The Neuroprotective Role of Coenzyme Q10 Against Lead Acetate-Induced Neurotoxicity Is Mediated by Antioxidant, Anti-Inflammatory and Anti-Apoptotic Activities

 , , and
, , and
Abstract
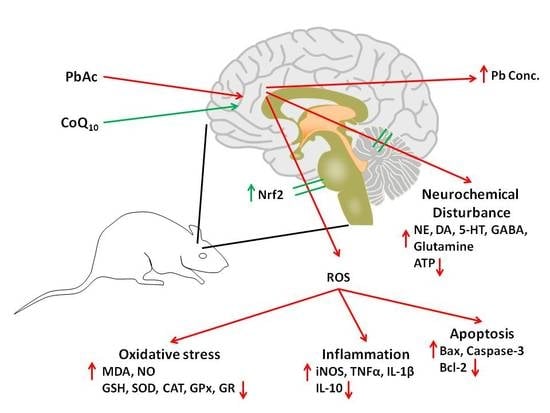
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Experimental Animals
2.3. Experimental Design
- (1)
- Control group injected intraperitoneally (i.p.) with 0.1 mL of saline containing 1% Tween 80 (v:v).
- (2)
- CoQ10 group injected i.p. daily at a dose of 10 mg/kg bwt, according to Fouad and Jresat [12].
- (3)
- (4)
- PbAc and CoQ10 injected group i.p. first with PbAc, then after 1 h injected with CoQ10 using the same mentioned doses.
2.4. Lead Concentration in the Cortical Tissue
2.5. Oxidative Stress Markers in the Cortical Tissue
2.6. Antioxidant Status of the Cortical Tissue
2.7. Inflammation Marker Assays
2.8. Quantitative Real Time PCR
2.9. Histological Changes
2.10. Apoptotic Proteins Quantification
2.11. Neurochemical Changes in the Cortical Tissue
2.12. Ethic Statement
2.13. Statistical Analysis
3. Results
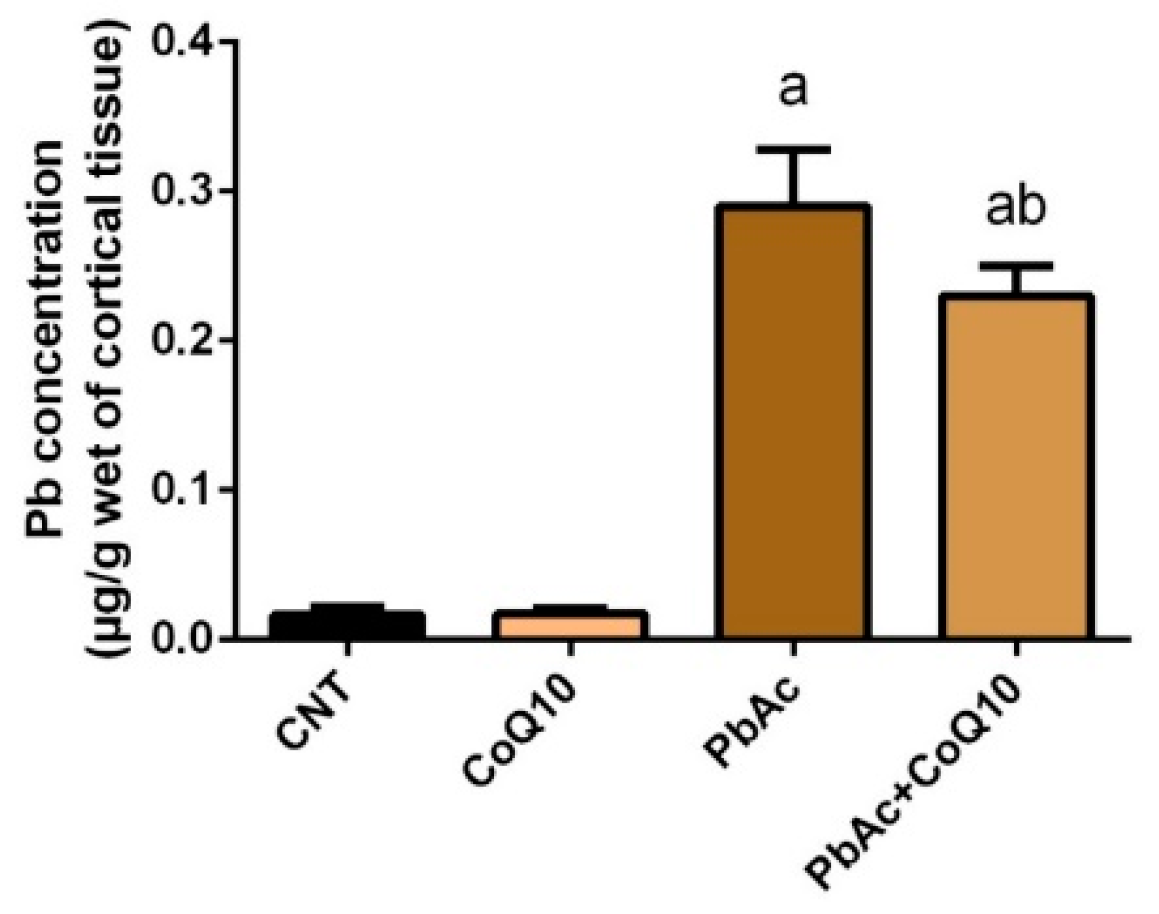
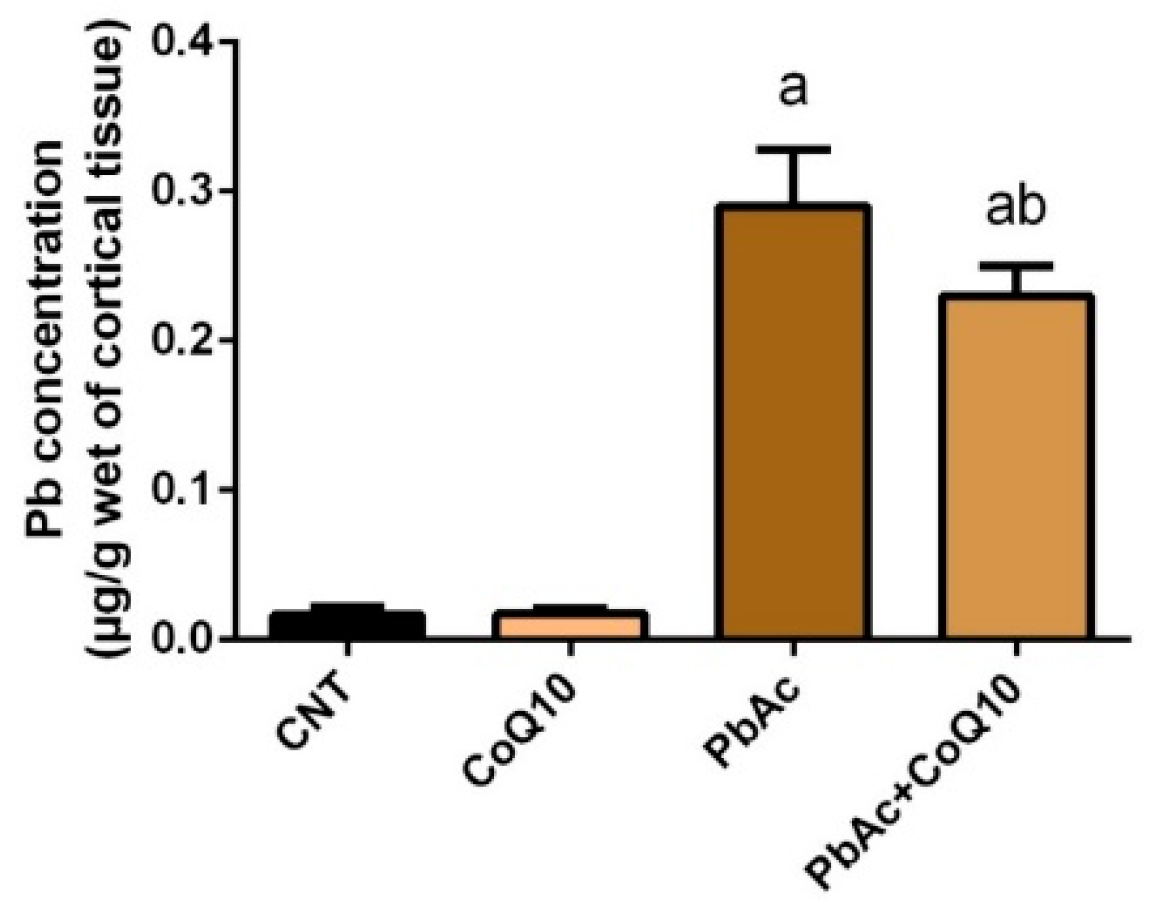
3.1. Pb Concentration in the Cortical Tissue
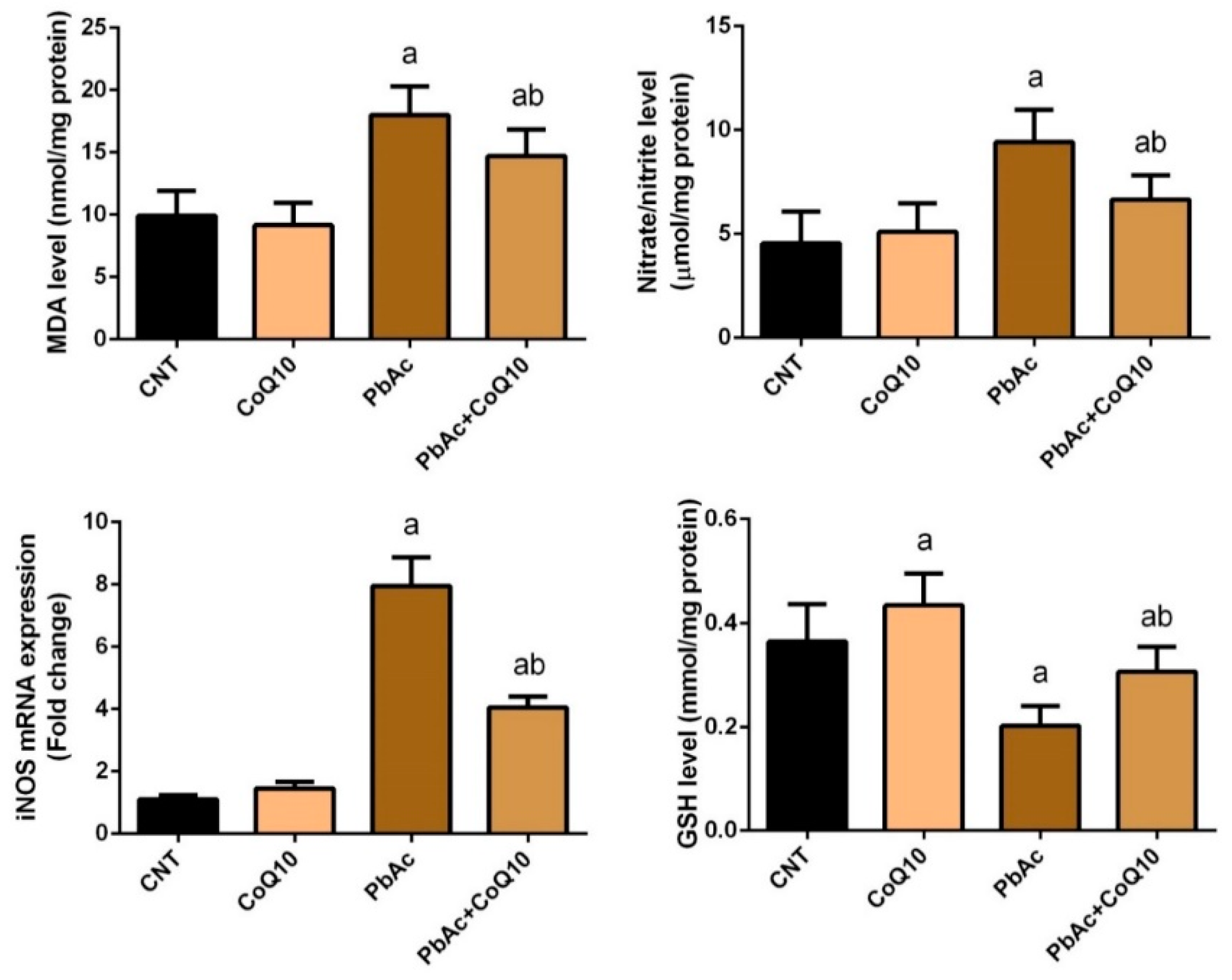
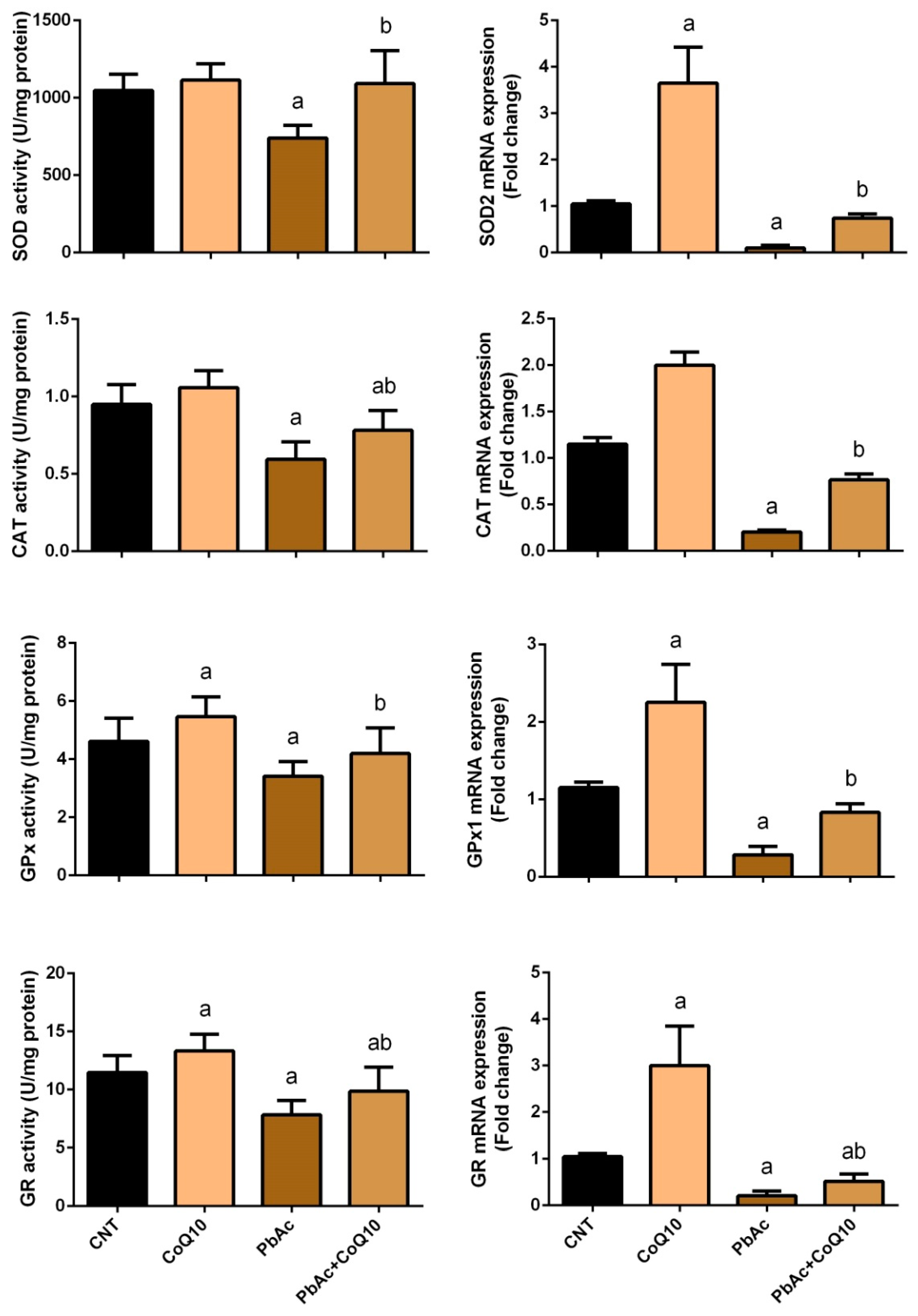
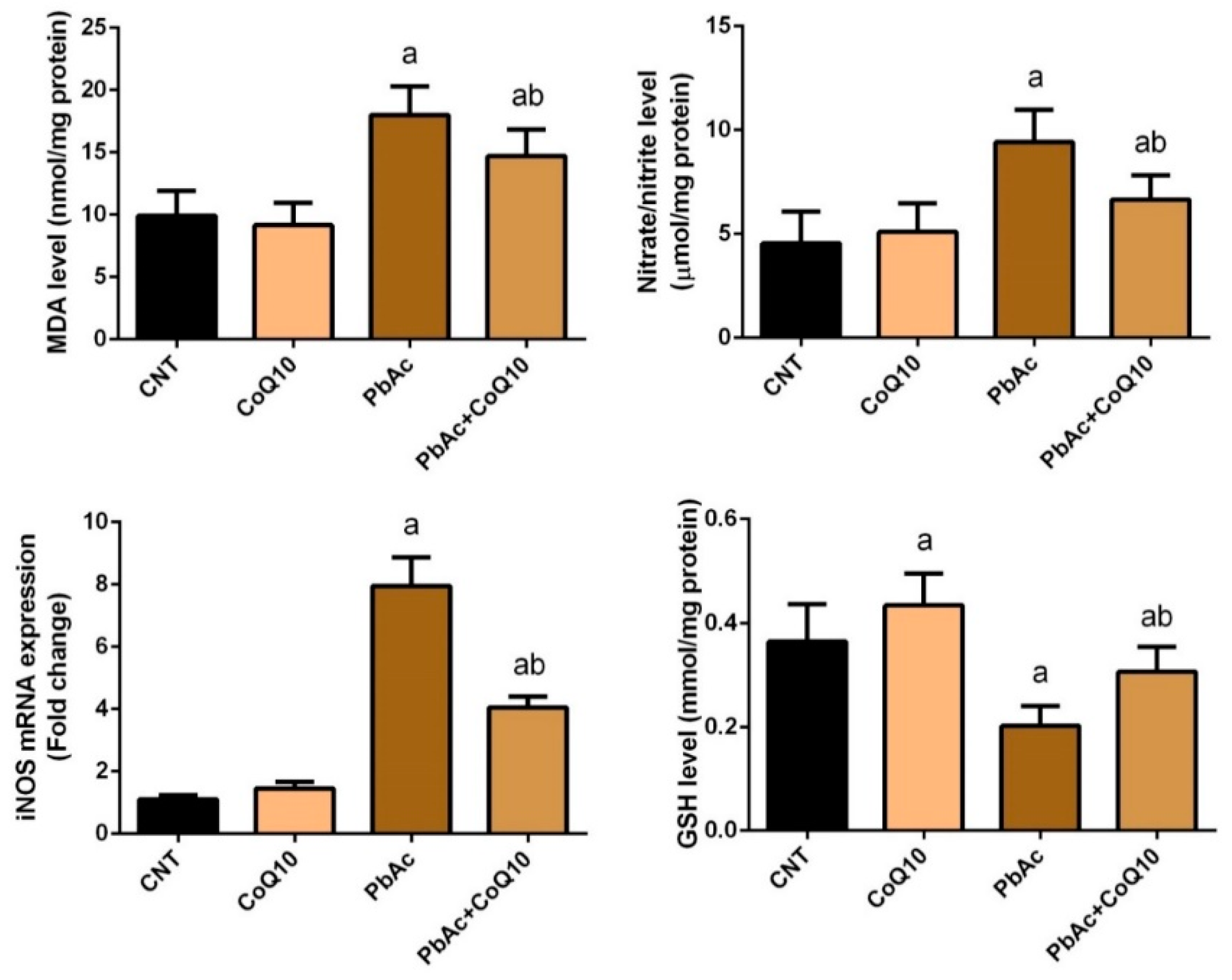
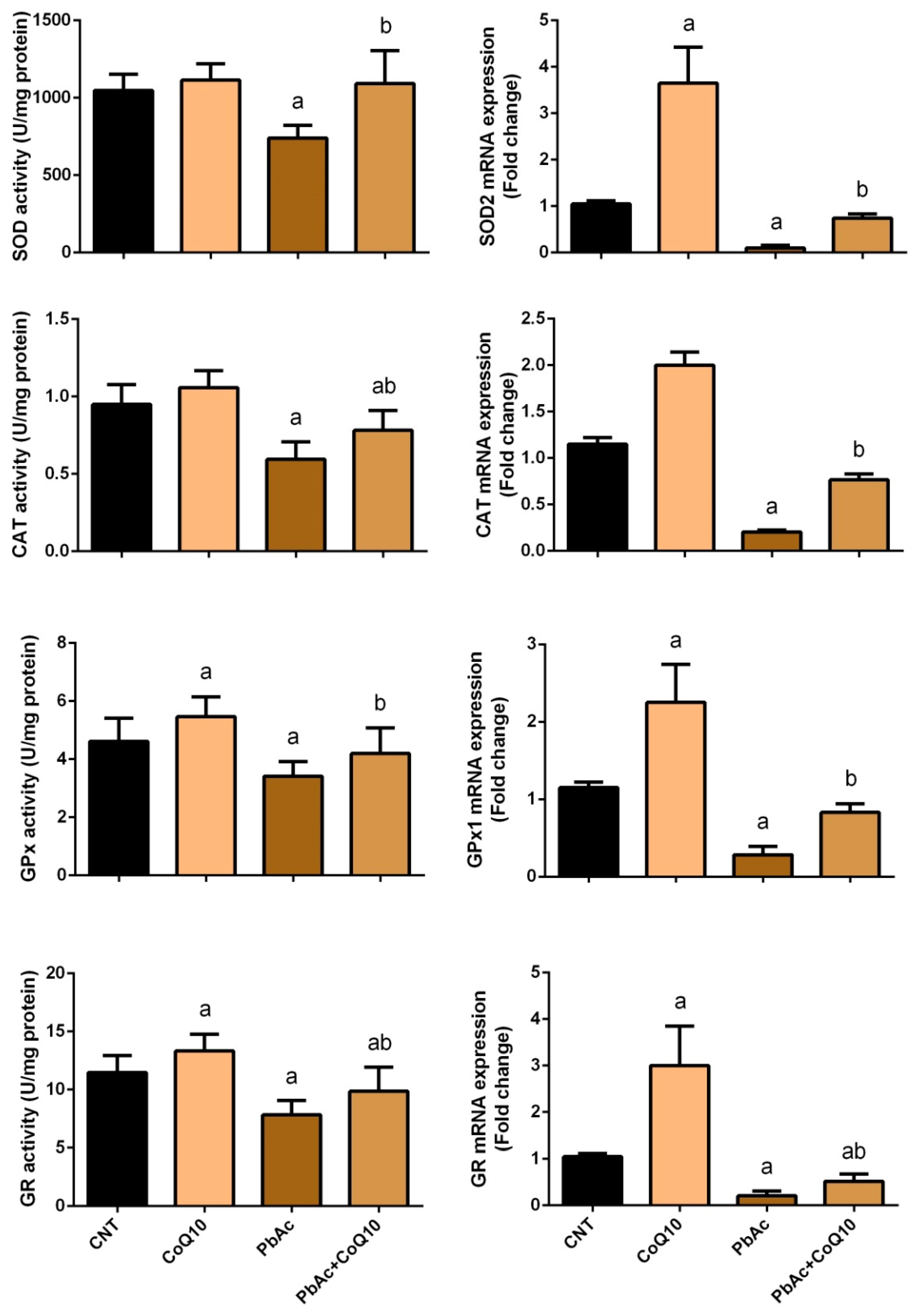
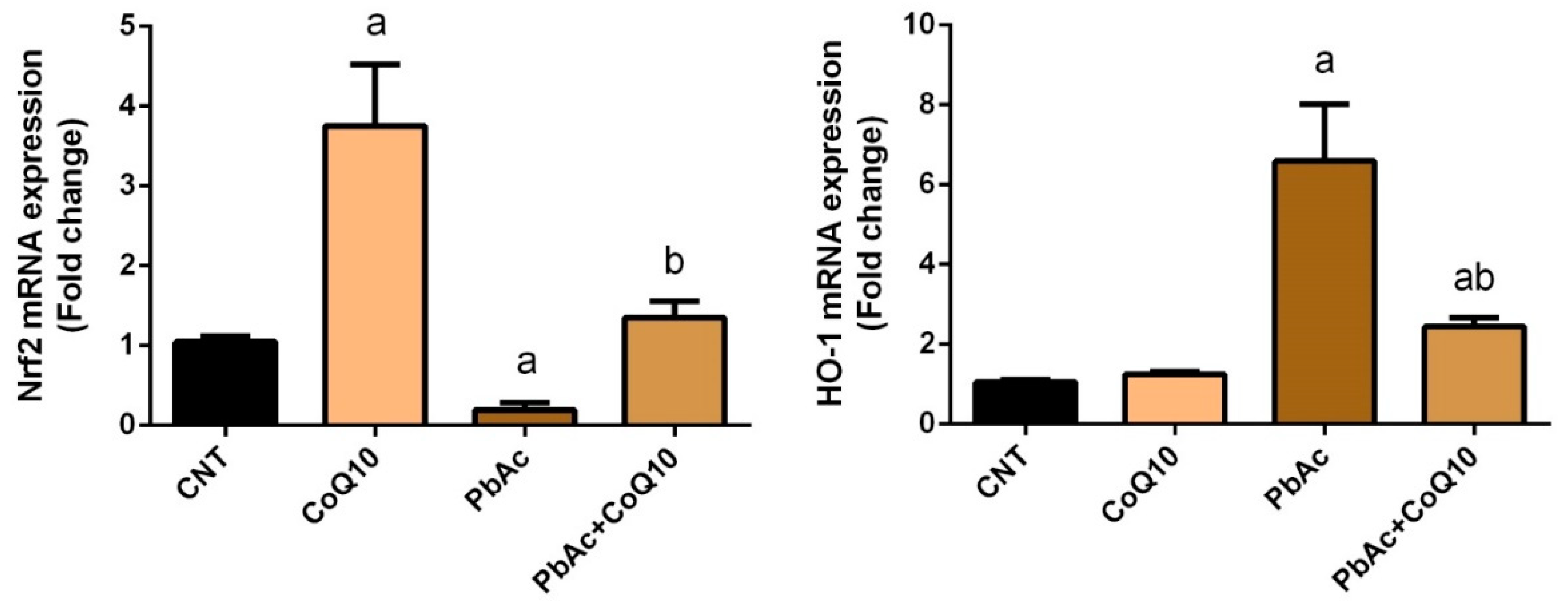
3.2. PbAc-Induced Oxidative Damage in the Cortical Tissue
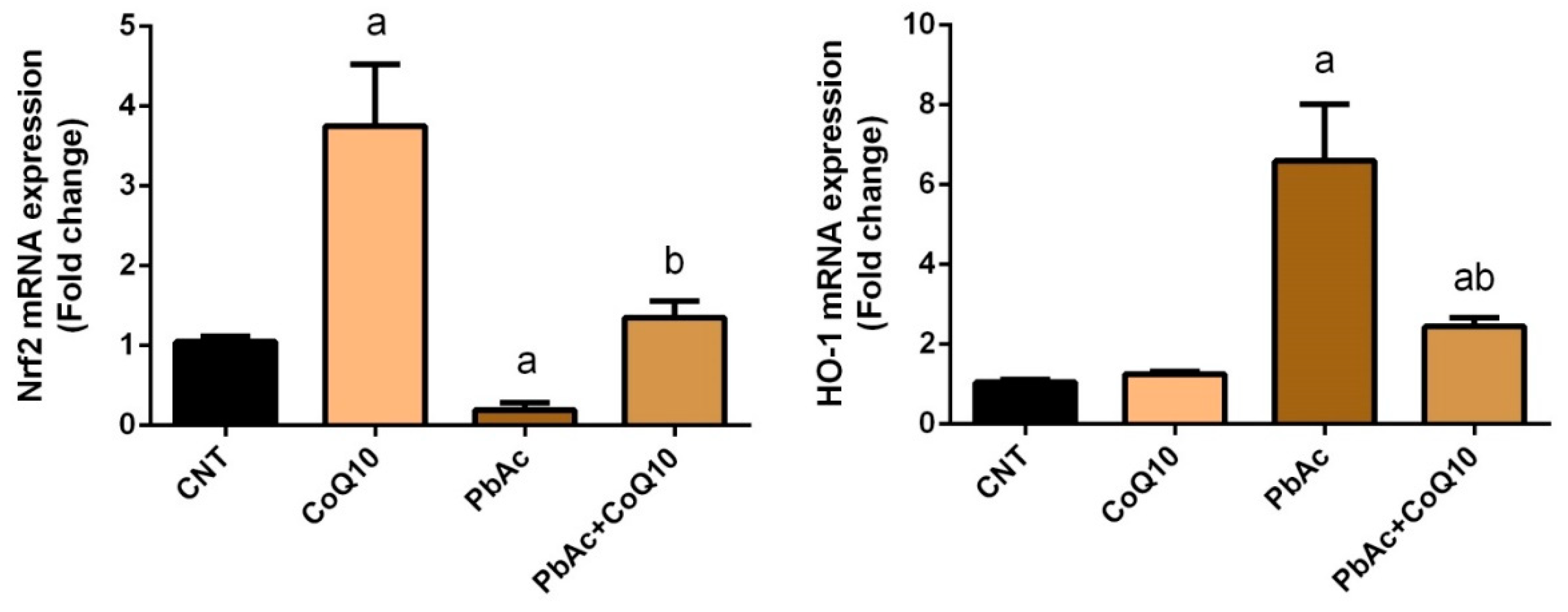
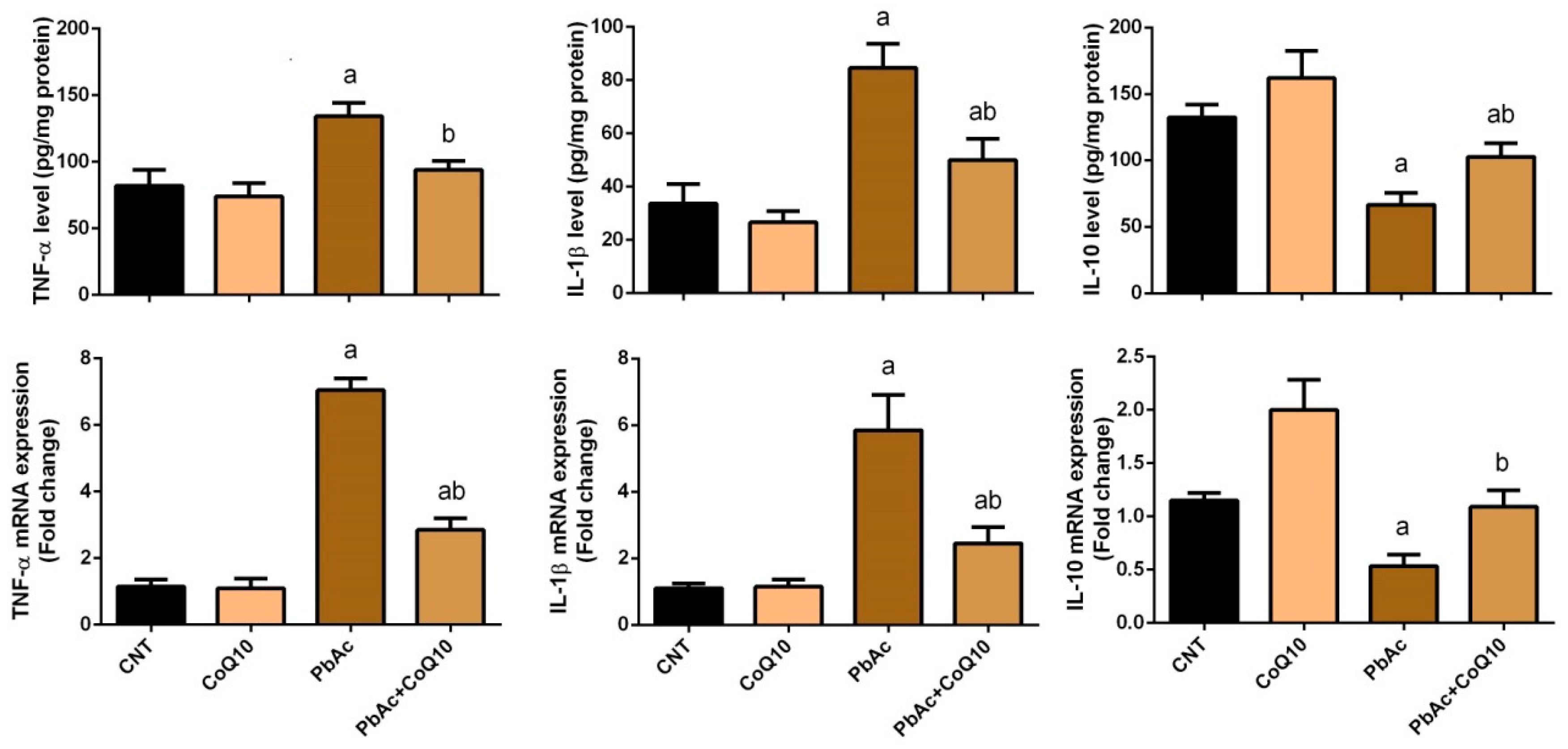
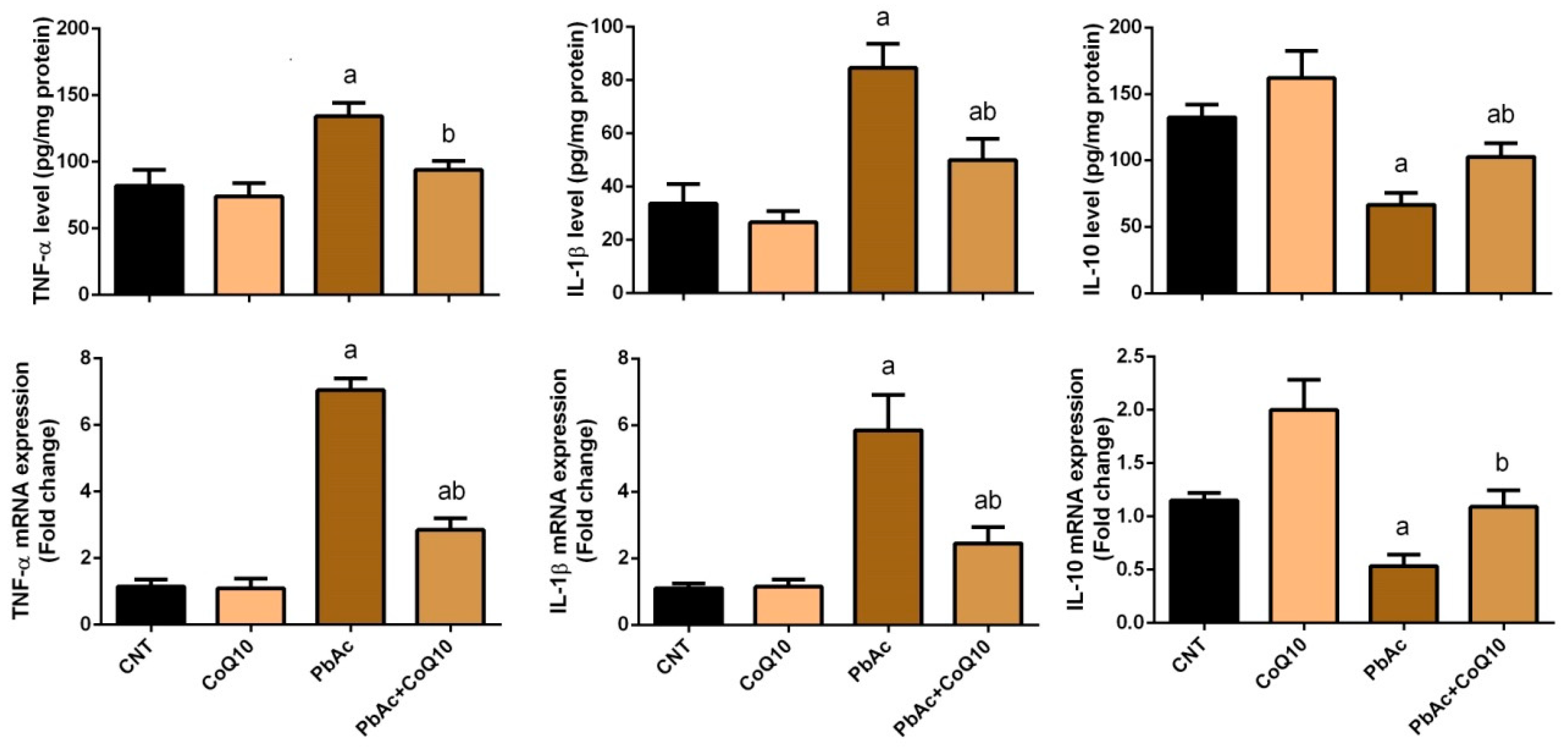
3.3. PbAc-Induced Inflammation in the Cortical Tissue
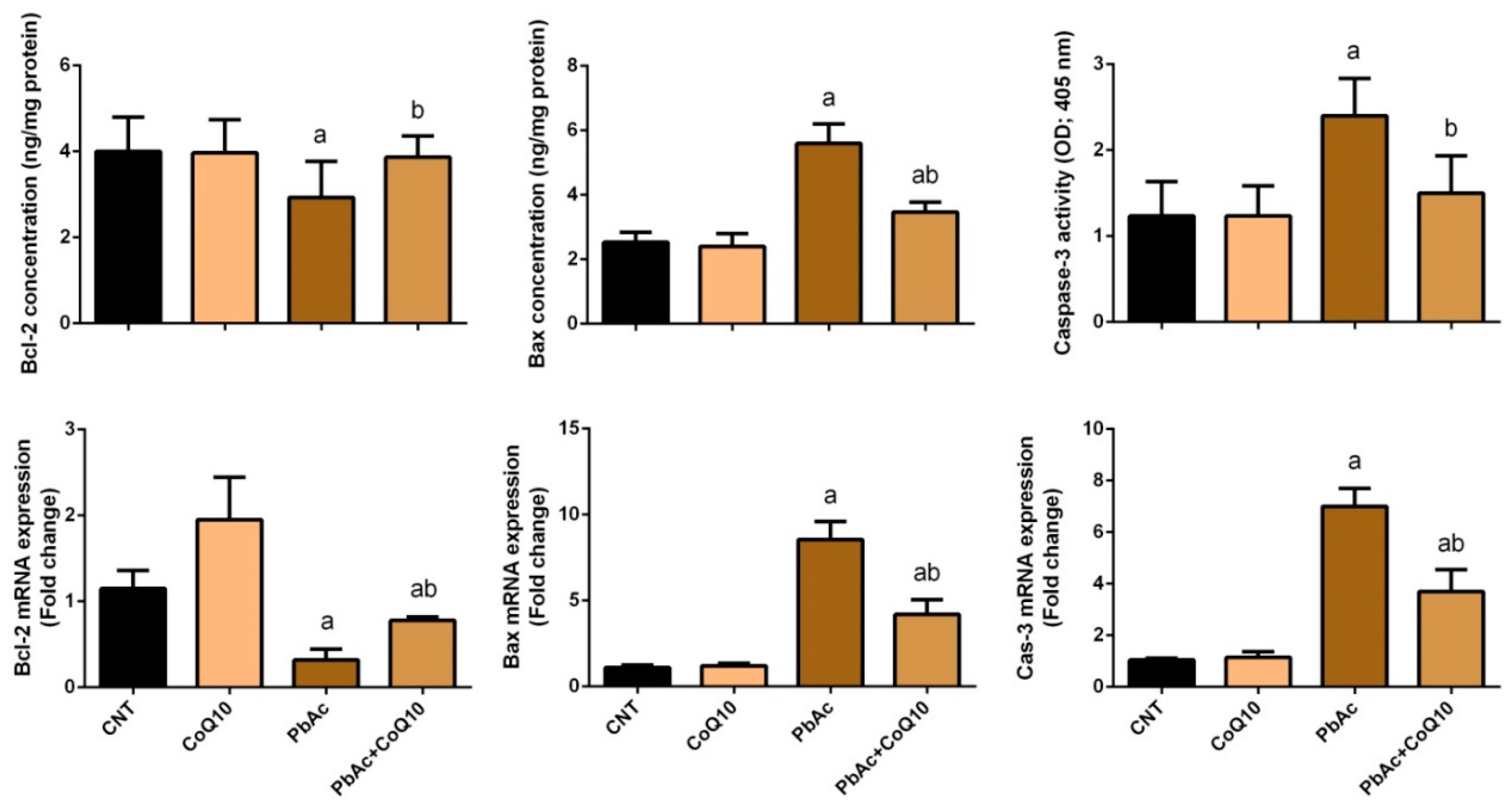
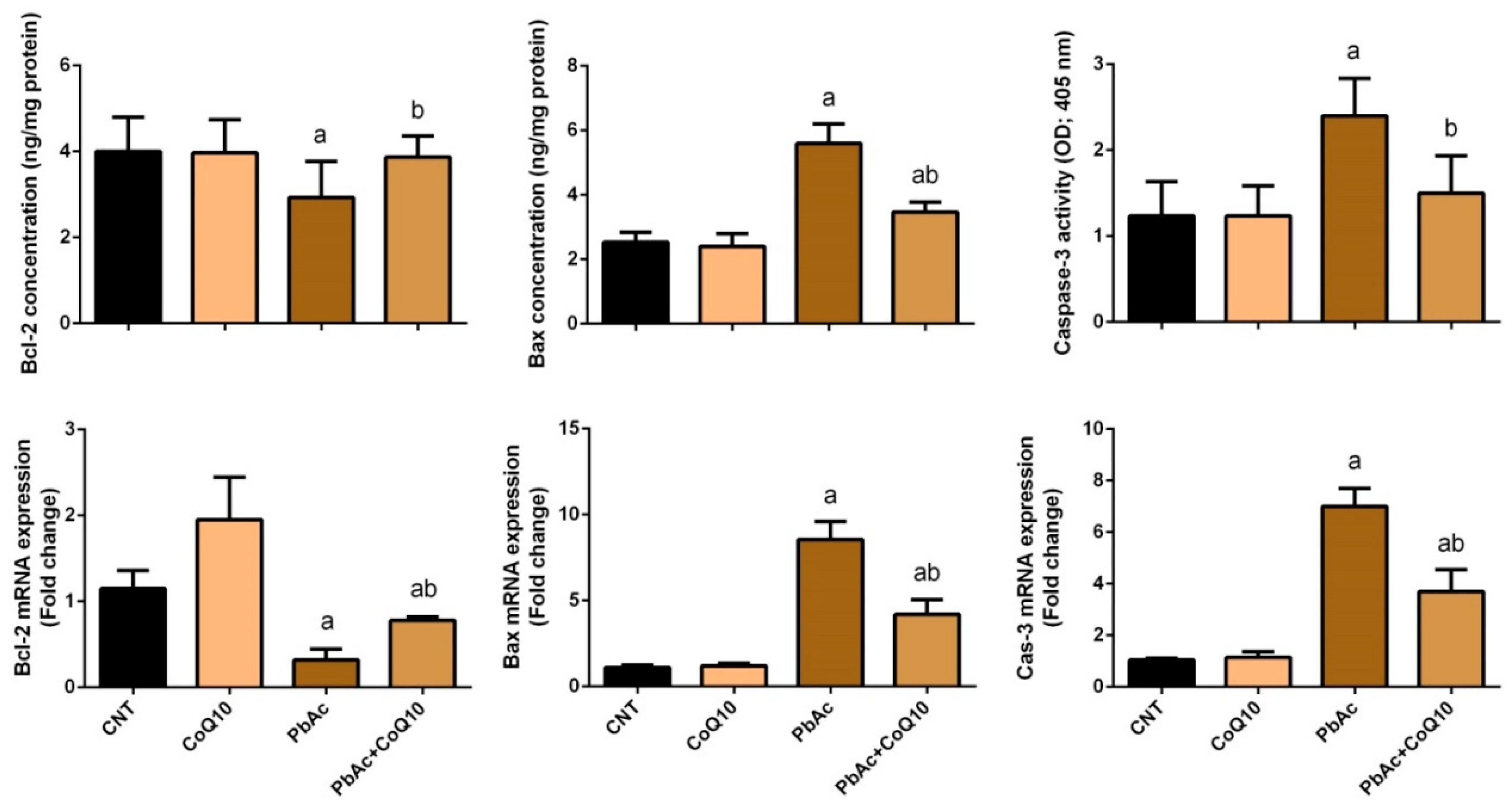
3.4. PbAc-Induced Apoptotic Cascade in the Cortical Tissue
3.5. PbAc-Induced Neurochemical Alterations in the Cortical Tissue
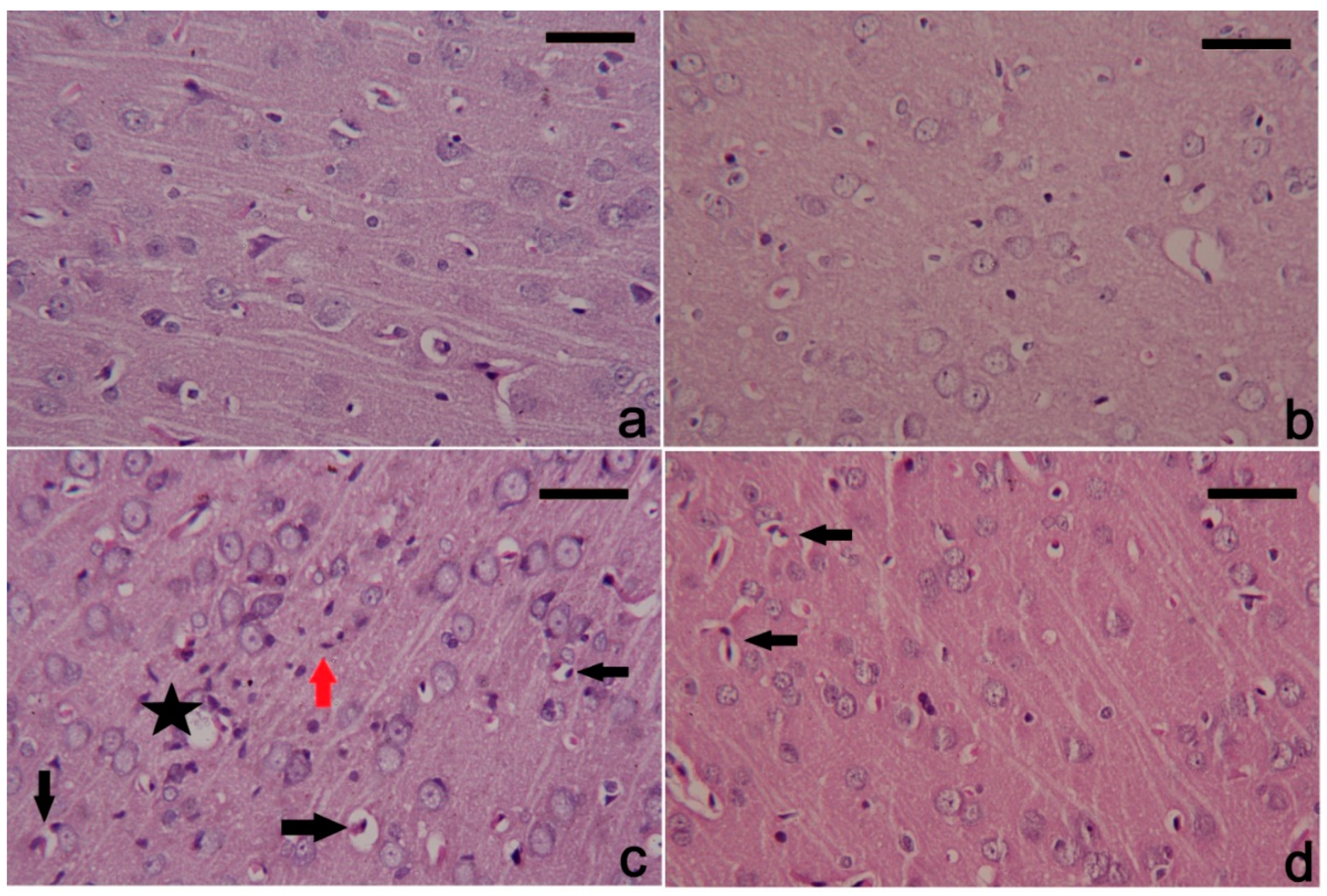
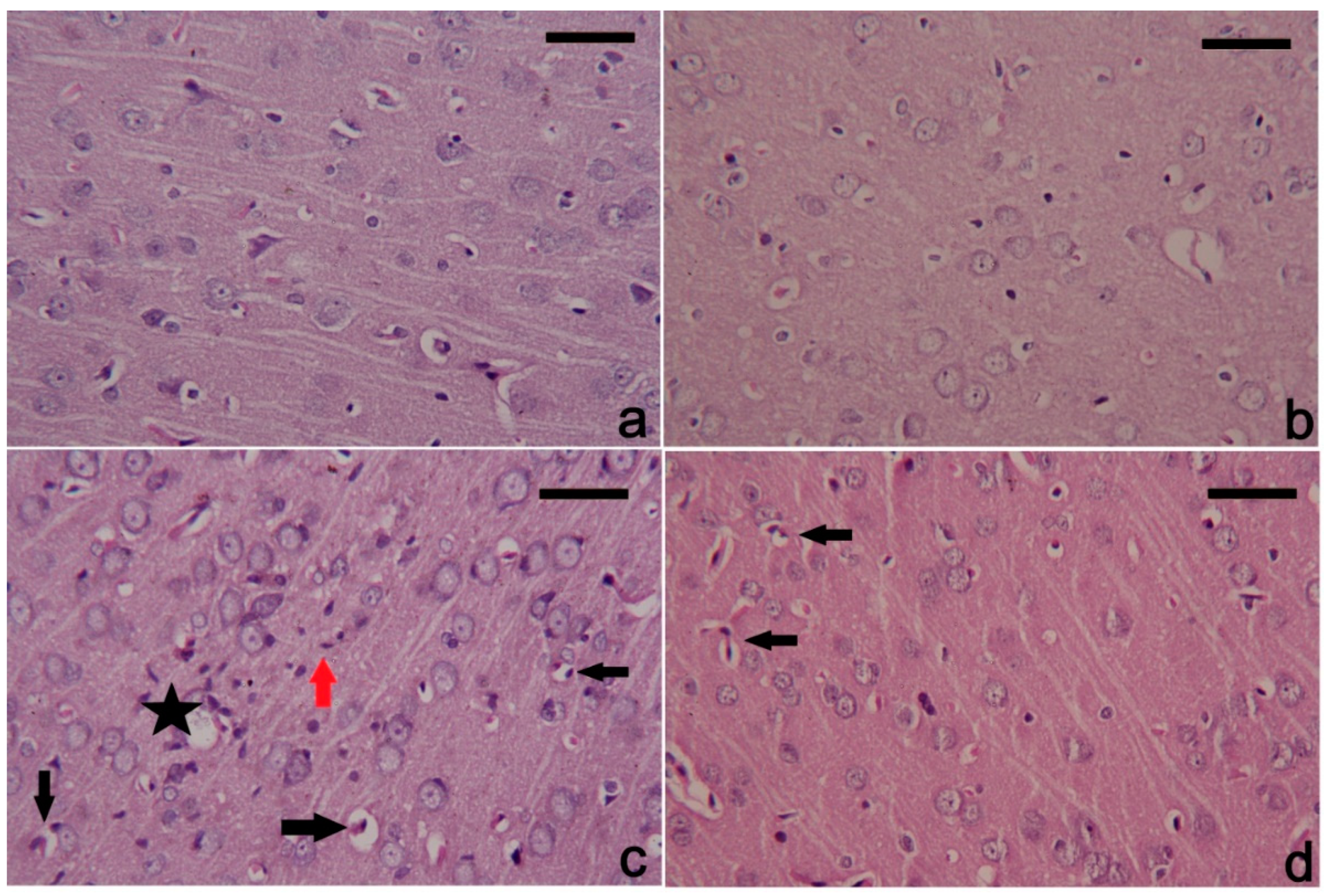
3.6. PbAc-Induced Histopathological Alterations in the Cortical Tissue
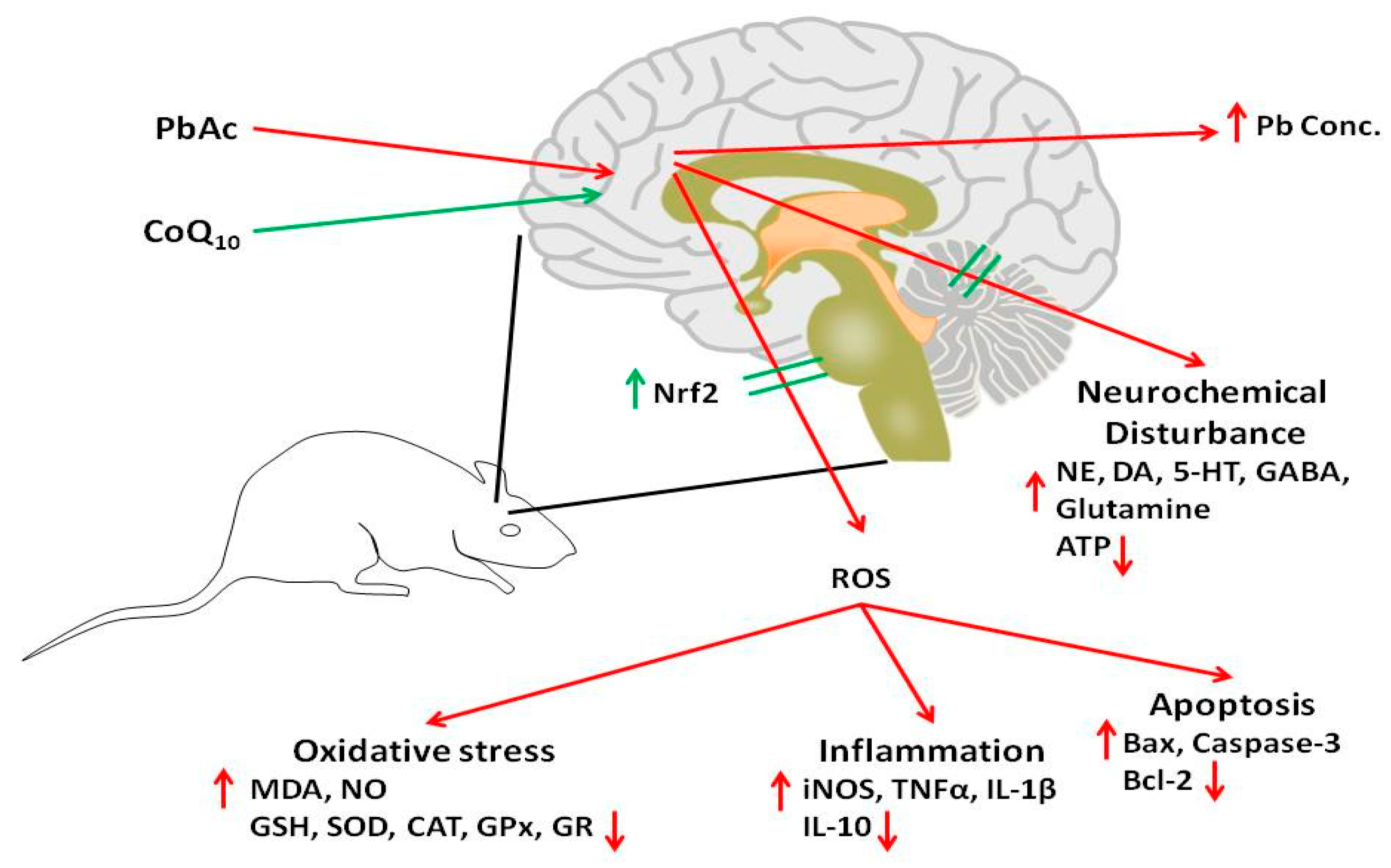
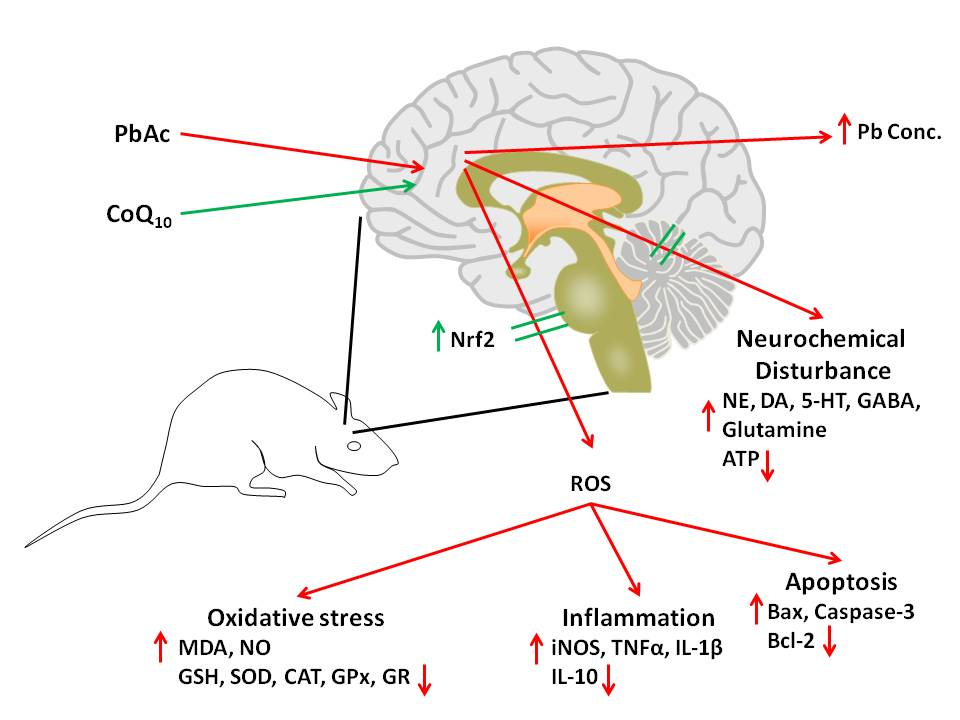
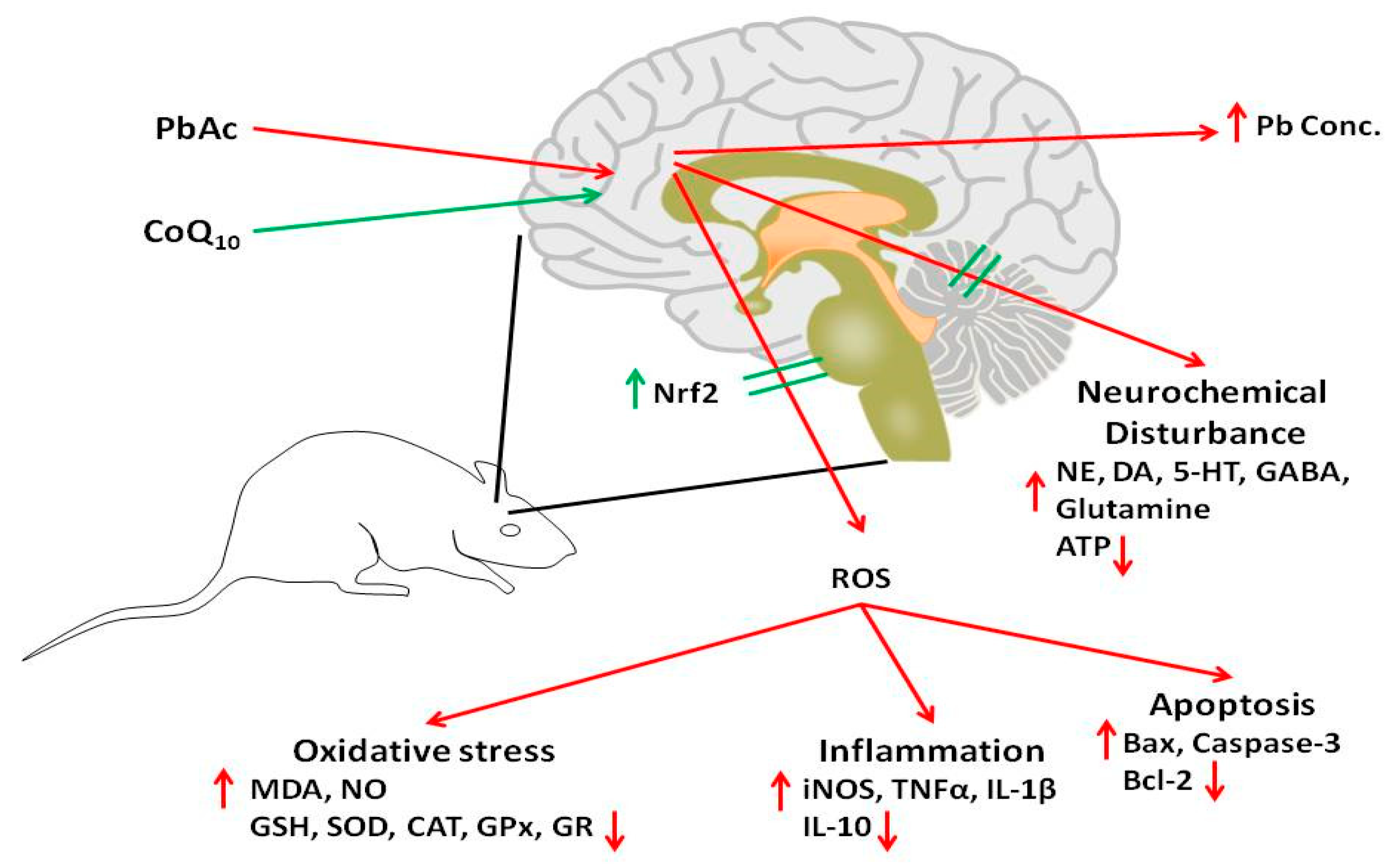
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, H.S.; Dsouza, S.A.; Menezes, G.; Venkatesh, T. Diagnosis, evaluation, and treatment of lead poisoning in general population. Indian J. Clin. Biochem. IJCB 2011, 26, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Molecular neurobiology of lead (pb(2+)): Effects on synaptic function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Bijoor, A.R.; Sudha, S.; Venkatesh, T. Neurochemical and neurobehavioral effects of low lead exposure on the developing brain. Indian J. Clin. Biochem. IJCB 2012, 27, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Kashala-Abotnes, E.; Mumbere, P.P.; Mishika, J.M.; Ndjukendi, A.O.; Mpaka, D.B.; Bumoko, M.G.; Kayembe, T.K.; Tshala-Katumbay, D.; Kazadi, T.K.; Okitundu, D.L. Lead exposure and early child neurodevelopment among children 12–24 months in kinshasa, the democratic republic of congo. Eur. Child Adolesc. Psychiatry 2016, 25, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Rodick, T.C.; Seibels, D.R.; Babu, R.; Huggins, K.W.; Ren, G.; Mathews, S.T. Potential role of coenzyme q10 in health and disease conditions. Nutr. Diet. Suppl. 2018, 10, 1. [Google Scholar] [CrossRef]
- Donnino, M.W.; Cocchi, M.N.; Salciccioli, J.D.; Kim, D.; Naini, A.B.; Buettner, C.; Akuthota, P. Coenzyme Q10 levels are low and may be associated with the inflammatory cascade in septic shock. Crit. Care 2011, 15, R189. [Google Scholar] [CrossRef]
- Hernandez-Camacho, J.D.; Bernier, M.; Lopez-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and heart failure: A state-of-the-art review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef]
- Sharma, A.; Kshetrimayum, C.; Sadhu, H.G.; Kumar, S. Arsenic-induced oxidative stress, cholinesterase activity in the brain of swiss albino mice, and its amelioration by antioxidants vitamin e and coenzyme Q10. Environ. Sci. Pollut. Res. Int. 2018, 25, 23946–23953. [Google Scholar] [CrossRef]
- Paunovic, M.G.; Matic, M.M.; Ognjanovic, B.I.; Saicic, Z.S. Antioxidative and haematoprotective activity of coenzyme Q10 and vitamin e against cadmium-induced oxidative stress in wistar rats. Toxicol. Ind. Health 2017, 33, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Fouad, A.A.; Jresat, I. Hepatoprotective effect of coenzyme Q10 in rats with acetaminophen toxicity. Environ. Toxicol. Pharmacol. 2012, 33, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Abdel Moneim, A.E. Flaxseed oil as a neuroprotective agent on lead acetate-induced monoamineric alterations and neurotoxicity in rats. Biol. Trace Elem. Res. 2012, 148, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Abdel Moneim, A.E.; Dkhil, M.A.; Al-Quraishy, S. Effects of flaxseed oil on lead acetate-induced neurotoxicity in rats. Biol. Trace Elem. Res. 2011, 144, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Jamshidzadeh, A.; Heidari, R.; Sun, Z.; Zamiri, M.J.; Khodaei, F.; Mousapour, S.; Ahmadi, F.; Javanmard, N.; Shirazi Yeganeh, B. Carnosine and histidine supplementation blunt lead-induced reproductive toxicity through antioxidative and mitochondria-dependent mechanisms. Biol. Trace Elem. Res. 2019, 187, 151–162. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Szkoda, J.; Zmudzki, J. Determination of lead and cadmium in biological material by graphite furnace atomic absorption spectrometry method. Bull. Vet. Inst. Pulawy 2005, 49, 89–92. [Google Scholar]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15n]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Almeer, R.S.; Abdel Moneim, A.E. Evaluation of the protective effect of olive leaf extract on cisplatin-induced testicular damage in rats. Oxid. Med. Cell. Longev. 2018, 2018, 11. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Pagel, P.; Blome, J.; Wolf, H.U. High-performance liquid chromatographic separation and measurement of various biogenic compounds possibly involved in the pathomechanism of parkinson’s disease. J. Chromatogr. B Biomed. Sci. Appl. 2000, 746, 297–304. [Google Scholar] [CrossRef]
- Heinrikson, R.L.; Meredith, S.C. Amino acid analysis by reverse-phase high-performance liquid chromatography: Precolumn derivatization with phenylisothiocyanate. Anal. Biochem. 1984, 136, 65–74. [Google Scholar] [CrossRef]
- Teerlink, T.; Hennekes, M.; Bussemaker, J.; Groeneveld, J. Simultaneous determination of creatine compounds and adenine nucleotides in myocardial tissue by high-performance liquid chromatography. Anal. Biochem. 1993, 214, 278–283. [Google Scholar] [CrossRef]
- Nam, S.M.; Seo, J.S.; Go, T.H.; Nahm, S.S.; Chang, B.J. Ascorbic acid supplementation prevents the detrimental effects of prenatal and postnatal lead exposure on the purkinje cell and related proteins in the cerebellum of developing rats. Biol. Trace Elem. Res. 2018, 190, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.S.; Hao, J.H.; Zeng, Y.; Dai, F.C.; Gu, P.Q. Neurotoxicity and biomarkers of lead exposure: A review. Chin. Med. Sci. J. 2013, 28, 178–188. [Google Scholar] [CrossRef]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic effects and biomarkers of lead exposure: A review. Rev. Environ. Health 2009, 24, 15–45. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpao, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols journey through blood-brain barrier towards neuronal protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Al-Quraishy, S.; Dkhil, M.A.; Ibrahim, S.R.; Abdel Moneim, A.E. Neuroprotective potential of Indigofera oblongifolia leaf methanolic extract against lead acetate-induced neurotoxicity. Neural Regen. Res. 2016, 11, 1797–1803. [Google Scholar]
- Maiti, A.K.; Saha, N.C.; More, S.S.; Panigrahi, A.K.; Paul, G. Neuroprotective efficacy of mitochondrial antioxidant mitoq in suppressing peroxynitrite-mediated mitochondrial dysfunction inflicted by lead toxicity in the rat brain. Neurotox. Res. 2017, 31, 358–372. [Google Scholar] [CrossRef]
- Shen, S.C.; Wu, M.S.; Lin, H.Y.; Yang, L.Y.; Chen, Y.H.; Chen, Y.C. Reactive oxygen species-dependent nitric oxide production in reciprocal interactions of glioma and microglial cells. J. Cell. Physiol. 2014, 229, 2015–2026. [Google Scholar] [CrossRef]
- Ahamed, M.; Siddiqui, M.K. Low level lead exposure and oxidative stress: Current opinions. Clin. Chim. Acta 2007, 383, 57–64. [Google Scholar] [CrossRef]
- Flora, S.J.; Saxena, G.; Mehta, A. Reversal of lead-induced neuronal apoptosis by chelation treatment in rats: Role of reactive oxygen species and intracellular Ca(2+). J. Pharmacol. Exp. Ther. 2007, 322, 108–116. [Google Scholar] [CrossRef]
- Dkhil, M.A.; Abdel Moneim, A.E.; Hafez, T.A.; Mubaraki, M.A.; Mohamed, W.F.; Thagfan, F.A.; Al-Quraishy, S. Myristica fragrans kernels prevent paracetamol-induced hepatotoxicity by inducing anti-apoptotic genes and Nrf2/HO-1 pathway. Int. J. Mol. Sci. 2019, 20, 993. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef]
- Ye, F.; Li, X.; Li, L.; Yuan, J.; Chen, J. T-bhq provides protection against lead neurotoxicity via Nrf2/HO-1 pathway. Oxid. Med. Cell. Longev. 2016, 2016, 2075915. [Google Scholar] [CrossRef]
- Xiaoyi, L.; Ye, F.; Li, L.; Chang, W.; Wu, X.; Chen, J. The role of HO-1 in protection against lead-induced neurotoxicity. Neurotoxicology 2016, 52, 1–11. [Google Scholar]
- Duberley, K.E.; Heales, S.J.R.; Abramov, A.Y.; Chalasani, A.; Land, J.M.; Rahman, S.; Hargreaves, I.P. Effect of coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in coenzyme q10 deficient human neuronal cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef]
- Sattarinezhad, E.; Shafaroodi, H.; Sheikhnouri, K.; Mousavi, Z.; Moezi, L. The effects of coenzyme Q10 on seizures in mice: The involvement of nitric oxide. Epilepsy Behav. E&B 2014, 37, 36–42. [Google Scholar]
- Erol, B.; Bozlu, M.; Hanci, V.; Tokgoz, H.; Bektas, S.; Mungan, G. Coenzyme Q10 treatment reduces lipid peroxidation, inducible and endothelial nitric oxide synthases, and germ cell-specific apoptosis in a rat model of testicular ischemia/reperfusion injury. Fertil. Steril. 2010, 93, 280–282. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Martin-Gronert, M.S.; McConnell, J.M.; Ozanne, S.E. Coenzyme Q10 prevents hepatic fibrosis, inflammation, and oxidative stress in a male rat model of poor maternal nutrition and accelerated postnatal growth. Am. J. Clin. Nutr. 2016, 103, 579–588. [Google Scholar] [CrossRef]
- Kassab, R.B.; Lokman, M.S.; Essawy, E.A. Neurochemical alterations following the exposure to di-n-butyl phthalate in rats. Metab. Brain Dis. 2018, 34, 235–244. [Google Scholar] [CrossRef]
- Sharma, P.; Chambial, S.; Shukla, K.K. Lead and neurotoxicity. Indian J. Clin. Biochem. IJCB 2015, 30, 1–2. [Google Scholar] [CrossRef]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of lead: A review with recent updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef]
- Leret, M.L.; Garcia-Uceda, F.; Antonio, M.T. Effects of maternal lead administration on monoaminergic, gabaergic and glutamatergic systems. Brain Res. Bull. 2002, 58, 469–473. [Google Scholar] [CrossRef]
- Gupta, V.; Gill, K.D. Lead and ethanol coexposure: Implications on the dopaminergic system and associated behavioral functions. Pharmacol. Biochem. Behav. 2000, 66, 465–474. [Google Scholar] [CrossRef]
- Saritha, S.; Davuljigari, C.B.; Kumar, K.P.; Reddy, G.R. Effects of arsenic and lead combined exposure on brain monoaminergic system and behavioral functions in rats: Reversal effect of miadmsa. Toxicol. Ind. Health 2019, 35, 89–108. [Google Scholar] [CrossRef]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. BioMed Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef]
- Braga, M.F.; Pereira, E.F.; Albuquerque, E.X. Nanomolar concentrations of lead inhibit glutamatergic and gabaergic transmission in hippocampal neurons. Brain Res. 1999, 826, 22–34. [Google Scholar] [CrossRef]
- Abuelezz, S.A.; Hendawy, N.; Magdy, Y. The potential benefit of combined versus monotherapy of coenzyme Q10 and fluoxetine on depressive-like behaviors and intermediates coupled to gsk-3beta in rats. Toxicol. Appl. Pharmacol. 2018, 340, 39–48. [Google Scholar] [CrossRef]
- Motawi, T.K.; Darwish, H.A.; Hamed, M.A.; El-Rigal, N.S.; Aboul Naser, A.F. Coenzyme Q10 and niacin mitigate streptozotocin- induced diabetic encephalopathy in a rat model. Metab. Brain Dis. 2017, 32, 1519–1527. [Google Scholar] [CrossRef]
- Kooncumchoo, P.; Sharma, S.; Porter, J.; Govitrapong, P.; Ebadi, M. Coenzyme Q(10) provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J. Mol. Neurosci. MN 2006, 28, 125–141. [Google Scholar] [CrossRef]
- Gao, H.L.; Yu, X.J.; Qi, J.; Yi, Q.Y.; Jing, W.H.; Sun, W.Y.; Cui, W.; Mu, J.J.; Yuan, Z.Y.; Zhao, X.F.; et al. Oral coQ10 attenuates high salt-induced hypertension by restoring neurotransmitters and cytokines in the hypothalamic paraventricular nucleus. Sci. Rep. 2016, 6, 30301. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Sena, C.; Nunes, E.; Seica, R.; Oliveira, C.R. CoQ10 therapy attenuates amyloid beta-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp. Neurol. 2005, 196, 112–119. [Google Scholar] [CrossRef]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of lead (pb) on inflammatory processes in the brain. Int. J. Mol. Sci. 2016, 17, 2140. [Google Scholar] [CrossRef]
- Struzynska, L.; Dabrowska-Bouta, B.; Koza, K.; Sulkowski, G. Inflammation-like glial response in lead-exposed immature rat brain. Toxicol. Sci. 2007, 95, 156–162. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Pabello, N.; Bolivar, V.J.; Lawrence, D.A. Developmental lead effects on behavior and brain gene expression in male and female balb/canntac mice. Neurotoxicology 2012, 33, 1005–1020. [Google Scholar] [CrossRef]
- Schmelzer, C.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Doring, F. Functions of coenzyme Q10 in inflammation and gene expression. Biofactors 2008, 32, 179–183. [Google Scholar] [CrossRef]
- Hassanzadeh, S.; Jameie, S.B.; Soleimani, M.; Farhadi, M.; Kerdari, M.; Danaei, N. Coenzyme Q10 influences on the levels of tnf-alpha and il-10 and the ratio of bax/bcl2 in a menopausal rat model following lumbar spinal cord injury. J. Mol. Neurosci. MN 2018, 65, 255–264. [Google Scholar] [CrossRef]
- Wong, M.L.; Bongiorno, P.B.; Rettori, V.; McCann, S.M.; Licinio, J. Interleukin (IL) 1beta, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: Pathophysiological implications. Proc. Natl. Acad. Sci. USA 1997, 94, 227–232. [Google Scholar] [CrossRef]
- Chao, S.L.; Moss, J.M.; Harry, G.J. Lead-induced alterations of apoptosis and neurotrophic factor mrna in the developing rat cortex, hippocampus, and cerebellum. J. Biochem. Mol. Toxicol. 2007, 21, 265–272. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, C.C.; Yang, Y.; Liu, J.W.; Yan, C.H. Gm1 ameliorates lead-induced cognitive deficits and brain damage through activating the SIRT1/CREB/BDNF pathway in the developing male rat hippocampus. Biol. Trace Elem. Res. 2018. [Google Scholar] [CrossRef]
- Shults, C.W.; Haas, R. Clinical trials of coenzyme Q10 in neurological disorders. Biofactors 2005, 25, 117–126. [Google Scholar] [CrossRef]
- Nasoohi, S.; Simani, L.; Khodagholi, F.; Nikseresht, S.; Faizi, M.; Naderi, N. Coenzyme Q10 supplementation improves acute outcomes of stroke in rats pretreated with atorvastatin. Nutr. Neurosci. 2017, 22, 264–272. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Gene Symbol and Accession No. | Sense (5’---3’) | Antisense (5’---3’) | Amplicon Size (bp) | PCR Efficiency (%) |
|---|---|---|---|---|---|
| β-actin | Actb: NM_031144.3 | GGCATCCTGACCCTGAAGTA | GGGGTGTTGAAGGTCTCAAA | 203 | 96.9% |
| SOD2 | Sod2: NM_017051.2 | AGCTGCACCACAGCAAGCAC | TCCACCACCCTTAGGGCTCA | 191 | 106.8% |
| CAT | Cat: NM_012520.2 | TCCGGGATCTTTTTAACGCCATTG | TCGAGCACGGTAGGGACAGTTCAC | 362 | 93.7% |
| GPx1 | Gpx1: NM_030826.4 | CGGTTTCCCGTGCAATCAGT | ACACCGGGGACCAAATGATG | 245 | 103.2% |
| GR | Gsr: NM_053906.2 | TGGCACTTGCGTGAATGTTG | CGAATGTTGCATAGCCGTGG | 233 | 116.0% |
| Nrf2 | Nfe2l2: NM_031789.2 | GGTTGCCCACATTCCCAAAC | GGCTGGGAATATCCAGGGC | 116 | 105.5% |
| HO-1 | Hmox1: NM_012580.2 | GCGAAACAAGCAGAACCCA | GCTCAGGATGAGTACCTCCCA | 185 | 105.2% |
| iNOS | Nos2: NM_012611.3 | GTTCCTCAGGCTTGGGTCTT | TGGGGGAACACAGTAATGGC | 825 | 97.7% |
| TNF-α | Tnfa: NM_012675.3 | AGAACTCAGCGAGGACACCAA | GCTTGGTGGTTTGCTACGAC | 461 | 104.7% |
| IL-1β | Il1b: NM_031512.2 | GACTTCACCATGGAACCCGT | GGAGACTGCCCATTCTCGAC | 104 | 109.3% |
| IL-10 | Il10: NM_012854.2 | TTGAACCACCCGGCATCTAC | CCAAGGAGTTGCTCCCGTTA | 91 | 106.8% |
| Bcl-2 | Bcl2: NM_016993.1 | ACTCTTCAGGGATGGGGTGA | TGACATCTCCCTGTTGACGC | 94 | 96.7% |
| Bax | Bax: NM_017059.2 | CTGAGCTGACCTTGGAGC | GACTCCAGCCACAAAGATG | 413 | 102.7% |
| Caspase-3 | Casp3: NM_012922.2 | GAGCTTGGAACGCGAAGAAA | TAACCGGGTGCGGTAGAGTA | 635 | 108.4% |
| Brain Areas | Experimental Groups | |||
|---|---|---|---|---|
| CNT | CoQ10 | PbAc | PbAc + CoQ10 | |
| Norepinephrine (μg/ g tissue) | 0.41 ± 0.003 | 0.39 ± 0.002 | 0.88 ± 0.002a | 0.62 ± 0.002ab |
| Dopamine (μg/g tissue) | 1.07 ± 0.007 | 1.14 ± 0.04 | 1.76 ± 0.03a | 1.38 ± 0.001b |
| Serotonin (μg/g tissue) | 0.48 ± 0.002 | 0.58 ± 0.002 | 0.93 ± 0.02a | 0.66 ± 0.002b |
| glutamate (μmol/g tissue) | 9.48 ± 0.06 | 9.17 ± 0.03a | 12.88 ± 0.03a | 10.72 ± 0.04b |
| GABA (μmol/g tissue) | 4.39 ± 0.02 | 4.22 ± 0.03 | 7.39 ± 0.04a | 5.39 ± 0.03b |
| ATP (μmol/g tissue) | 5.03 ± 0.03 | 7.93 ± 0.03a | 2.39 ± 0.03 | 6.48 ± 0.04b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Yousef, A.O.; A. Fahad, A.; Abdel Moneim, A.E.; Metwally, D.M.; El-khadragy, M.F.; Kassab, R.B. The Neuroprotective Role of Coenzyme Q10 Against Lead Acetate-Induced Neurotoxicity Is Mediated by Antioxidant, Anti-Inflammatory and Anti-Apoptotic Activities. Int. J. Environ. Res. Public Health 2019, 16, 2895. https://doi.org/10.3390/ijerph16162895
S. Yousef AO, A. Fahad A, Abdel Moneim AE, Metwally DM, El-khadragy MF, Kassab RB. The Neuroprotective Role of Coenzyme Q10 Against Lead Acetate-Induced Neurotoxicity Is Mediated by Antioxidant, Anti-Inflammatory and Anti-Apoptotic Activities. International Journal of Environmental Research and Public Health. 2019; 16(16):2895. https://doi.org/10.3390/ijerph16162895
Chicago/Turabian StyleS. Yousef, Al Omar, Alkhuriji A. Fahad, Ahmed E. Abdel Moneim, Dina M. Metwally, Manal F. El-khadragy, and Rami B. Kassab. 2019. "The Neuroprotective Role of Coenzyme Q10 Against Lead Acetate-Induced Neurotoxicity Is Mediated by Antioxidant, Anti-Inflammatory and Anti-Apoptotic Activities" International Journal of Environmental Research and Public Health 16, no. 16: 2895. https://doi.org/10.3390/ijerph16162895