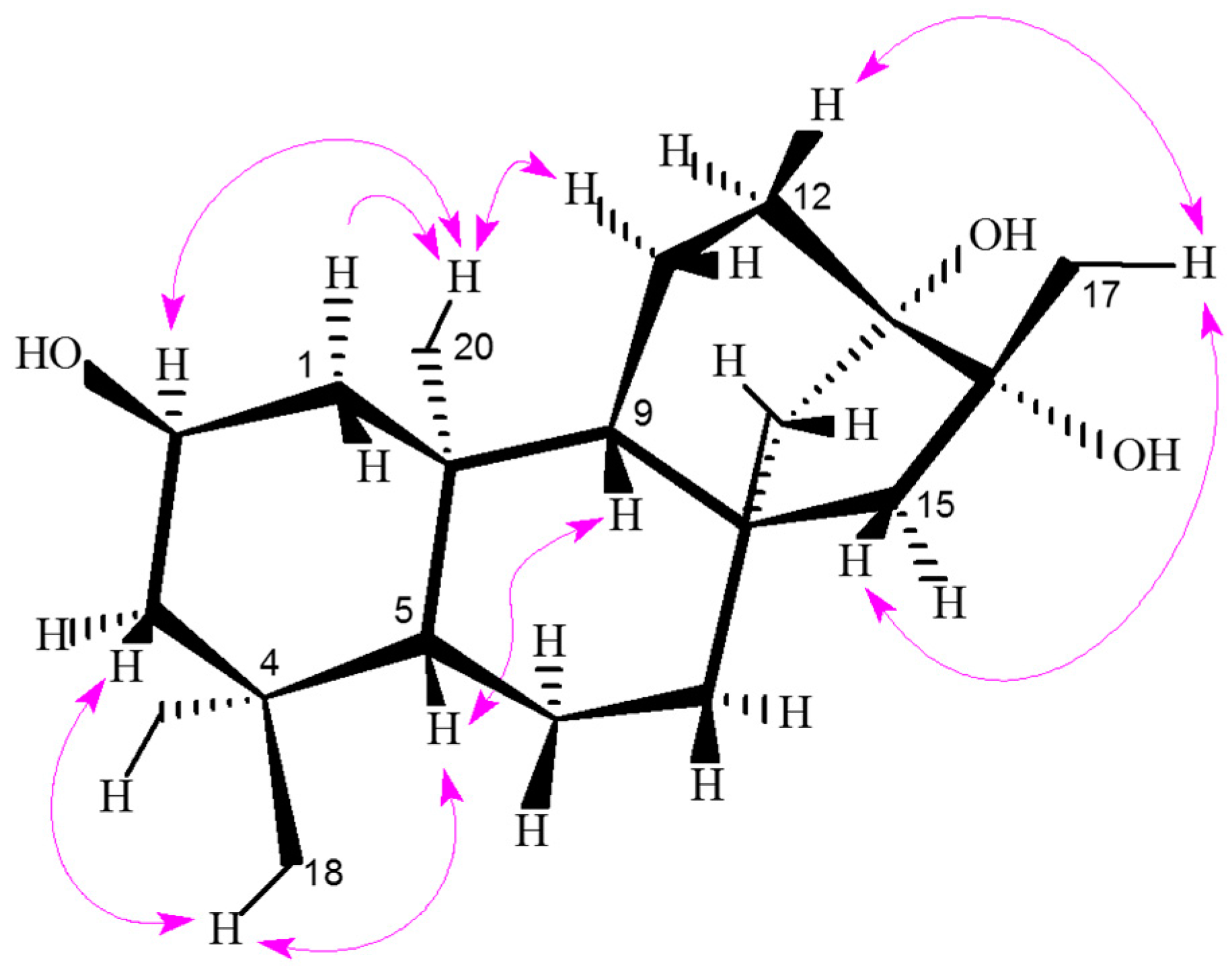
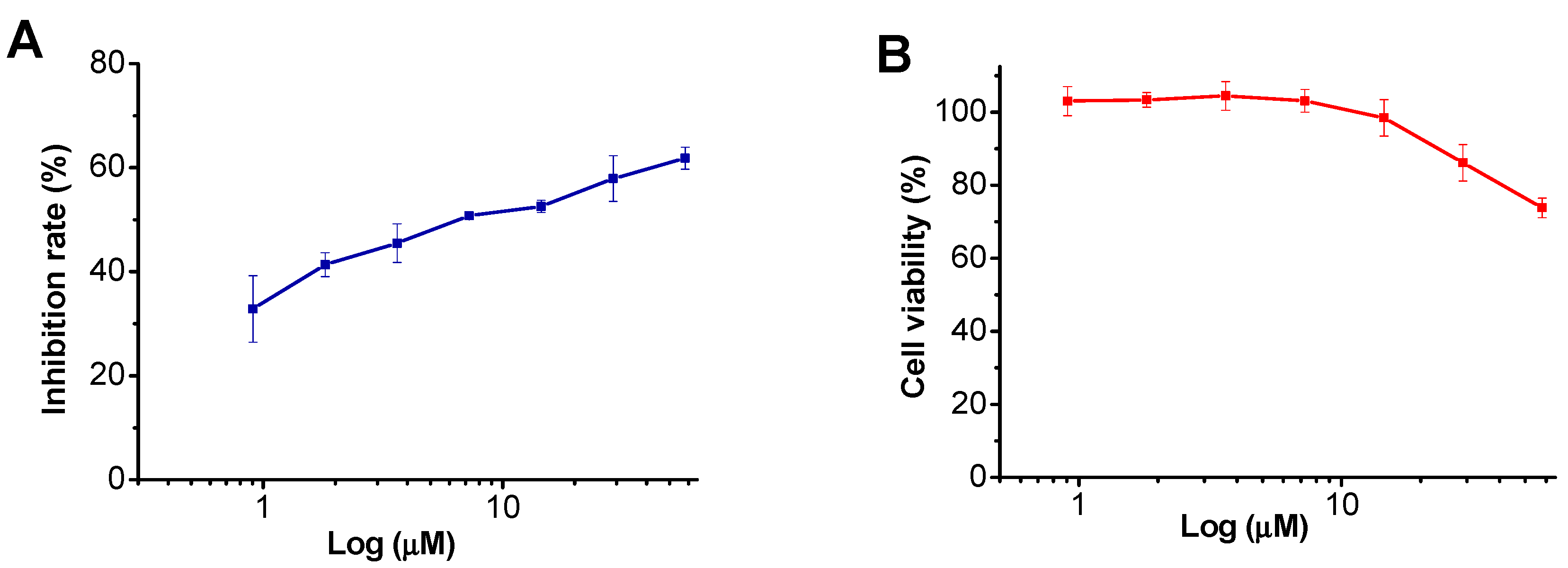
2.2. Biological Activity
| Compounds | Growth Inhibition Rate (%) | ||
|---|---|---|---|
| Bacterial Biofilm Formation | Fungus | ||
| S. mutans | S. sobrinus | T. rubrum | |
| Henrin A (1) a | 15.55 ± 5.66 | 11.20 ± 2.23 | 9.73 ± 8.75 |
| Penicillin G b | 80.44 ± 3.14 | 76.82 ± 3.93 | - |
| Chlorhexidine c | 77.26 ± 6.30 | 71.49 ± 5.21 | 83.12 ± 0.47 |
| Miconazole a | - | - | 80.69 ± 0.10 |

| Compound | IC50 (µM) | CC50 (µM) | SI |
|---|---|---|---|
| Henrin A | 9.1 | 110.5 | 12.2 |
| AZT (zidovudine) | 0.03 | >100 | >3333 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. X-ray Data of Henrin A (1)
3.5. Biological Activity of Henrin A (1)
3.5.1. Cytotoxicity Assay
3.5.2. Anti-Biofilm Activity Test
3.5.3. Antifungal Microtiter Assay
3.5.4. Inhibitory HIV Activity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, S.X.; Xiao, W.L.; Li, L.M.; Li, S.H.; Zhou, Y.; Ding, L.S.; Lou, L.G.; Sun, H.D. Bisrubescensins A–C: Three new dimeric ent-kauranoids isolated from Isodon rubescens. Org. Lett. 2006, 8, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.D.; Xu, Y.L.; Jiang, B. Diterpenoids from Isodon Species; SciPress, Ltd.: Beijing, China, 2001. [Google Scholar]
- Sun, H.D.; Huang, S.X.; Han, Q.B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gao, J.; Halicka, H.D.; Huang, X.; Traganos, F.; Darzynkiewicz, Z. The cytostatic and cytotoxic effects of oridonin (Rubescenin), a diterpenoid from Rabdosia rubescens, on tumor cells of different lineage. Int. J. Oncol. 2005, 26, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Zhong, Z.F.; Wan, J.B.; Tan, W.; Wu, G.S.; Chen, M.W.; Wang, Y.T. Oridonin induces apoptosis, inhibits migration and invasion on highly-metastatic human breast cancer cells. Am. J. Chin. Med. 2013, 41, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Yang, Y.; Bandobashi, K.; Saito, T.; Takemoto, S.; Machida, H.; Togitani, K.; Koeffler, H.P.; Taguchi, H. Oridonin, a diterpenoid purified from Rabdosia rubescens, inhibits the proliferation of cells from lymphoid malignancies in association with blockade of the NF-κB signal pathways. Mol. Cancer Ther. 2005, 4, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Fuji, K.; Xu, H.J.; Tatsumi, H.; Imahori, H.; Ito, N.; Node, M.; Inaba, M. Design and synthesis of antitumor compounds based on the cytotoxic diterpenoids from the genus Rabdosia. Chem. Pharm. Bull. 1991, 39, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Maehashi, H.; Tanaka, N.; Satake, T.; Kuraishi, T.; Komazawa, Y.; Saiki, Y.; Chen, C.M. Chemical and chemotaxonomical studies on filices. LV. Studies on the constituents of several species of Pteris. Yakugaku Zasshi 1985, 105, 640–648. [Google Scholar]
- Murakami, T.; Iida, H.; Tanaka, N.; Saiki, Y.; Chen, C.M.; Iitaka, Y. Chemical and chemotaxonomic study of filicales. 33. Chemical studies of the components of Pteris longipes Don. Chem. Pharm. Bull. 1981, 29, 657–662. [Google Scholar] [CrossRef]
- Liu, Q.F.; Qin, M.Z. Chemical studies of the components of Pteris multifida Poir. Chin. Tradit. Herb. Drugs (Zhong Cao Yao) 2002, 33, 113–114. [Google Scholar]
- Tanaka, N.; Hata, M.; Murakami, T.; Saiki, Y.; Chen, C.M. Chemical and chemotaxonomic studies of Pteris and related genera (Pteridaceae). XIII. Additional components of Pteris dispar Kunze. Chem. Pharm. Bull. 1976, 24, 1965–1966. [Google Scholar] [CrossRef]
- Murakami, T.; Tanaka, N.; Komazawa, Y.; Saiki, Y.; Chen, C.M. Chemical and chemotaxonomic studies of ferns. XLI. Additional contents of Pteris purpureorachis Copel. Chem. Pharm. Bull. 1983, 31, 1502–1504. [Google Scholar] [CrossRef]
- Tanaka, N.; Murakami, T.; Saiki, Y.; Chen, C.M.; Iitaka, Y. Chemical and chemo taxonomic study of filices. XXXIV. chemical studies of the components of Pteris purpureorachis Copel. Chem. Pharm. Bull. 1981, 29, 663–666. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.H.; He, C.W.; Tanaka, N. Study on the diterpenoid constituents and anticancer action of Pteris semipinnata. Chin. Pharm. J. 1999, 34, 512–514. [Google Scholar]
- Cheng, H.E.; Liang, N.; Li, M.O. Inhibitory effect of compound 6F isolated from Pteris semipinnata L. on biosynthesis of DNA, RNA and protein in HL-60 cells. J. Guangdong Med. Coll. 2002, 20, 247–250. [Google Scholar]
- Zhu, Y.L.; Song, L.D. The research of Pteris genus in China. China Prac. Med. 2014, 9, 256. [Google Scholar]
- Murakami, T.; Tanaka, N. Occurrence, structure and taxonomic implications of fern constituents. In Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Tamm, C., Eds.; Springer Vienna: Tokyo, Japan, 1988; pp. 1–310. [Google Scholar]
- The Editorial Committee of Flora of Guizhou. Flora of Guizhou (Guizhou Zhiwu Zhi Volume 8); Guizhou People Press: Guiyang, China, 1986; pp. 491–492. [Google Scholar]
- Pan, L.T.; Zhao, J.H.; Sun, Q.W. Medicinal Pteridophytes of Guizhou (Guizhou Juelei Zhiwu zhi); Guizhou Science and Technology Press: Guiyang, China, 2012; p. 113. [Google Scholar]
- Flack, H.D. On enantiomorph-polarity estimation. Act. Crystallogr. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.R. The tetracyclic diterpenes. In International Series of Monographs in Organic Chemistry; Pergamon Press: London, UK, 1968; Volume 9, p. 8. [Google Scholar]
- Huang, S.X. Tetracyclic diterpenes. In Chemistry of Diterpenes; Sun, H.D., Ed.; Chemical Industry Press: Beijing, China, 2011; pp. 203–227. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products, A Biosynthetic Approach; John Wiley & Sons Ltd.: West Sussex, UK, 2009; pp. 228–229. [Google Scholar]
- Rumschlag-Booms, E.; Zhang, H.J.; Soejarto, D.D.; Fong, H.H.; Rong, L.J. Development of an antiviral screening protocol: One-Stone-Two-Birds. J. Antivir. Antiretrovir. 2011, 3, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, S.; Bruno, M.; Maggio, A.; Bellone, G.; Chen, T.H.; Bastow, K.F.; Lee, K.H. Cytotoxic activity of some natural and synthetic ent-kauranes. J. Nat. Prod. 2007, 70, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Shi, Q.; Fujioka, T.; Zhang, D.C.; Hu, C.Q.; Jin, J.Q.; Kilkuskie, R.E.; Lee, K.H. Anti-AIDS agents, 4. Tripterifordin, a novel anti-HIV principle from Tripterygium wilfordii: Isolation and structural elucidation. J. Nat. Prod. 1992, 55, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.R.; Yang, P.Y.; Lin, J.Y.; Lee, K.H.; Wu, Y.C. Bioactive kaurane diterpenoids from Annona glabra. J. Nat. Prod. 1998, 61, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Hung, Y.C.; Chang, F.R.; Cosentino, M.; Wang, H.K.; Lee, K.H. Identification of ent-16β,17-dihydroxykauran-19-oic acid as an anti-HIV principle and isolation of the new diterpenoids annosquamosins A and B from Annona squamosal. J. Nat. Prod. 1996, 59, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.D. Diterpenoid Chemistry (Ertie Huaxue); Chemical Industry Press: Beijing, China, 2011; p. 206. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, Program for X-ray Crystal Structure Refinement; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Zhang, H.J.; Ma, C.Y.; Hung, N.V.; Cuong, N.M.; Tan, G.T.; Santarsiero, B.D.; Mesecar, A.D.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H.S. Miliusanes, a class of cytotoxic agents from Miliusa sinensis. J. Med. Chem. 2006, 49, 693–708. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.A.; Kolter, R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signaling pathways: A genetic analysis. Mol. Microbiol. 1998, 28, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Koneman, E.W.; Allen, S.D.; Janda, W.M.; Schreckenberger, P.C.; Winn, W.C. Color Atlas And Textbook of Diagnostic Microbiology, 5th ed.; Lippincott: Philadelphia, PA, USA, 1997; pp. 803–841. [Google Scholar]
- Yang, H.C.; Mikami, Y.; Yazawa, K.; Taguchi, H.; Nishimura, K.; Miyaji, M.; Branchini, M.L.; Aoki, F.H.; Yamamoto, K. Colorimetric MTT assessment of antifungal activity of D0870 against fluconazole-resistant Candida albicans. Mycoses 1998, 41, 477–480. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi, Approved Standard, CLSI document M38-A2, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- He, J.; Choe, S.; Walker, R.; di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type I viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [PubMed]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.-F.; Wang, J.; Zhang, J.-J.; Song, X.; Ku, C.-F.; Zou, J.; Li, J.-X.; Rong, L.-J.; Pan, L.-T.; Zhang, H.-J. Henrin A: A New Anti-HIV Ent-Kaurane Diterpene from Pteris henryi. Int. J. Mol. Sci. 2015, 16, 27978-27987. https://doi.org/10.3390/ijms161126071
Li W-F, Wang J, Zhang J-J, Song X, Ku C-F, Zou J, Li J-X, Rong L-J, Pan L-T, Zhang H-J. Henrin A: A New Anti-HIV Ent-Kaurane Diterpene from Pteris henryi. International Journal of Molecular Sciences. 2015; 16(11):27978-27987. https://doi.org/10.3390/ijms161126071
Chicago/Turabian StyleLi, Wan-Fei, Juan Wang, Jing-Jie Zhang, Xun Song, Chuen-Fai Ku, Juan Zou, Ji-Xin Li, Li-Jun Rong, Lu-Tai Pan, and Hong-Jie Zhang. 2015. "Henrin A: A New Anti-HIV Ent-Kaurane Diterpene from Pteris henryi" International Journal of Molecular Sciences 16, no. 11: 27978-27987. https://doi.org/10.3390/ijms161126071