Maternal–Fetal Nutrient Transport in Pregnancy Pathologies: The Role of the Placenta

Abstract
:1. Introduction
2. Placenta Nutrient Transport
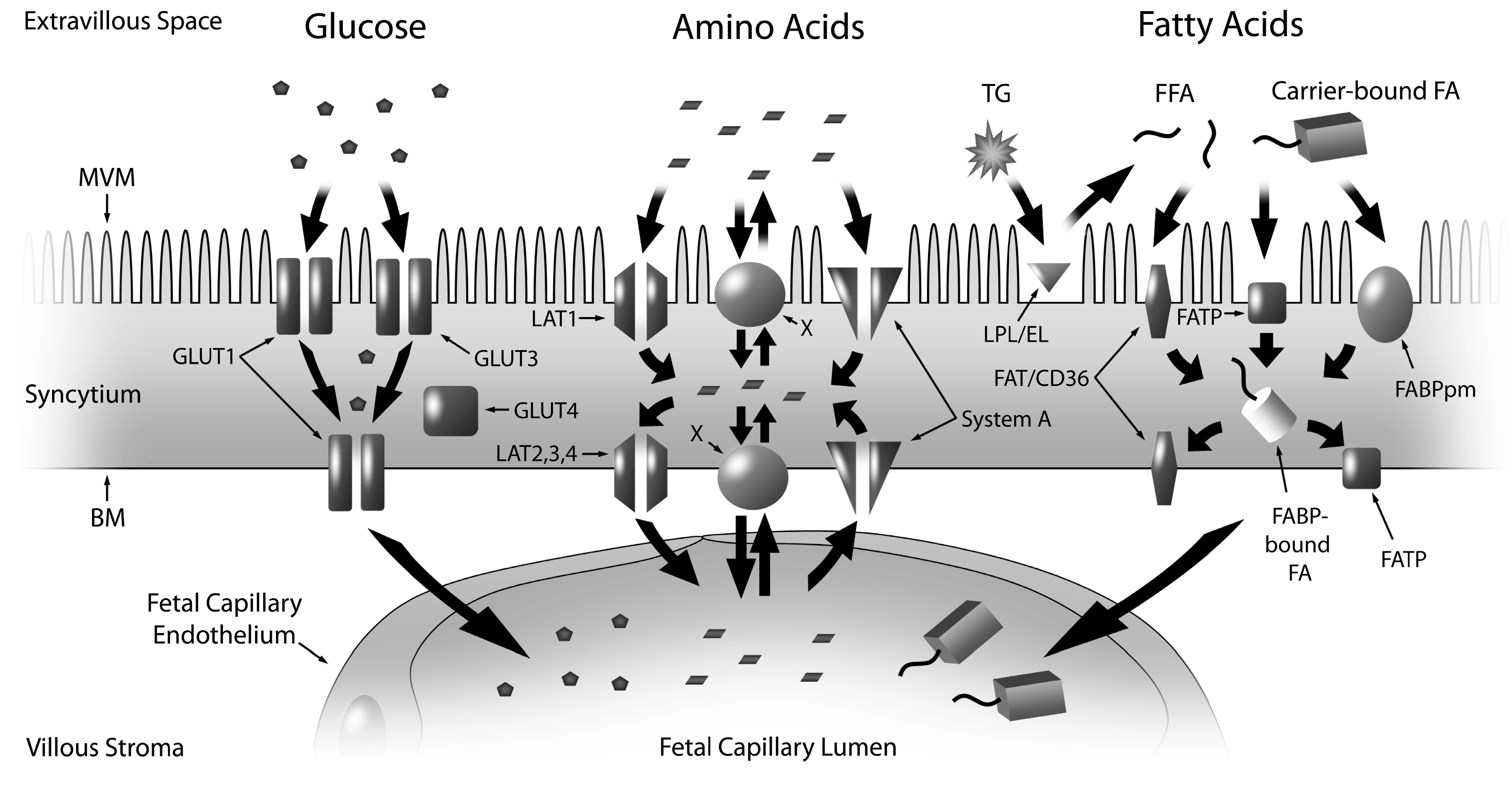
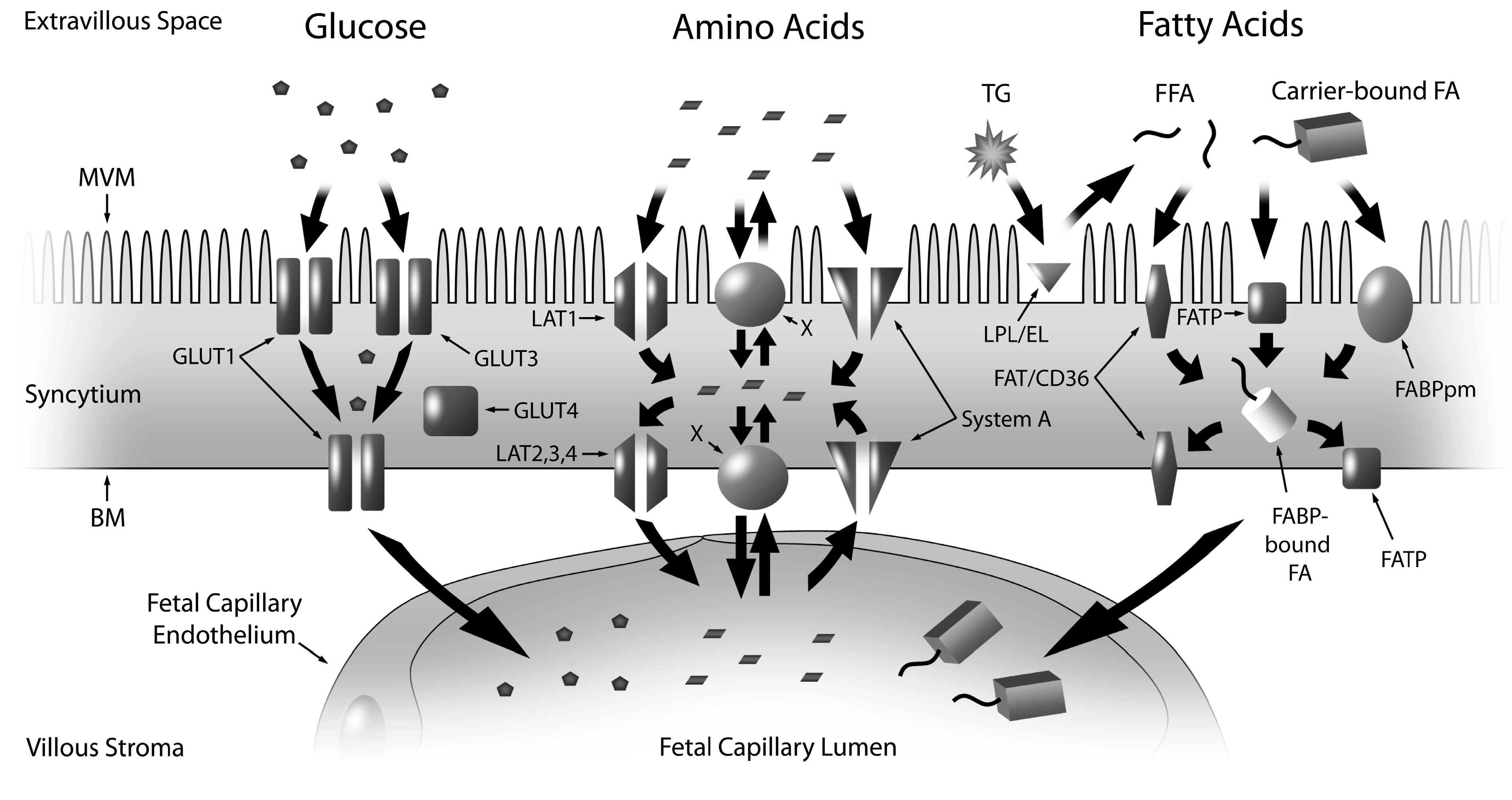
2.1. Glucose
2.2. Amino Acids
2.3. Fatty Acids
2.4. Cholesterol/Lipoproteins
3. Placental Nutrient Transport in Altered Fetal Growth

{kind=link}
{kind=link}
| Nutrient Transporter | IUGR—Placental Dysfunction | Type 1 Diabetes | GDM | Obesity |
|---|---|---|---|---|
| GLUT1 | ▬ [43,81] | ▲* (BM) (birth weight > control) [82] ▬ (MVM) (birth weight > control) [82] | ▬ (with and without LGA) [83]. ▲ (normal weight mothers; insulin controlled; no difference in birth weight) [84] ▬ (normal weight mothers; diet controlled; no difference in birth weight) [84] ▬ (obese mothers; diet or insulin controlled; no fetal overgrowth) [84] | ▬ (no fetal over growth) [84] |
| GLUT3 | ▲ [85] | |||
| GLUT4 | ▬ [85] | ▼ (normal weight mothers; insulin controlled; no difference in birth weight) [84] ▼ mRNA (obese mothers; diet or insulin controlled; no fetal overgrowth) [84] | ▼ mRNA (no fetal over growth) [84] | |
| System A (SNAT1,2,4) | ▼* (MVM) [58,86] ▬* (BM) [81] | ▼* (MVM) (macrosomic) (no maternal BMI) [87] ▲* (MVM) (independent of fetal growth, similar maternal BMI) [88] | ▲* (MVM) (independent of fetal over growth) [81] | ▼* SNAT4 (no difference in birth weight) [89] ▬ SNAT1, SNAT2 (no difference in birth weight) [89] ▬* (no difference in birth weight) [90] * positive correlation to birth weight [90] ▲ SNAT2 (no difference in birth weight; positive correlation to birth weight) [90] |
| System L (LAT1-4) | ▼* [91] | ▼* (MVM) (fetal overgrowth) [81] | ▬* (no difference in birth weight) [90] | |
| LPL | ▼* (preterm) [92] ▲ mRNA (preterm) [93] | ▲* (macrosomic) [92] ▬ (macrosomic) [92] ▼ mRNA (birth weight > control; not macrosomic) [94] | ▬ * ( macrosomic) [92] ▼ mRNA (birth weight > control; not macrosomic) [94] | ▲* (no difference in birth weight) [95] ▬ mRNA (birth weight > control; not macrosomic) [96] |
| Endothelial Lipase | ▲ mRNA (preterm) [93] | ▲ (birth weight > control; not macrosomic) [97]. | ▬ (no fetal over growth) [98] ▲ (with obesity) (no fetal over growth) [98] | |
| FATP4 | ▼ (no difference in birth weight) [95] ▬ mRNA (birth weight > control; not macrosomic) [96] | |||
| FAT/CD36 | ▲ (no difference in birth weight) [95] ▼ mRNA (male) (birth weight > control; not macrosomic) [96] ▬ mRNA (female) (birth weight > control; not macrosomic) [96] | |||
| FABP1 | ▲ (macrosomic) [92] | ▲ ( macrosomic) [92] | ▼(no difference in birth weight) [95] | |
| FABP3 | ▬ (no difference in birth weight) [99] ▼(no difference in birth weight) [95] | |||
| FABP4 | ▲ mRNA (birth weight > control; not macrosomic) [94] | ▲ mRNA (birth weight > control; not macrosomic) [94] | ▬ (no difference in birth weight) [99] ▲ (with diabetes) (birth weight > control; not macrosomic) [99] ▬ mRNA (birth weight > control; not macrosomic) [96] | |
| FABP5 | ▲ mRNA (birth weight > control; not macrosomic) [94] | ▲ mRNA (with diabetes) (birth weight > control; not macrosomic) [99] ▬ (no difference in birth weight) [99] ▼ mRNA (male) (birth weight > control; not macrosomic) [96] ▬ mRNA (female) (birth weight > control; not macrosomic) [96] | ||
| FABPpm | ▬ (no difference in birth weight) [99] ▬ mRNA (birth weight > control; not macrosomic) [96] |
| Nutrient Transporter | IUGR—Nutrient Restriction | Maternal Diet |
|---|---|---|
| GLUT1 | Mice §▼(no change in fetal weight) [100] Mice ¥▲(reduced fetal weight) [100] Sheep § ▲(reduced fetal weight) [101] Baboon ¥ ▼(reduced fetal weight) [102] | Mice ▲ (high fat) (increased fetal weight) [103] |
| GLUT3 | Mice ▲ (high fat, high sugar) (§ reduced fetal weight; ¥ no change in fetal weight) [104] | |
| System A (SNAT1, 2, 4) | Mice ¥ ▲SNAT1(reduced fetal weight) [100] Mice ¥ ▼ (reduced fetal weight) SNAT4 [100] Sheep § (reduced fetal weight) [101] Baboon ¥ ▼SNAT2 (reduced fetal weight) [102] | Mice ▲ SNAT2 (high fat) (increased fetal weight) [103] Mice (male) ▲ SNAT2 (“cafeteria” diet) (no change in fetal weight) [105] Mice (female) ▲ SNAT4 (“cafeteria” diet) (no change in fetal weight) [105] Mice ▲ SNAT2 (high fat, high sugar) (§ reduced fetal weight; ¥ no change in fetal weight) [104] |
| System L (LAT1–4) | Baboon ¥ ▼ LAT1/2 (reduced fetal weight) [102] | |
| FATP4 | Sheep § ▲(reduced fetal weight) [101] | |
| FAT/CD36 | Sheep § ▲ (reduced fetal weight) [101] |
3.1. Intrauterine Growth Restriction
3.1.1. IUGR—Placental Dysfunction
3.1.2. IUGR—Maternal Nutrient Restriction—Evidence from Animal Models
3.2. Fetal Overgrowth
3.2.1. Diabetes
3.2.2. Obesity
4. The Impact of Maternal Diet
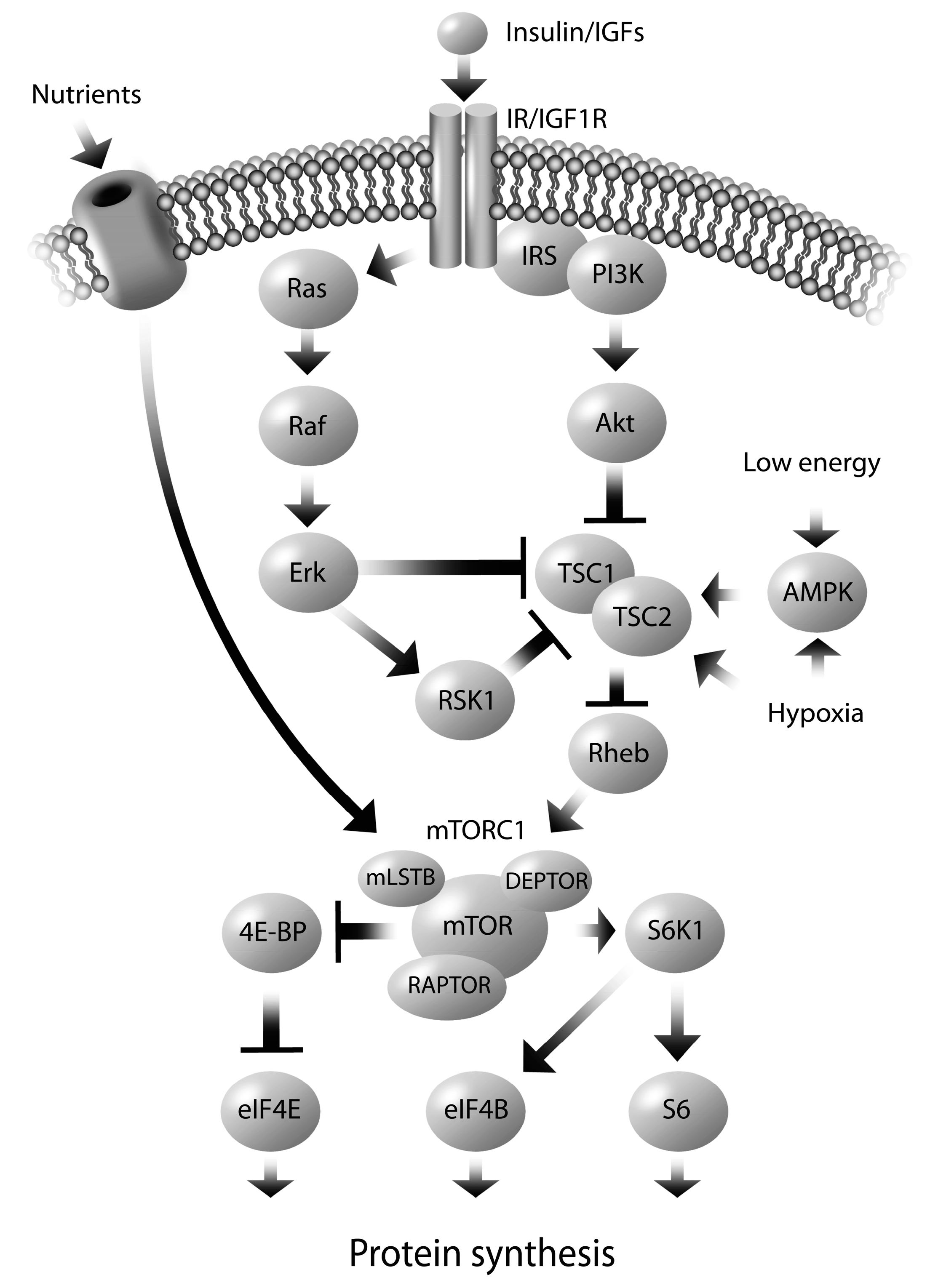
5. Molecular Mechanisms Regulating Altered Nutrient Transport
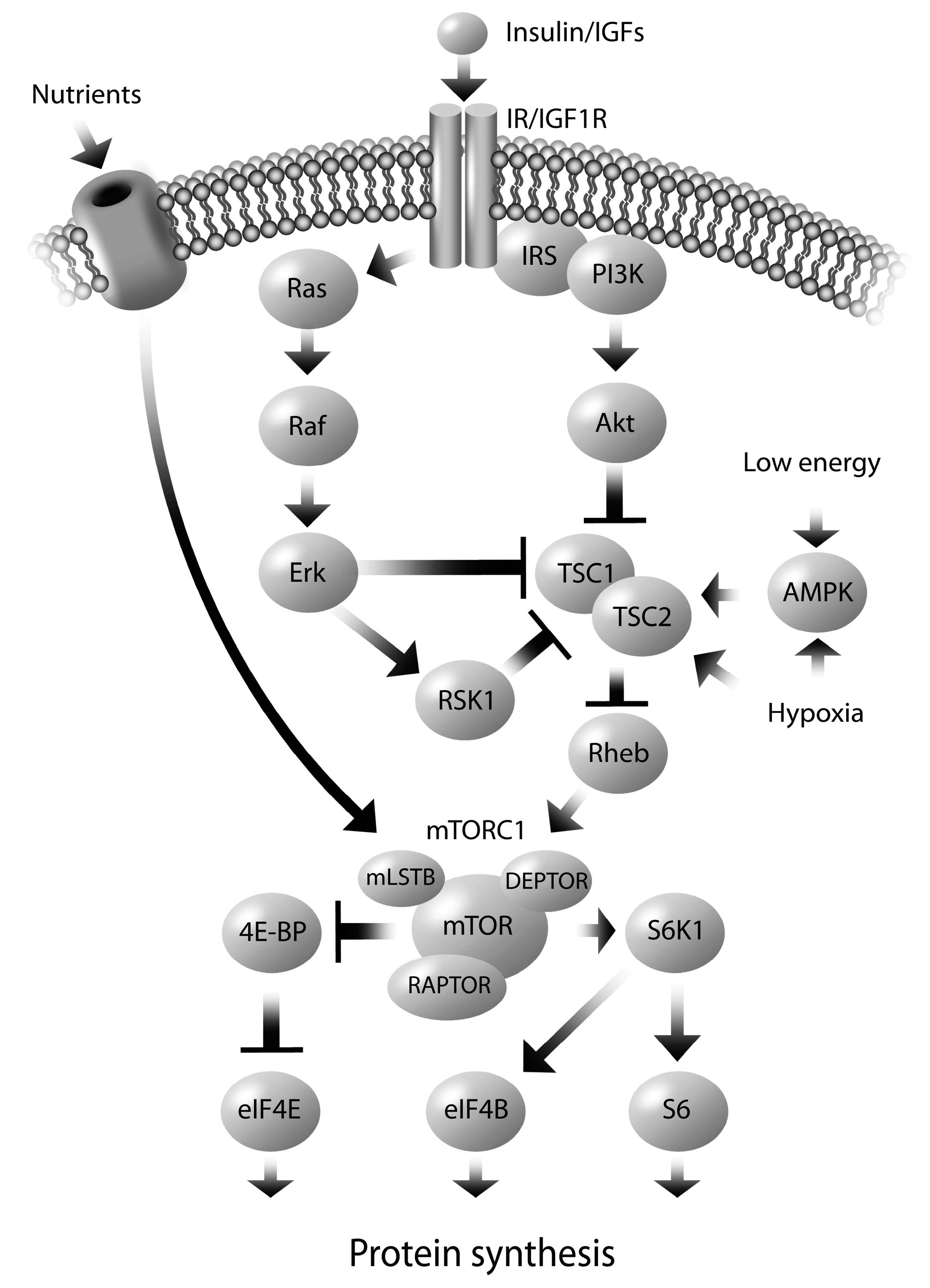
6. Sex Dependent Regulation of Fetal Programming
7. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Otten, J.; Pitzi Hellig, J.; Meyers, L. Dietary Reference Intakes: The Essential Guide to Nutrient Requirements; National Academic Press: Washington, DC, USA, 2006. [Google Scholar]
- Marconi, A.M.; Paolini, C.; Buscaglia, M.; Zerbe, G.; Battaglia, F.C.; Pardi, G. The impact of gestational age and fetal growth on the maternal–fetal glucose concentration difference. Obstet. Gynecol. 1996, 87, 937–942. [Google Scholar] [CrossRef]
- Baumann, M.U.; Deborde, S.; Illsley, N.P. Placental glucose transfer and fetal growth. Endocrine 2002, 19, 13–22. [Google Scholar] [CrossRef]
- Freinkel, N. Banting Lecture 1980. Of pregnancy and progeny. Diabetes 1980, 29, 1023–1035. [Google Scholar]
- Poissonnet, C.M.; Burdi, A.R.; Garn, S.M. The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum. Dev. 1984, 10, 1–11. [Google Scholar] [CrossRef]
- Haggarty, P. Fatty acid supply to the human fetus. Annu. Rev. Nutr. 2010, 30, 237–255. [Google Scholar] [CrossRef]
- Sparks, J.W.; Girard, J.R.; Battaglia, F.C. An estimate of the caloric requirements of the human fetus. Biol. Neonate 1980, 38, 113–119. [Google Scholar] [CrossRef]
- Gerretsen, G.; Huisjes, H.J.; Elema, J.D. Morphological changes of the spiral arteries in the placental bed in relation to pre-eclampsia and fetal growth retardation. Br. J. Obstet. Gynaecol. 1981, 88, 876–881. [Google Scholar] [CrossRef]
- Khong, T.Y.; de Wolf, W.B.; Robertson, W.B.; Brosens, I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br. J. Obstet. Gynaecol. 1986, 93, 1049–1059. [Google Scholar] [CrossRef]
- Clandinin, M.T.; Chappell, J.E.; Heim, T.; Swyer, P.R.; Chance, G.W. Fatty acid utilization in perinatal de novo synthesis of tissues. Early Hum. Dev. 1981, 5, 355–366. [Google Scholar] [CrossRef]
- Clandinin, M.T.; Chappell, J.E.; Heim, T.; Swyer, P.R.; Chance, G.W. Fatty acid accretion in fetal and neonatal liver: Implications for fatty acid requirements. Early Hum. Dev. 1981, 5, 7–14. [Google Scholar] [CrossRef]
- Hull, H.R.; Thornton, J.C.; Ji, Y.; Paley, C.; Rosenn, B.; Mathews, P.; Navder, K.; Yu, A.; Dorsey, K.; Gallagher, D. Higher infant body fat with excessive gestational weight gain in overweight women. Am. J. Obstet. Gynecol. 2011, 205, 211–217. [Google Scholar] [CrossRef]
- Sewell, M.F.; Huston-Presley, L.; Super, D.M.; Catalano, P. Increased neonatal fat mass, not lean body mass, is associated with maternal obesity. Am. J. Obstet. Gynecol. 2006, 195, 1100–1103. [Google Scholar] [CrossRef]
- Josefson, J.L.; Hoffmann, J.A.; Metzger, B.E. Excessive weight gain in women with a normal pre-pregnancy BMI is associated with increased neonatal adiposity. Pediatr. Obes. 2013, 8, e33–e36. [Google Scholar] [CrossRef]
- Jansson, T.; Illsley, N.P. Osmotic water permeabilities of human placental microvillous and basal membranes. J. Membr. Biol. 1993, 132, 147–155. [Google Scholar] [CrossRef]
- Johnson, L.W.; Smith, C.H. Glucose transport across the basal plasma membrane of human placental syncytiotrophoblast. Biochim. Biophys. Acta 1985, 815, 44–50. [Google Scholar] [CrossRef]
- Firth, J.A.; Leach, L. Not trophoblast alone: A review of the contribution of the fetal microvasculature to transplacental exchange. Placenta 1996, 17, 89–96. [Google Scholar] [CrossRef]
- Eaton, B.M.; Leach, L.; Firth, J.A. Permeability of the fetal villous microvasculature in the isolated perfused term human placenta. J. Physiol. 1993, 463, 141–155. [Google Scholar]
- Jansson, T.; Myatt, L.; Powell, T.L. The role of trophoblast nutrient and ion transporters in the development of pregnancy complications and adult disease. Curr. Vasc. Pharmacol. 2009, 7, 521–533. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef]
- Sibley, C.P.; Birdsey, T.J.; Brownbill, P.; Clarson, L.H.; Doughty, I.; Glazier, J.D.; Greenwood, S.L.; Hughes, J.; Jansson, T.; Mylona, P.; et al. Mechanisms of maternofetal exchange across the human placenta. Biochem. Soc. Trans. 1998, 26, 86–91. [Google Scholar]
- Smith, C.H.; Moe, A.J.; Ganapathy, V. Nutrient transport pathways across the epithelium of the placenta. Annu. Rev. Nutr. 1992, 12, 183–206. [Google Scholar] [CrossRef]
- Lager, S.; Powell, T.L. Regulation of nutrient transport across the placenta. J. Pregnancy 2012, 2012, 179827. [Google Scholar]
- Larque, E.; Ruiz-Palacios, M.; Koletzko, B. Placental regulation of fetal nutrient supply. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 292–297. [Google Scholar] [CrossRef]
- Fowden, A.L.; Ward, J.W.; Wooding, F.P.; Forhead, A.J.; Constancia, M. Programming placental nutrient transport capacity. J. Physiol. 2006, 572, 5–15. [Google Scholar]
- Higgins, L.; Greenwood, S.L.; Wareing, M.; Sibley, C.P.; Mills, T.A. Obesity and the placenta: A consideration of nutrient exchange mechanisms in relation to aberrant fetal growth. Placenta 2011, 32, 1–7. [Google Scholar]
- Roland, M.C.; Friis, C.M.; Voldner, N.; Godang, K.; Bollerslev, J.; Haugen, G.; Henriksen, T. Fetal growth versus birthweight: The role of placenta versus other determinants. PLoS One 2012, 7, e39324. [Google Scholar] [CrossRef]
- Wallace, J.M.; Horgan, G.W.; Bhattacharya, S. Placental weight and efficiency in relation to maternal body mass index and the risk of pregnancy complications in women delivering singleton babies. Placenta 2012, 33, 611–618. [Google Scholar] [CrossRef]
- Naeye, R.L. Do placental weights have clinical significance? Hum Pathol. 1987, 18, 387–391. [Google Scholar]
- Wilson, M.E.; Ford, S.P. Comparative aspects of placental efficiency. Reprod. Suppl. 2001, 58, 223–332. [Google Scholar]
- Fowden, A.L.; Sferruzzi-Perri, A.N.; Coan, P.M.; Constancia, M.; Burton, G.J. Placental efficiency and adaptation: Endocrine regulation. J. Physiol. 2009, 587, 3459–3472. [Google Scholar] [CrossRef]
- Salafia, C.M.; Zhang, J.; Miller, R.K.; Charles, A.K.; Shrout, P.; Sun, W. Placental growth patterns affect birth weight for given placental weight. Birth Defects Res. A 2007, 79, 281–288. [Google Scholar] [CrossRef]
- Jansson, N.; Pettersson, J.; Haafiz, A.; Ericsson, A.; Palmberg, I.; Tranberg, M.; Ganapathy, V.; Powell, T.L.; Jansson, T. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J. Physiol. 2006, 576, 935–946. [Google Scholar] [CrossRef]
- Jansson, T.; Cetin, I.; Powell, T.L.; Desoye, G.; Radaelli, T.; Ericsson, A.; Sibley, C.P. Placental transport and metabolism in fetal overgrowth—A workshop report. Placenta 2006, 27, S109–S113. [Google Scholar] [CrossRef]
- Johansson, M.; Karlsson, L.; Wennergren, M.; Jansson, T.; Powell, T.L. Activity and protein expression of Na+/K+ ATPase are reduced in microvillous syncytiotrophoblast plasma membranes isolated from pregnancies complicated by intrauterine growth restriction. J. Clin. Endocrinol. Metab. 2003, 88, 2831–2837. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. IFPA 2005 Award in Placentology Lecture. Human placental transport in altered fetal growth: Does the placenta function as a nutrient sensor?—A review. Placenta 2006, 27, S91–S97. [Google Scholar]
- Malandro, M.S.; Beveridge, M.J.; Kilberg, M.S.; Novak, D.A. Effect of low-protein diet-induced intrauterine growth retardation on rat placental amino acid transport. Am. J. Physiol. 1996, 271, C295–C303. [Google Scholar]
- Godfrey, K.M.; Matthews, N.; Glazier, J.; Jackson, A.; Wilman, C.; Sibley, C.P. Neutral amino acid uptake by the microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J. Clin. Endocrinol. Metab. 1998, 83, 3320–3326. [Google Scholar]
- Ogura, K.; Sakata, M.; Yamaguchi, M.; Kurachi, H.; Murata, Y. High concentration of glucose decreases glucose transporter-1 expression in mouse placenta in vitro and in vivo. J. Endocrinol. 1999, 160, 443–452. [Google Scholar] [CrossRef]
- Jones, H.N.; Powell, T.L.; Jansson, T. Regulation of placental nutrient transport—A review. Placenta 2007, 28, 763–774. [Google Scholar] [CrossRef]
- Kalhan, S.; Parimi, P. Gluconeogenesis in the fetus and neonate. Semin. Perinatol. 2000, 24, 94–106. [Google Scholar] [CrossRef]
- Illsley, N.P. Glucose transporters in the human placenta. Placenta 2000, 21, 14–22. [Google Scholar] [CrossRef]
- Jansson, T.; Wennergren, M.; Illsley, N.P. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1993, 77, 1554–1562. [Google Scholar]
- Chiesa, C.; Osborn, J.F.; Haass, C.; Natale, F.; Spinelli, M.; Scapillati, E.; Spinelli, A.; Pacifico, L. Ghrelin, leptin, IGF-1,IGFBP-3,and insulin concentrations at birth: Is there a relationship with fetal growth and neonatal anthropometry? Clin Chem. 2008, 54, 550–558. [Google Scholar]
- Baumann, M.U.; Schneider, H.; Malek, A.; Palta, V.; Surbek, D.V.; Sager, R.; Zamudio, S.; Illsley, N.P. Regulation of human trophoblast GLUT1 glucose transporter by insulin-like growth factor I (IGF-I). PLoS One 2014, 9, e106037. [Google Scholar] [CrossRef]
- Brown, K.; Heller, D.S.; Zamudio, S.; Illsley, N.P. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 2011, 32, 1041–1049. [Google Scholar] [CrossRef]
- Ericsson, A.; Hamark, B.; Powell, T.L.; Jansson, T. Glucose transporter isoform 4 is expressed in the syncytiotrophoblast of first trimester human placenta. Hum. Reprod. 2005, 20, 521–530. [Google Scholar] [CrossRef]
- Cetin, I.; Marconi, A.M.; Corbetta, C.; Lanfranchi, A.; Baggiani, A.M.; Battaglia, F.C.; Pardi, G. Fetal amino acids in normal pregnancies and in pregnancies complicated by intrauterine growth retardation. Early Hum. Dev. 1992, 29, 183–186. [Google Scholar] [CrossRef]
- Jansson, T. Amino acid transporters in the human placenta. Pediatr. Res. 2001, 49, 141–147. [Google Scholar] [CrossRef]
- Hoeltzli, S.D.; Smith, C.H. Alanine transport systems in isolated basal plasma membrane of human placenta. Am. J. Physiol. 1989, 256, C630–C637. [Google Scholar]
- Desforges, M.; Mynett, K.J.; Jones, R.L.; Greenwood, S.L.; Westwood, M.; Sibley, C.P.; Glazier, J.D. The SNAT4 isoform of the system A amino acid transporter is functional in human placental microvillous plasma membrane. J. Physiol. 2009, 587, 61–72. [Google Scholar]
- Jansson, N.; Greenwood, S.L.; Johansson, B.R.; Powell, T.L.; Jansson, T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J. Clin. Endocrinol. Metab. 2003, 88, 1205–1211. [Google Scholar] [CrossRef]
- Jones, H.N.; Jansson, T.; Powell, T.L. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am. J. Physiol. Cell Physiol. 2009, 297, C1228–C1235. [Google Scholar]
- Roos, S.; Lagerlof, O.; Wennergren, M.; Powell, T.L.; Jansson, T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am. J. Physiol. Cell Physiol. 2009, 297, C723–C731. [Google Scholar] [CrossRef]
- Verrey, F.; System, L. Heteromeric exchangers of large, neutral amino acids involved in directional transport. Pflugers Arch. 2003, 445, 529–533. [Google Scholar]
- Cleal, J.K.; Glazier, J.D.; Ntani, G.; Crozier, S.R.; Day, P.E.; Harvey, N.C.; Robinson, S.M.; Cooper, C.; Godfrey, K.M.; Hanson, M.A. Facilitated transporters mediate net efflux of amino acids to the fetus across the basal membrane of the placental syncytiotrophoblast. J. Physiol. 2011, 589, 987–997. [Google Scholar] [CrossRef]
- Kudo, Y.; Boyd, C.A. Characterisation of l-tryptophan transporters in human placenta: A comparison of brush border and basal membrane vesicles. J. Physiol. 2001, 531, 405–416. [Google Scholar] [CrossRef]
- Glazier, J.D.; Cetin, I.; Perugino, G.; Ronzoni, S.; Grey, A.M.; Mahendran, D.; Marconi, A.M.; Pardi, G.; Sibley, C.P. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr. Res. 1997, 42, 514–519. [Google Scholar] [CrossRef]
- King, J.C. Maternal obesity, metabolism, and pregnancy outcomes. Annu. Rev. Nutr. 2006, 26, 271–291. [Google Scholar] [CrossRef]
- Duttaroy, A.K. Transport of fatty acids across the human placenta: A review. Prog. Lipid Res. 2009, 48, 52–61. [Google Scholar] [CrossRef]
- Jaye, M.; Lynch, K.J.; Krawiec, J.; Marchadier, D.; Maugeais, C.; Doan, K.; South, V.; Amin, D.; Perrone, M.; Rader, D.J. A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 1999, 21, 424–428. [Google Scholar] [CrossRef]
- Lindegaard, M.L.; Olivecrona, G.; Christoffersen, C.; Kratky, D.; Hannibal, J.; Petersen, B.L.; Zechner, R.; Damm, P.; Nielsen, L.B. Endothelial and lipoprotein lipases in human and mouse placenta. J. Lipid Res. 2005, 46, 2339–2346. [Google Scholar] [CrossRef]
- Waterman, I.J.; Emmison, N.; Dutta-Roy, A.K. Characterisation of triacylglycerol hydrolase activities in human placenta. Biochim. Biophys. Acta 1998, 1394, 169–176. [Google Scholar] [CrossRef]
- McCoy, M.G.; Sun, G.S.; Marchadier, D.; Maugeais, C.; Glick, J.M.; Rader, D.J. Characterization of the lipolytic activity of endothelial lipase. J. Lipid Res. 2002, 43, 921–999. [Google Scholar]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef]
- Schaiff, W.T.; Bildirici, I.; Cheong, M.; Chern, P.L.; Nelson, D.M.; Sadovsky, Y. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor signaling regulate fatty acid uptake by primary human placental trophoblasts. J. Clin. Endocrinol. Metab. 2005, 90, 4267–4275. [Google Scholar] [CrossRef]
- Larque, E.; Demmelmair, H.; Klingler, M.; de Jonge, S; Bondy, B.; Koletzko, B. Expression pattern of fatty acid transport protein-1 (FATP-1), FATP-4 and heart-fatty acid binding protein (H-FABP) genes in human term placenta. Early Hum. Dev. 2006, 82, 697–701. [Google Scholar]
- Campbell, F.M.; Bush, P.G.; Veerkamp, J.H.; Dutta-Roy, A.K. Detection and cellular localization of plasma membrane-associated and cytoplasmic fatty acid-binding proteins in human placenta. Placenta 1998, 19, 409–415. [Google Scholar] [CrossRef]
- Cunningham, P.; McDermott, L. Long chain PUFA transport in human term placenta. J. Nutr. 2009, 139, 636–639. [Google Scholar] [CrossRef]
- Mousiolis, A.V.; Kollia, P.; Skentou, C.; Messinis, I.E. Effects of leptin on the expression of fatty acid-binding proteins in human placental cell cultures. Mol. Med. Rep. 2012, 5, 497–502. [Google Scholar]
- Magnusson-Olsson, A.L.; Hamark, B.; Ericsson, A.; Wennergren, M.; Jansson, T.; Powell, T.L. Gestational and hormonal regulation of human placental lipoprotein lipase. J. Lipid Res. 2006, 47, 2551–2561. [Google Scholar]
- Lager, S.; Jansson, N.; Olsson, A.L.; Wennergren, M.; Jansson, T.; Powell, T.L. Effect of IL-6 and TNF-α on fatty acid uptake in cultured human primary trophoblast cells. Placenta 2011, 32, 121–127. [Google Scholar] [CrossRef]
- Herrera, E. Lipid metabolism in pregnancy and its consequences in the fetus and newborn. Endocrine 2002, 19, 43–55. [Google Scholar]
- Woollett, L.A. Review: Transport of maternal cholesterol to the fetal circulation. Placenta 2011, 32, S218–S221. [Google Scholar] [CrossRef]
- Wittmaack, F.M.; Gafvels, M.E.; Bronner, M.; Matsuo, H.; McCrae, K.R.; Tomaszewski, J.E.; Robinson, S.L.; Strickland, D.K.; Strauss, J.F., 3rd. Localization and regulation of the human very low density lipoprotein/apolipoprotein-E receptor: Trophoblast expression predicts a role for the receptor in placental lipid transport. Endocrinology 1995, 136, 340–348. [Google Scholar]
- Furuhashi, M.; Seo, H.; Mizutani, S.; Narita, O.; Tomoda, Y.; Matsui, N. Expression of low density lipoprotein receptor gene in human placenta during pregnancy. Mol. Endocrinol. 1989, 3, 1252–1256. [Google Scholar] [CrossRef]
- Wadsack, C.; Hammer, A.; Levak-Frank, S.; Desoye, G.; Kozarsky, K.F.; Hirschmugl, B.; Sattler, W.; Malle, E. Selective cholesteryl ester uptake from high density lipoprotein by human first trimester and term villous trophoblast cells. Placenta 2003, 24, 131–143. [Google Scholar] [CrossRef]
- Stefulj, J.; Panzenboeck, U.; Becker, T.; Hirschmugl, B.; Schweinzer, C.; Lang, I.; Marsche, G.; Sadjak, A.; Lang, U.; Desoye, G.; et al. Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1. Circ. Res. 2009, 104, 600–608. [Google Scholar] [CrossRef]
- Aye, I.L.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Placental ABCA1 and ABCG1 transporters efflux cholesterol and protect trophoblasts from oxysterol induced toxicity. Biochim. Biophys. Acta 2010, 1801, 1013–1024. [Google Scholar] [CrossRef]
- Nikitina, L.; Wenger, F.; Baumann, M.; Surbek, D.; Korner, M.; Albrecht, C. Expression and localization pattern of ABCA1 in diverse human placental primary cells and tissues. Placenta 2011, 32, 420–430. [Google Scholar] [CrossRef]
- Jansson, T.; Ylven, K.; Wennergren, M.; Powell, T.L. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta 2002, 23, 392–399. [Google Scholar] [CrossRef]
- Jansson, T.; Wennergren, M.; Powell, T.L. Placental glucose transport and GLUT 1 expression in insulin-dependent diabetes. Am. J. Obstet. Gynecol. 1999, 180, 163–168. [Google Scholar] [CrossRef]
- Jansson, T.; Ekstrand, Y.; Wennergren, M.; Powell, T.L. Placental glucose transport in gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2001, 184, 111–116. [Google Scholar] [CrossRef]
- Colomiere, M.; Permezel, M.; Riley, C.; Desoye, G.; Lappas, M. Defective insulin signaling in placenta from pregnancies complicated by gestational diabetes mellitus. Eur. J. Endocrinol. 2009, 160, 567–578. [Google Scholar] [CrossRef]
- Janzen, C.; Lei, M.Y.; Cho, J.; Sullivan, P.; Shin, B.C.; Devaskar, S.U. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta 2013, 34, 1072–1078. [Google Scholar] [CrossRef]
- Mahendran, D.; Donnai, P.; Glazier, J.D.; D’Souza, S.W.; Boyd, R.D.; Sibley, C.P. Amino acid (system A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr. Res. 1993, 34, 661–665. [Google Scholar] [CrossRef]
- Kuruvilla, A.G.; D’Souza, S.W.; Glazier, J.D.; Mahendran, D.; Maresh, M.J.; Sibley, C.P. Altered activity of the system A amino acid transporter in microvillous membrane vesicles from placentas of macrosomic babies born to diabetic women. J. Clin. Investig. 1994, 94, 689–695. [Google Scholar] [CrossRef]
- Jansson, T.; Ekstrand, Y; Bjorn, C.; Wennergren, M.; Powell, T.L. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes 2002, 51, 2214–2219. [Google Scholar]
- Farley, D.M.; Choi, J.; Dudley, D.J.; Li, C.; Jenkins, S.L.; Myatt, L.; Nathanielsz, P.W. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta 2010, 31, 718–724. [Google Scholar] [CrossRef]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef]
- Jansson, T.; Scholtbach, V.; Powell, T.L. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr. Res. 1998, 44, 532–537. [Google Scholar] [CrossRef]
- Magnusson, A.L.; Waterman, I.J.; Wennergren, M.; Jansson, T.; Powell, T.L. Triglyceride hydrolase activities and expression of fatty acid binding proteins in the human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4607–4614. [Google Scholar] [CrossRef]
- Gauster, M.; Hiden, U.; Blaschitz, A.; Frank, S.; Lang, U.; Alvino, G.; Cetin, I.; Desoye, G.; Wadsack, C. Dysregulation of placental endothelial lipase and lipoprotein lipase in intrauterine growth-restricted pregnancies. J. Clin. Endocrinol. Metab. 2007, 92, 2256–2263. [Google Scholar]
- Radaelli, T.; Lepercq, J.; Varastehpour, A.; Basu, S.; Catalano, P.M.; Hauguel-de, M.S. Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 201, 209. [Google Scholar] [CrossRef]
- Dube, E.; Gravel, A.; Martin, C.; Desparois, G.; Moussa, I.; Ethier-Chiasson, M.; Forest, J.C.; Giguère, Y.; Masse, A.; Lafond, J. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol. Reprod. 2012, 14, 14–11. [Google Scholar] [CrossRef]
- Brass, E.; Hanson, E.; O’Tierney-Ginn, P.F. Placental oleic acid uptake is lower in male offspring of obese women. Placenta 2013, 34, 503–509. [Google Scholar] [CrossRef]
- Lindegaard, M.L.; Damm, P.; Mathiesen, E.R.; Nielsen, L.B. Placental triglyceride accumulation in maternal type 1 diabetes is associated with increased lipase gene expression. J. Lipid Res. 2006, 47, 2581–2588. [Google Scholar] [CrossRef]
- Gauster, M.; Hiden, U.; van Poppel, M.; Frank, S.; Wadsack, C.; Hauguel-de Mouzon, S.; Desoye, G. Dysregulation of placental endothelial lipase in obese women with gestational diabetes mellitus. Diabetes 2011, 60, 2457–2464. [Google Scholar] [CrossRef]
- Scifres, C.M.; Chen, B.; Nelson, D.M.; Sadovsky, Y. Fatty acid binding protein 4 regulates intracellular lipid accumulation in human trophoblasts. J. Clin. Endocrinol. Metab. 2011, 96, E1083–E1091. [Google Scholar] [CrossRef]
- Coan, P.M.; Vaughan, O.R.; Sekita, Y.; Finn, S.L.; Burton, G.J.; Constancia, M.; Fowden, A.L. Adaptations in placental phenotype support fetal growth during undernutrition of pregnant mice. J. Physiol. 2010, 588, 527–538. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, M.J.; Uthlaut, A.B.; Nijland, M.J.; Nathanielsz, P.W.; Hess, B.W.; Ford, S.P. Upregulation of growth signaling and nutrient transporters in cotyledons of early to mid-gestational nutrient restricted ewes. Placenta 2011, 32, 255–263. [Google Scholar]
- Kavitha, J.V.; Rosario, F.J.; Nijland, M.J.; McDonald, T.J.; Wu, G.; Kanai, Y.; Powell, T.L.; Nathanielsz, P.W.; Jansson, T. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014, 28, 1294–1305. [Google Scholar] [CrossRef]
- Jones, H.N.; Woollett, L.A.; Barbour, N.; Prasad, P.D.; Powell, T.L.; Jansson, T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2009, 23, 271–278. [Google Scholar] [CrossRef]
- Sferruzzi-Perri, A.N.; Vaughan, O.R.; Haro, M.; Cooper, W.N.; Musial, B.; Charalambous, M.; Pestana, D.; Ayyar, S.; Ferguson-Smith, A.C.; et al. An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J. 2013, 27, 3928–3937. [Google Scholar]
- King, V.; Hibbert, N.; Seckl, J.R.; Norman, J.E.; Drake, A.J. The effects of an obesogenic diet during pregnancy on fetal growth and placental gene expression are gestation dependent. Placenta 2013, 34, 1087–1090. [Google Scholar] [CrossRef]
- Prada, J.A.; Tsang, R.C. Biological mechanisms of environmentally induced causes of IUGR. Eur. J. Clin. Nutr. 1998, 52, S21–S27. [Google Scholar]
- Radulescu, L.; Munteanu, O.; Popa, F.; Cirstoiu, M. The implications and consequences of maternal obesity on fetal intrauterine growth restriction. J. Med. Life 2013, 6, 292–298. [Google Scholar]
- Rajasingam, D.; Seed, P.T.; Briley, A.L.; Shennan, A.H.; Poston, L. A prospective study of pregnancy outcome and biomarkers of oxidative stress in nulliparous obese women. Am. J. Obstet. Gynecol. 2009, 200, 395–399. [Google Scholar] [CrossRef]
- Perlow, J.H.; Morgan, M.A.; Montgomery, D.; Towers, C.V.; Porto, M. Perinatal outcome in pregnancy complicated by massive obesity. Am. J. Obstet. Gynecol. 1992, 167, 958–962. [Google Scholar] [CrossRef]
- Sibley, C.P.; Turner, M.A.; Cetin, I.; Ayuk, P.; Boyd, C.A.; D’Souza, S.W.; Glazier, J.D.; Greenwood, S.L.; Jansson, T.; Powell, T. Placental phenotypes of intrauterine growth. Pediatr. Res. 2005, 58, 827–832. [Google Scholar] [CrossRef]
- Sibley, C.P. Understanding placental nutrient transfer—Why bother? New biomarkers of fetal growth. J. Physiol. 2009, 587, 3431–3440. [Google Scholar] [CrossRef]
- Norberg, S.; Powell, T.L.; Jansson, T. Intrauterine growth restriction is associated with a reduced activity of placental taurine transporters. Pediatr. Res. 1998, 44, 233–238. [Google Scholar] [CrossRef]
- Wadsack, C.; Tabano, S.; Maier, A.; Hiden, U.; Alvino, G.; Cozzi, V.; Hüttinger, M.; Schneider, W.J.; Lang, U.; Cetin, I.; et al. Intrauterine growth restriction is associated with alterations in placental lipoprotein receptors and maternal lipoprotein composition. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E476–E484. [Google Scholar] [CrossRef]
- Roseboom, T.J.; Painter, R.C.; de Rooij, S.R.; van Abeelen, A.F.; Veenendaal, M.V.; Osmond, C.; Barker, D.J. Effects of famine on placental size and efficiency. Placenta 2011, 32, 395–399. [Google Scholar] [CrossRef]
- Ford, S.P.; Hess, B.W.; Schwope, M.M.; Nijland, M.J.; Gilbert, J.S.; Vonnahme, K.A.; Means, W.J.; Han, H.; Nathanielsz, P.W. Maternal undernutrition during early to mid-gestation in the ewe results in altered growth, adiposity, and glucose tolerance in male offspring. J. Anim. Sci. 2007, 85, 1285–1294. [Google Scholar] [CrossRef]
- Catalano, P.M.; Presley, L.; Minium, J.; Hauguel-de, M.S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef]
- Luo, Z.C.; Nuyt, A.M.; Delvin, E.; Audibert, F.; Girard, I.; Shatenstein, B.; Cloutier, A.; Cousineau, J.; Djemli, A.; Deal, C.; et al. Maternal and fetal IGF-I and IGF-II levels, fetal growth, and gestational diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1720–1728. [Google Scholar] [CrossRef]
- Dube, E.; Ethier-Chiasson, M.; Lafond, J. Modulation of cholesterol transport by insulin-treated gestational diabetes mellitus in human full term placenta. Biol. Reprod. 2013, 88, 16. [Google Scholar] [CrossRef]
- Kuhl, C. Etiology and pathogenesis of gestational diabetes. Diabetes Care 1998, 21, B19–B26. [Google Scholar]
- Hull, H.R.; Dinger, M.K.; Knehans, A.W.; Thompson, D.M.; Fields, D.A. Impact of maternal body mass index on neonate birthweight and body composition. Am. J. Obstet. Gynecol. 2008, 198, 416. [Google Scholar] [CrossRef]
- Visiedo, F.; Bugatto, F.; Sanchez, V.; Cozar-Castellano, I.; Bartha, J.L.; Perdomo, G. High glucose levels reduce fatty acid oxidation and increase triglyceride accumulation in human placenta. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E205–E212. [Google Scholar] [CrossRef]
- Desforges, M.; Greenwood, S.L.; Glazier, J.D.; Westwood, M.; Sibley, C.P. The contribution of SNAT1 to system A amino acid transporter activity in human placental trophoblast. Biochem. Biophys. Res. Commun. 2010, 398, 130–134. [Google Scholar] [CrossRef]
- Zhu, M.J.; Ma, Y.; Long, N.M.; Du, M.; Ford, S.P. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1224–R1231. [Google Scholar] [CrossRef]
- Clifton, V.L. Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival. Placenta 2010, 31, S33–S39. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta 2013, 34, 1183–1189. [Google Scholar] [CrossRef]
- Lin, Y.; Zhuo, Y.; Fang, Z.F.; Che, L.Q.; Wu, D. Effect of maternal dietary energy types on placenta nutrient transporter gene expressions and intrauterine fetal growth in rats. Nutrition 2012, 28, 1037–1043. [Google Scholar] [CrossRef]
- Roos, S.; Powell, T.L.; Jansson, T. Placental mTOR links maternal nutrient availability to fetal growth. Biochem. Soc. Trans. 2009, 37, 295–298. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Jones, H.N.; Jansson, T.; Powell, T.L. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino acid transport in human primary trophoblast cells. Diabetes 2010, 59, 1161–1170. [Google Scholar] [CrossRef]
- Von Versen-Hoynck, F.; Rajakumar, A.; Parrott, M.S.; Powers, R.W. Leptin affects system A amino acid transport activity in the human placenta: Evidence for STAT3 dependent mechanisms. Placenta 2009, 30, 361–367. [Google Scholar]
- Nelson, D.M.; Smith, S.D.; Furesz, T.C.; Sadovsky, Y.; Ganapathy, V.; Parvin, C.A.; Smith, C.H. Hypoxia reduces expression and function of system A amino acid transporters in cultured term human trophoblasts. Am. J. Physiol. Cell Physiol. 2003, 284, C310–C315. [Google Scholar] [CrossRef]
- Rosario, F.J.; Schumacher, M.A.; Jiang, J.; Kanai, Y.; Powell, T.L.; Jansson, T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J. Physiol. 2012, 590, 1495–1509. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef]
- Roos, S.; Jansson, N.; Palmberg, I.; Saljo, K.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Roos, S.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am. J. Physiol. Cell Physiol. 2009, 296, C142–C150. [Google Scholar]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J. Physiol. 2013, 591, 609–625. [Google Scholar] [CrossRef]
- Proud, C.G. Amino acids and mTOR signalling in anabolic function. Biochem. Soc. Trans. 2007, 35, 1187–1190. [Google Scholar] [CrossRef]
- Fang, J.; Mao, D.; Smith, C.H.; Fant, M.E. IGF regulation of neutral amino acid transport in the BeWo choriocarcinoma cell line (b30 clone): Evidence for MAP kinase-dependent and MAP kinase-independent mechanisms. Growth Horm. IGF Res. 2006, 16, 318–325. [Google Scholar] [CrossRef]
- Yung, H.W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef]
- Rosario, F.J.; Jansson, N.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 2011, 152, 1119–1129. [Google Scholar] [CrossRef]
- Gaccioli, F.; White, V.; Capobianco, E.; Powell, T.L.; Jawerbaum, A.; Jansson, T. Maternal overweight induced by a diet with high content of saturated fat activates placental mTOR and eIF2 alpha signaling and increases fetal growth in rats. Biol. Reprod. 2013, 89, 96. [Google Scholar] [CrossRef]
- Zhu, M.J.; Du, M.; Nijland, M.J.; Nathanielsz, P.W.; Hess, B.W.; Moss, G.E.; Ford, S.P. Down-regulation of growth signaling pathways linked to a reduced cotyledonary vascularity in placentomes of over-nourished, obese pregnant ewes. Placenta 2009, 30, 405–410. [Google Scholar]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Di Renzo, G.C.; Rosati, A.; Sarti, R.D.; Cruciani, L.; Cutuli, A.M. Does fetal sex affect pregnancy outcome? Gend. Med. 2007, 4, 19–30. [Google Scholar]
- Eriksson, J.G.; Kajantie, E.; Osmond, C.; Thornburg, K.; Barker, D.J. Boys live dangerously in the womb. Am. J. Hum. Biol. 2010, 22, 330–335. [Google Scholar] [CrossRef]
- Aiken, C.E.; Ozanne, S.E. Sex differences in developmental programming models. Reproduction 2013, 145, R1–R13. [Google Scholar]
- Van Abeelen, A.F.; de Rooij, S.R.; Osmond, C.; Painter, R.C.; Veenendaal, M.V.; Bossuyt, P.M.; Elias, S.G.; Grobbee, D.E.; van der Schouw, Y.T. The sex-specific effects of famine on the association between placental size and later hypertension. Placenta 2011, 32, 694–698. [Google Scholar]
- Tarrade, A.; Rousseau-Ralliard, D.; Aubriere, M.C.; Peynot, N.; Dahirel, M.; Bertrand-Michel, J.; Aguirre-Lavin, T.; Morel, O.; Beaujean, N.; Duranthon, V.; et al. Sexual dimorphism of the feto-placental phenotype in response to a high fat and control maternal diets in a rabbit model. PLoS One 2013, 8, e83458. [Google Scholar]
- Cox, L.A.; Li, C.; Glenn, J.P.; Lange, K.; Spradling, K.D.; Nathanielsz, P.W.; Jansson, T. Expression of the placental transcriptome in maternal nutrient reduction in baboons is dependent on fetal sex. J. Nutr. 2013, 143, 1698–1708. [Google Scholar] [CrossRef]
- Walker, S.P.; Ugoni, A.M.; Lim, R.; Lappas, M. Inverse relationship between gestational weight gain and glucose uptake in human placenta from female foetuses. Pediatr. Obes. 2014, 9, e73–e76. [Google Scholar] [CrossRef]
- Lewis, R.M.; Greenwood, S.L.; Cleal, J.K.; Crozier, S.R.; Verrall, L.; Inskip, H.M.; Cameron, I.T.; Cooper, C.; Sibley, C.P.; Hanson, M.A.; et al. Maternal muscle mass may influence system A activity in human placenta. Placenta 2010, 31, 418–422. [Google Scholar] [CrossRef]
- Kim, S.Y.; Sharma, A.J.; Sappenfield, W.; Wilson, H.G.; Salihu, H.M. Association of maternal body mass index, excessive weight gain, and gestational diabetes mellitus with large-for-gestational-age births. Obstet. Gynecol. 2014, 123, 737–744. [Google Scholar] [CrossRef]
- Ferraro, Z.M.; Barrowman, N.; Prud’homme, D.; Walker, M.; Wen, S.W.; Rodger, M.; Adamo, K.B. Excessive gestational weight gain predicts large for gestational age neonates independent of maternal body mass I ndex. J. Matern. Fetal Neonatal Med. 2012, 25, 538–542. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brett, K.E.; Ferraro, Z.M.; Yockell-Lelievre, J.; Gruslin, A.; Adamo, K.B. Maternal–Fetal Nutrient Transport in Pregnancy Pathologies: The Role of the Placenta. Int. J. Mol. Sci. 2014, 15, 16153-16185. https://doi.org/10.3390/ijms150916153
Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB. Maternal–Fetal Nutrient Transport in Pregnancy Pathologies: The Role of the Placenta. International Journal of Molecular Sciences. 2014; 15(9):16153-16185. https://doi.org/10.3390/ijms150916153
Chicago/Turabian StyleBrett, Kendra Elizabeth, Zachary Michael Ferraro, Julien Yockell-Lelievre, Andrée Gruslin, and Kristi Bree Adamo. 2014. "Maternal–Fetal Nutrient Transport in Pregnancy Pathologies: The Role of the Placenta" International Journal of Molecular Sciences 15, no. 9: 16153-16185. https://doi.org/10.3390/ijms150916153