TERribly Difficult: Searching for Telomerase RNAs in Saccharomycetes


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phylogenomics of Ascomycotes
2.2. Ascomycote Telomerase RNAs
2.3. Synteny-Based Homology Search
2.4. Search for Candidates Using Telomere Template Sequences
2.5. BLAST Pipeline
3. Results
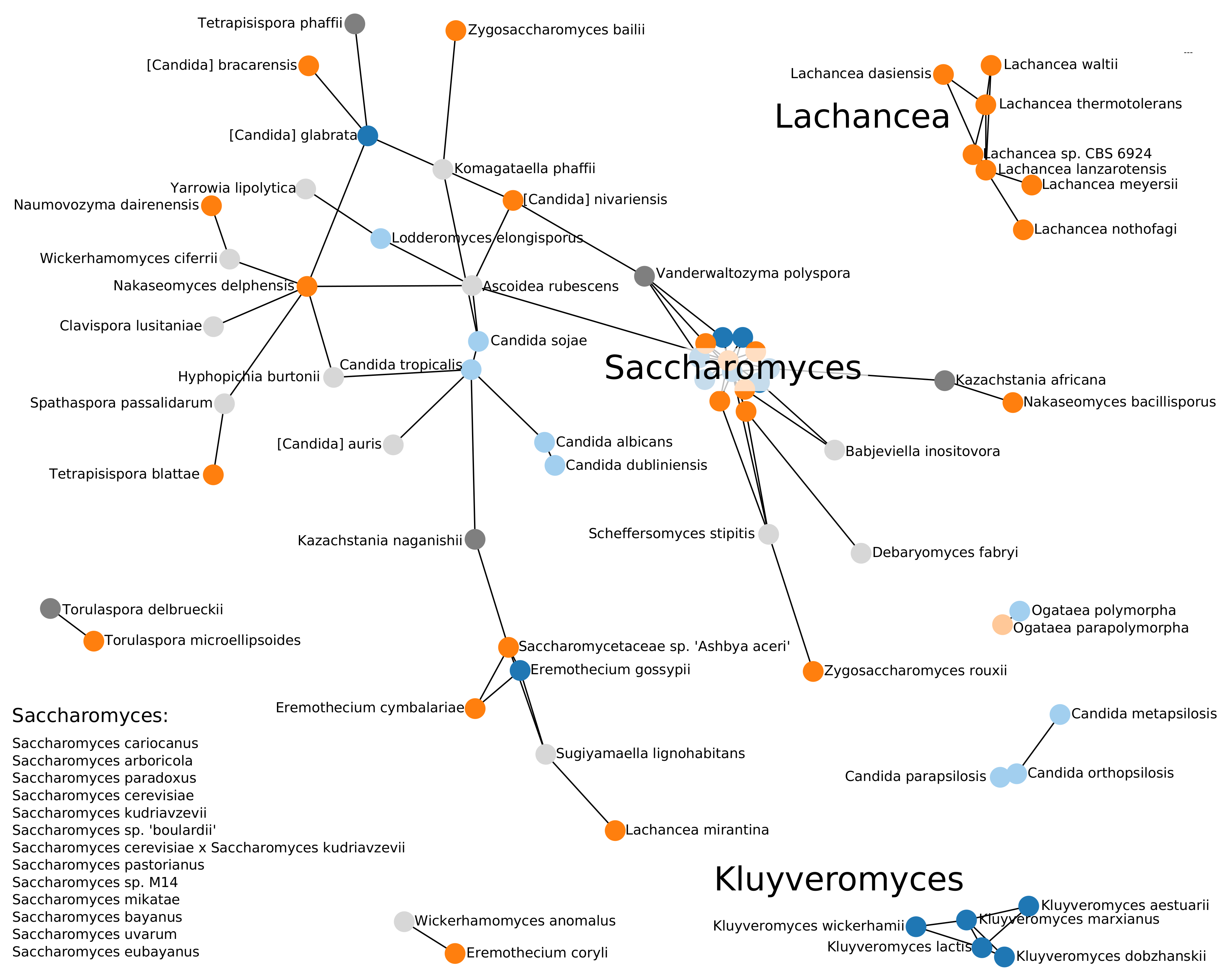
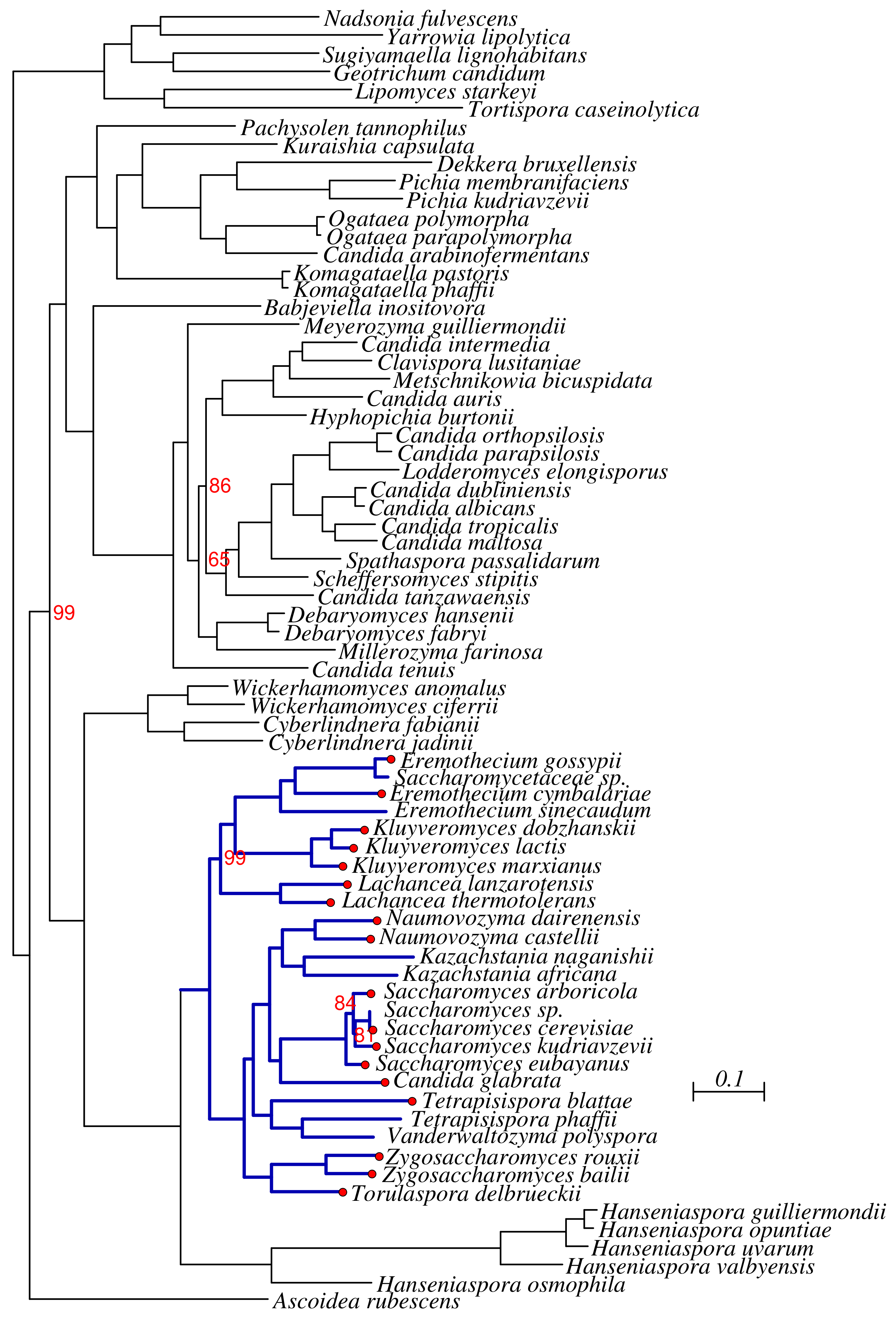
3.1. Phylogenomics of Saccharomycotina
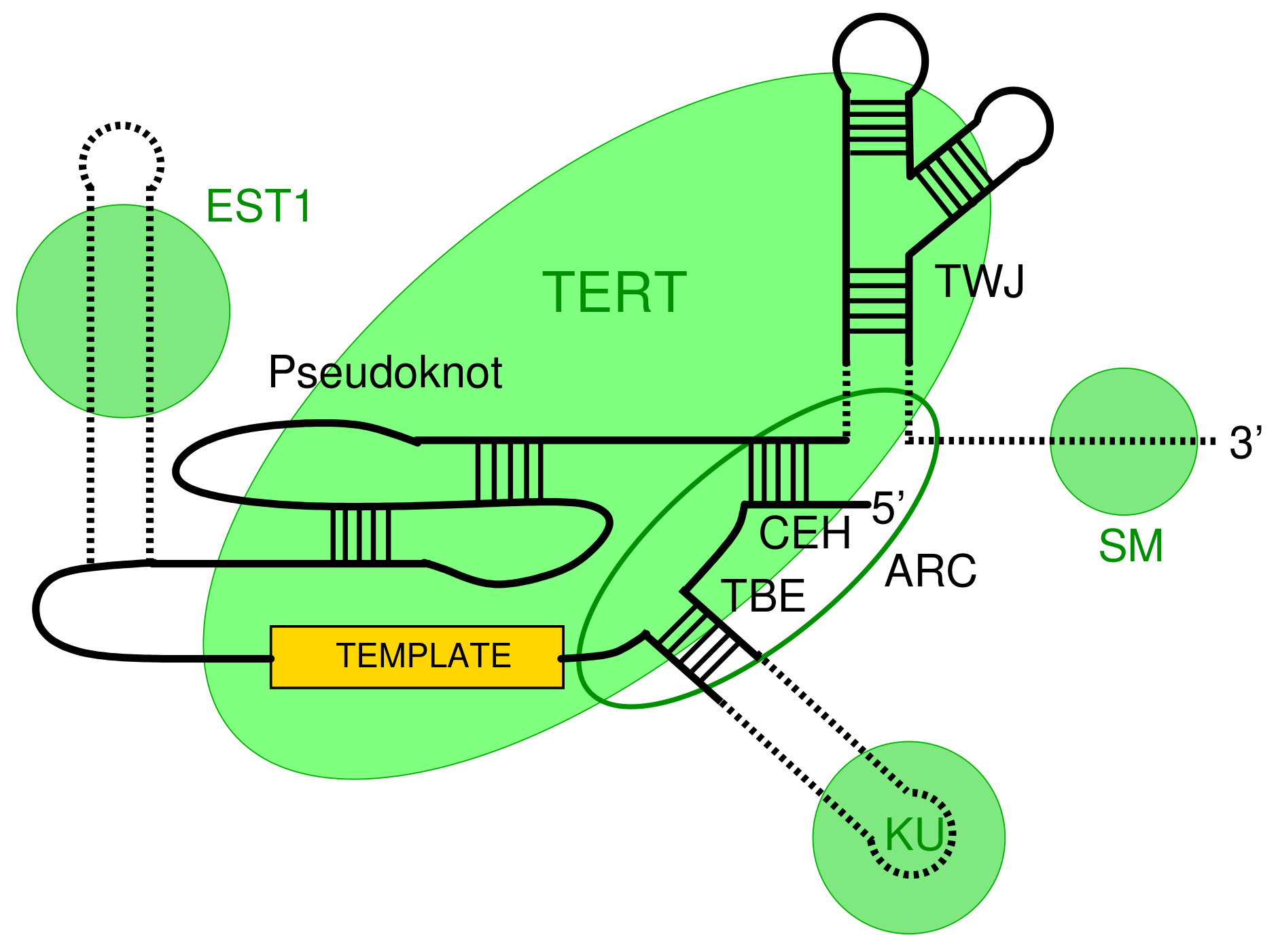
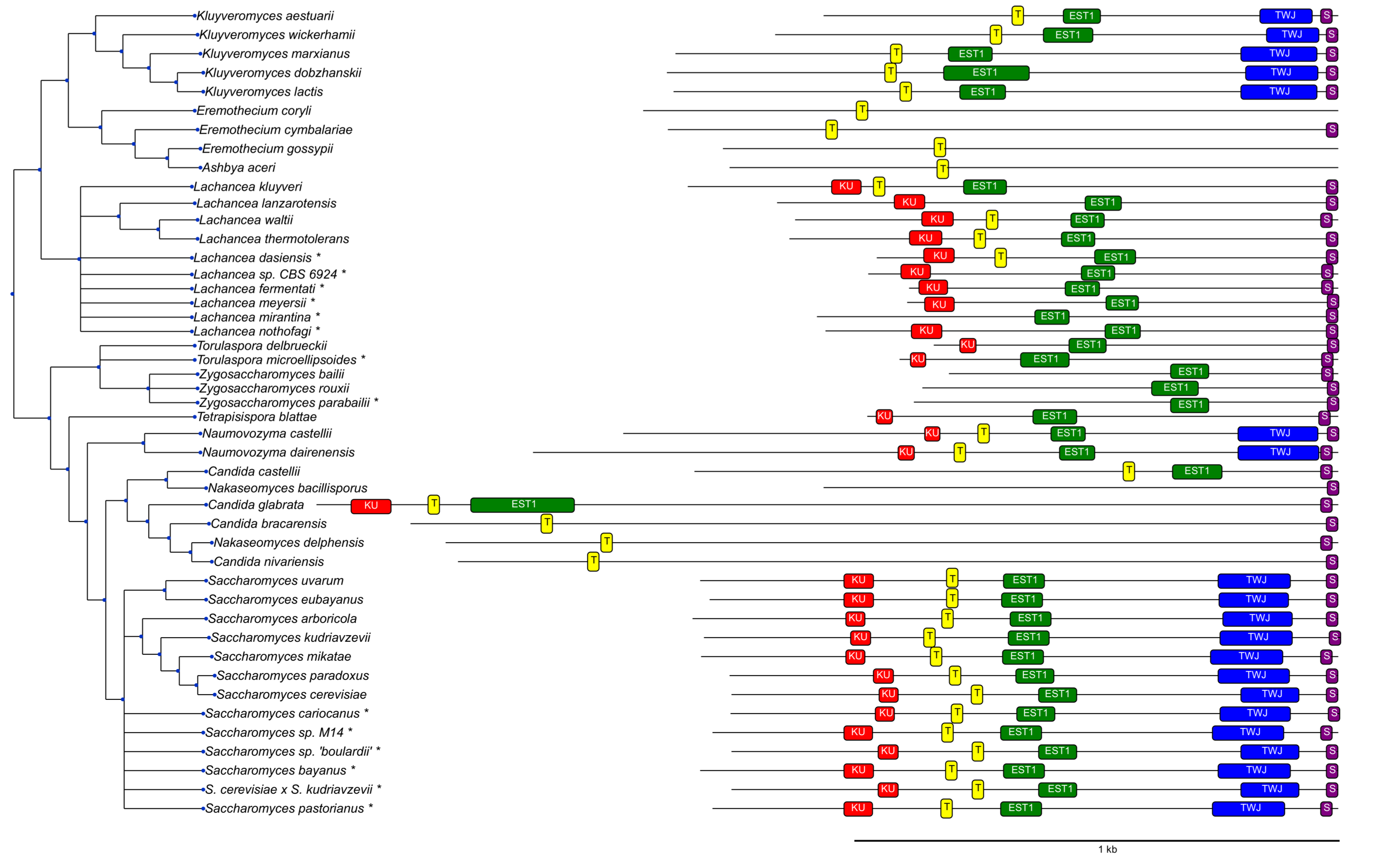
3.2. Survey of Telomerase RNA Genes in Saccharomycotina
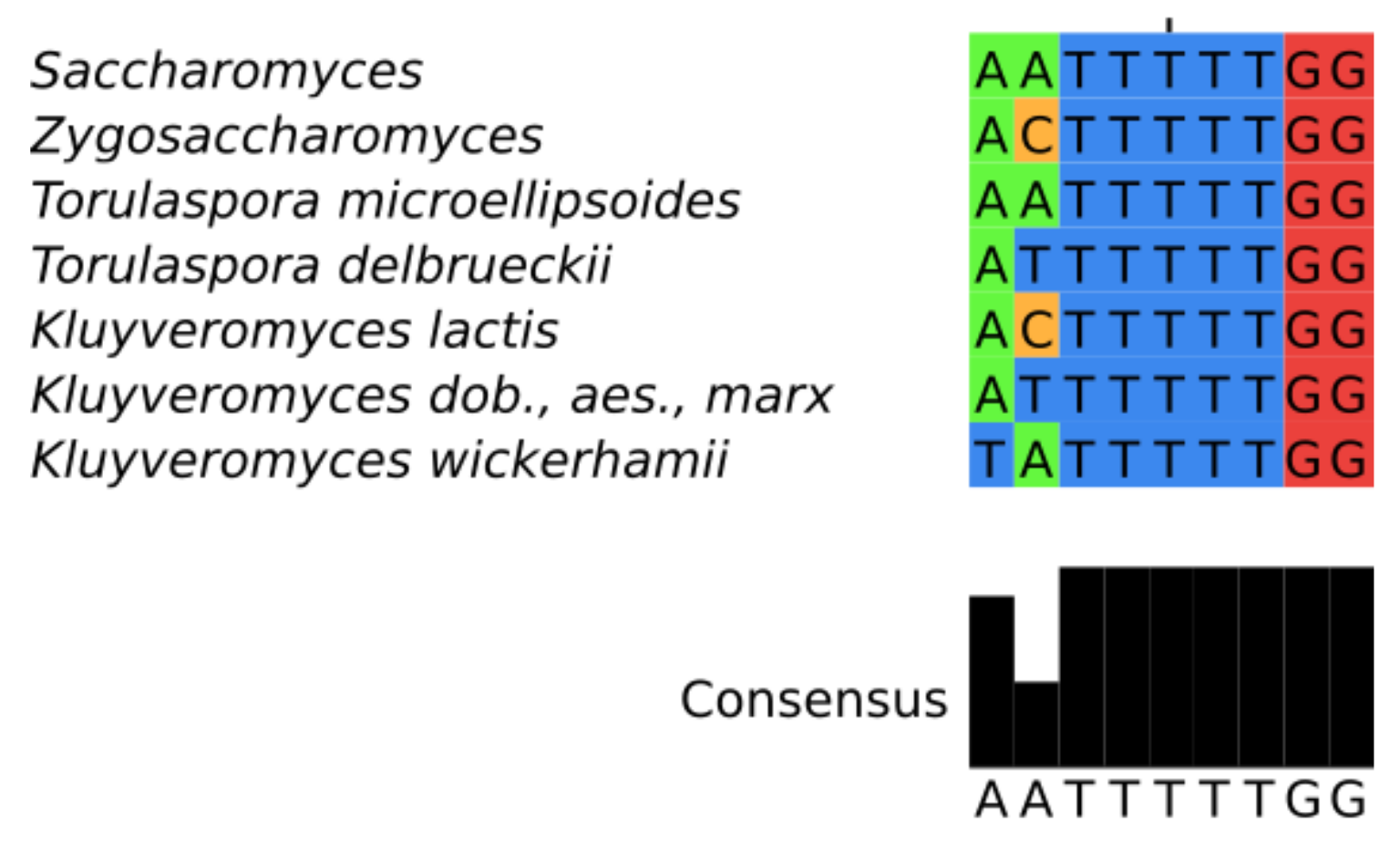
3.3. Features of Telomerase RNA in Saccharomycetacea
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Phylogenomics of the Saccharomycetales

References
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Mason, J.M.; Reddy, H.M.; Frydrychova, R.C. Telomere maintenance in organisms without telomerase. In DNA Replication—Current Advances; Seligmann, H., Ed.; InTech: Rijeka, Croatia, 2011; Chapter 15. [Google Scholar]
- Pardue, M.; Rashkova, S.; Casacuberta, E.; DeBaryshe, P.G.; George, J.A.; Traverse, K. Two retrotransposons maintain telomeres in Drosophila. Chromosome Res. 2005, 13, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlevsky, J.D.; Chen, J. Evolutionary perspectives of telomerase RNA structure and function. RNA Biol. 2016, 13, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, C.; Jansson, L.I.; Stone, M.D. New perspectives on telomerase RNA structure and function. Wiley Interdiscip. Rev. RNA 2018, 9, e1456. [Google Scholar] [CrossRef] [PubMed]
- Mefford, M.A.; Rafiq, Q.; Zappulla, D.C. RNA connectivity requirements between conserved elements in the core of the yeast telomerase RNP. EMBO J. 2013, 32, 2980–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Greider, C.W. An emerging consensus for telomerase RNA structure. Proc. Natl. Acad. Sci. USA 2004, 101, 14683–14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mefford, M.A.; Zappulla, D.C. Physical connectivity mapping by circular permutation of human telomerase RNA reveals new regions critical for activity and processivity. Mol. Cell. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brown, Y.; Abraham, M.; Pearl, S.; Kabaha, M.M.; Elboher, E.; Tsfati, Y. A critical three-way junction is conserved in budding yeast and vertebrate telomerase RNAs. Nucleic Acids Res. 2007, 35, 6280–6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livengood, A.J.; Zaug, A.J.; Cech, T.R. Essential regions of Saccharomyces cerevisiae telomerase RNA: Separate elements for Est1p and Est2p interaction. Mol. Cell. Biol. 2002, 22, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Zappulla, D.C.; Goodrich, K.; Cech, T.R. A miniature yeast telomerase RNA functions in vivo and reconstitutes activity in vitro. Nat. Struct. Mol. Biol. 2005, 12, 1072. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.A.; Upton, H.E.; Vogan, J.M.; Collins, K. Telomerase mechanism of telomere synthesis. Annu. Rev. Biochem. 2017, 86, 439–460. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.J.; Zakian, V.A. Telomerase RNA is more than a DNA template. RNA Biol. 2016, 13, 683–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.S.; Gottschling, D.E. TLC1: Template RNA component of Saccharomyces cerevisiae telomerase. Science 1994, 266, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Dandjinou, A.T.; Lévesque, N.; Larose, S.; Lucier, J.F.; Abou Elela, S.; Wellinger, R.J. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr. Biol. 2004, 14, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Bosoy, D.; Peng, Y.P.; Mian, I.S.; Lue, N.F. Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J. Biol. Chem. 2003, 278, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Kachouri-Lafond, R.; Dujon, B.; Gilson, E.; Westhof, E.; Fairhead, C.; Teixeira, M.T. Large telomerase RNA, telomere length heterogeneity and escape from senescence in Candida glabrata. FEBS Lett. 2009, 583, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Kannan, K.; Tseng, L.; Shippen, D.E. Two RNA subunits and POT1a are components of Arabidopsis telomerase. Proc. Natl. Acad. Sci. USA 2011, 108, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, M.A.; Brinegar, A.E.; Shippen, D.E. Evolution of the Arabidopsis telomerase RNA. Front. Genet. 2012, 3, 188. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Kolet, L.; Doniger, T.; Biswas, V.K.; Unger, R.; Tzfati, Y.; Michaeli, S. The Trypanosoma brucei telomerase RNA (TER) homologue binds core proteins of the C/D snoRNA family. FEBS Lett. 2013, 587, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, R.; Sanford, S.; Basu, S.; Park, M.; Pandya, U.M.; Li, B.; Chakrabarti, K. A trans-spliced telomerase RNA dictates telomere synthesis in Trypanosoma brucei. Cell Res. 2013, 23, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, K.; Pearson, M.; Grate, L.; Sterne-Weiler, T.; Deans, J.; Donohue, J.P.; Ares, M., Jr. Structural RNAs of known and unknown function identified in malaria parasites by comparative genomics and RNA analysis. RNA 2007, 13, 1923–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Religa, A.A.; Ramesar, J.; Janse, C.J.; Scherf, A.; Waters, A.P. P. berghei telomerase subunit TERT is essential for parasite survival. PLoS ONE 2014, 9, e108930. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Mosig, A.; Qi, X.; Li, Y.; Stadler, P.F.; Chen, J.J.L. Size variation and structural conservation of vertebrate telomerase RNA. J. Biol. Chem. 2008, 283, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Marz, M.; Qi, X.; Podlevsky, J.D.; Hoffmann, S.; Stadler, P.F.; Chen, J.J.L. Identification of Purple Sea urchin telomerase RNA using a next-generation sequencing based approach. RNA 2013, 19, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Podlevsky, J.D.; Li, Y.; Chen, J.J. Structure and function of echinoderm telomerase RNA. RNA 2016, 22, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, Y.; Honda, S.; Hoffmann, S.; Marz, M.; Mosig, A.; Podlevskya, J.D.; Stadler, P.F.; Selker, E.U.; Chen, J.J.L. The common ancestral core of vertebrate and fungal telomerase RNAs. Nucleic Acids Res. 2013, 41, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Kuprys, P.V.; Davis, S.M.; Hauer, T.M.; Meltser, M.; Tzfati, Y.; Kirk, K.E. Identification of telomerase RNAs from filamentous fungi reveals conservation with vertebrates and yeasts. PLoS ONE 2013, 8, e58661. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Rand, D.P.; Podlevsky, J.D.; Li, Y.; Mosig, A.; Stadler, P.F.; Chen, J.J. Prevalent and distinct spliceosomal 3′-end processing mechanisms for fungal telomerase RNA. Nat. Commun. 2015, 6, 6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, A.G.; Livengood, A.J.; Tzfati, Y.; Blackburn, E.H.; Cech, T.R. A bulged stem tethers Est1p to telomerase RNA in budding yeast. Genes Dev. 2002, 16, 2800–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEachern, M.J.; Blackburn, E.H. Runaway telomere elongation caused by telomerase RNA gene mutations. Nature 1995, 376, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; McEachern, M.J.; Dandjinou, A.T.; Tzfati, Y.; Orr, E.; Blackburn, E.H.; Lue, N.F. Telomerase core components protect Candida telomeres from aberrant overhang accumulation. Proc. Natl. Acad. Sci. USA 2007, 104, 11682–11687. [Google Scholar] [CrossRef] [PubMed]
- Gunisova, S.; Elboher, E.; Nosek, J.; Gorkovoy, V.; Brown, Y.; Jean-François, L.; Laterreur, N.; Wellinger, R.J.; Tzfati, Y.; Tomaska, L. Identification and comparative analysis of telomerase RNAs from Candida species reveal conservation of functional elements. RNA 2009, 15, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Smekalova, E.M.; Malyavko, A.N.; Zvereva, M.I.; Mardanov, A.V.; Ravin, N.V.; Skryabin, K.G.; Westhof, E.; Dontsova, O.A. Specific features of telomerase RNA from Hansenula polymorpha. RNA 2013, 19, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, A.E. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev. 2003, 17, 2384–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappulla, D.C.; Goodrich, K.J.; Arthur, J.R.; Gurski, L.A.; Denham, E.M.; Stellwagen, A.E.; Cech, T.R. Ku can contribute to telomere lengthening in yeast at multiple positions in the telomerase RNP. RNA 2011, 17, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Mosig, A.; Chen, J.L.; Stadler, P.F. Homology Search with Fragmented Nucleic Acid Sequence Patterns. In Lecture Notes in Computer Science, Proceedings of the International Workshop on Algorithms in Bioinformatics (WABI 2007), Philadelphia, PA, USA, 8–9 September 2007; Giancarlo, R., Hannenhalli, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4645, pp. 335–345. [Google Scholar]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappulla, D.C.; Cech, T.R. Yeast telomerase RNA: A flexible scaffold for protein subunits. Proc. Natl. Acad. Sci. USA 2004, 101, 10024–10029. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L. A new dawn for the naming of fungi: Impacts of decisions made in Melbourne in July 2011 on the future publication and regulation of fungal names. IMA Fungus 2011, 2, 155–162. [Google Scholar] [CrossRef] [PubMed]
- McNeill, J.; Barrie, F.R.; Buck, W.R.; Demoulin, V.; Greuter, W.; Hawksworth, D.L.; Herendeen, P.S.; Knapp, S.; Marhold, K.; Prado, J.; et al. (Eds.) International Code of Nomenclature for algae, fungi and plants. In Regnum Vegetabile; Koeltz Scientific Books: Oberreifenberg, Germany, 2012; Volume 154. [Google Scholar]
- Shen, X.X.; Zhou, X.; Kominek, J.; Kurtzman, C.P.; Hittinger, C.T.; Rokas, A. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 (Bethesda) 2016, 6, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Hernandez-Rosales, M.; Doerr, D.; Wieseke, N.; Thévenin, A.; Stoye, J.; Hartmann, R.K.; Prohaska, S.J.; Stadler, P.F. Orthology detection combining clustering and synteny for very large datasets. PLoS ONE 2014, 9, e105015. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.C.J.; Gonnet, G.H.; Dessimoz, C. Algorithm of OMA for large-scale orthology inference. BMC Bioinform. 2008, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Gil, M.; Gonnet, G.H.; Dessimoz, C. Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS ONE 2013, 8, e53786. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Podlevsky, J.D.; Bley, C.J.; Omana, R.V.; Qi, X.; Chen, J.J.L. The telomerase database. Nucleic Acids Res. 2007, 36, D339–D343. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.C.; De Grave, K. Fast neighborhood subgraph pairwise distance Kernel. In Proceedings of the 27th International Conference on International Conference on Machine Learning (ICML’10), Haifa, Israel, 21–24 June 2010; Fürnkranz, J., Joachims, T., Eds.; Omnipress: Madison, WI, USA, 2010; pp. 255–262. [Google Scholar]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comp. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xue, J.; Churikov, D.; Hass, E.P.; Shi, S.; Lemon, L.D.; Luciano, P.; Bertuch, A.A.; Zappulla, D.C.; Géli, V.; et al. Structural insights into yeast telomerase recruitment to telomeres. Cell 2018, 172, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, F.; Hofacker, I.L.; Höner zu Siederdissen, C. CMCompare webserver: Comparing RNA families via covariance models. Nucleic Acids Res. 2012, 41, W499–W503. [Google Scholar] [CrossRef] [PubMed]
- Wolfinger, M.T.; Fallmann, J.; Eggenhofer, F.; Amman, F. ViennaNGS: A toolbox for building efficient next-generation sequencing analysis pipelines. F1000Research 2015, 4, 50. [Google Scholar] [PubMed]
- Teixeira, M.T.; Gilson, E. Telomere maintenance, function and evolution: The yeast paradigm. Chromosome Res. 2005, 13, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Wendland, J.; Walther, A. Genome evolution in the Eremothecium clade of the Saccharomyces complex revealed by comparative genomics. Genes Genomes Genet. 2011, 1, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Kabaha, M.M.; Zhitomirsky, B.; Schwartz, I.; Tzfati, Y. The 5′ arm of Kluyveromyces lactis telomerase RNA is critical for telomerase function. Mol. Cell. Biol. 2008, 28, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Telomerase Database-Secondary Structures. Available online: http://telomerase.asu.edu/structures.html#secondary (accessed on 30 April 2018).
- Dietrich, F.S. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 2004, 304, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Genome Resources for Yeast Chromosomes Database-TLC1 (Telomerase RNA Template). Available online: http://gryc.inra.fr/index.php?page=locus&seqid=SAKL0D04356r (accessed on 30 April 2018).
- Ortiz-Merino, R.A.; Kuanyshev, N.; Braun-Galleani, S.; Byrne, K.P.; Porro, D.; Branduardi, P.; Wolfe, K.H. Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol. 2017, 15, e2002128. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.E.; Stellwagen, A.E.; Diede, S.J.; Singer, M.S.; Haimberger, Z.W.; Johnson, C.O.; Tzoneva, M.; Gottschling, D.E. The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat. Genet. 2001, 27, 64. [Google Scholar] [CrossRef] [PubMed]
- Box, J.A.; Bunch, J.T.; Tang, W.; Baumann, P. Spliceosomal cleavage generates the 3′ end of telomerase RNA. Nature 2008, 456, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Helston, R.M.; Dannebaum, R.O.; Baumann, P. Diverse mechanisms for spliceosome-mediated 3′ end processing of telomerase RNA. Nat. Commun. 2015, 6, 6104. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, B.; Laterreur, N.; Perederina, A.; Noël, J.F.; Dubois, M.L.; Krasilnikov, A.S.; Wellinger, R.J. Active yeast telomerase shares subunits with ribonucleoproteins RNase P and RNase MRP. Cell 2016, 165, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Wang, Z.; Koo, B.K.; Patel, A.; Cascio, D.; Collins, K.; Feigon, J. Structural basis for telomerase RNA recognition and RNP assembly by the holoenzyme La family protein p65. Mol. Cell 2012, 47, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Stadler, P.F. Evolutionary clues in lncRNAs. Wiley Interdiscip. Rev. RNA 2017, 8, e1376. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession | Strand | TER Coordinates | Ku Binding Site | Template Region | Est1 Binding Site | TWJ | SM1 |
|---|---|---|---|---|---|---|---|---|
| K. aestuarii | AEAS01000245.1 | neg | 16,338–17,322 [32] | 16,940–16,966 | 16,794–16,862 [32] | 16,378–16,485 [9] | 16,350–16,359 | |
| K. wickerhamii | AEAV01000432.1 | pos | 250–1327 [32] | 662–693 | 765–858 [32] | 1183–1290 [9] | 1307–1316 | |
| K. marxianus | NC_036029.1 | pos | 506,443–507,711 [32] | 506,855–506,888 | 506,967–507,049 [32] | 507,518–50,7671 [9] | 507,691–507,700 | |
| K. dobzhanskii | CCBQ010000012.1 | pos | 461,805–463,090 [32] | 462,224–462,257 | 462,337–462,499 [32] | 462,905–463,051 [9] | 463,070–463,079 | |
| K. lactis | NC_006038.1 | pos | 611,456–612,727 [33] | absent [63] | 611,890–611,919 [64] | 612,006–612,090 [32] | 612,532–612,687 [9] | 612,708–612,716 [64] |
| E. coryli | AZAH01000001.1 | neg | 269,038–270,368 | 269,938–269,968 | ||||
| E. cymbalariae | NC_016454.1 | pos | 54,147–54,960 | 54,451–54,480 | ||||
| E. gossypii | NC_005782.2 | neg | 677,871–679,048 [65] | 678,276–678,305 [35] | ||||
| Ashbya aceri | CP006020.1 | neg | 693,543–694,708 | 693,942–693,973 | ||||
| L. kluyveri | CM000690.1 | pos | 348,600–349,844 | 348,876–348,930 | 348,957–348,982 [66] | 349,129–349,208 | 349,825–349,833 | |
| L. lanzarotensis | NW_019212880.1 | pos | 854,162–855,236 | 854,389–854,444 | 854,754–854,820 | 855,217–855,225 | ||
| L. waltii | AADM01000270.1 | neg | 134,961–136,000 | 135,698–135,756 [19] | 135,613–135,636 | 135,409–135,470 | 134,973–134,981 | |
| L. thermotolerans | NC_013079.1 | pos | 702,500–703,549 | 702,730–702,791 [19] | 702,853–702,876 | 703,022–703,083 | 703,530–703,538 | |
| L. dasiensis | LT598456.1 | pos | 682,034–682,916 | 682,124–682,181 | 682,261–682,283 | 682,900–682,905 | ||
| L. sp. CBS 6924 | LT598470.1 | neg | 441,802–442,700 | 442,582–442,638 | 442,229–442,292 | 441,811–441,820 | ||
| L. fermentati | LT598488.1 | neg | 306,329–307,150 | 307,076–307,129 | 306,786–306,850 | 306,339–306,348 | ||
| L. meyersii | LT598477.1 | pos | 575,851–576,676 | 575,886–575,941 | 576,233–576,294 | 576,657–576,666 | ||
| L. mirantina | LT598468.1 | pos | 690,800–691,797 | 691,218–691,282 | 691,777–691,786 | |||
| L. nothofagi | LT598449.1 | pos | 388,401–389,382 | 388,567–388,624 | 388,937–389,004 | 389,362–389,371 | ||
| T. delbrueckii | NC_016504.1 | pos | 709,007–709,780 | 709,057–709,086 | 709,267–709,336 | 709,761–709,770 | ||
| T. microellipsoides | FYBL01000005.1 | neg | 426,211–427,050 | 427,000–427,028 | 426,726–426,817 | 426,221–426,229 | ||
| Z. bailii | HG316456.1 | neg | 712,655–713,400 | 712,902–712,974 | 712,665–712,673 | |||
| Z. rouxii | NC_012990.1 | pos | 297,087–297,883 | 297,527–297,616 | 297,865–297,873 | |||
| Z. parabailii | CP019499.1 | pos | 455,564–455,975 [67] | 455,656–455,728 | 455,957–455,965 | |||
| T. blattae | NC_020193.1 | neg | 404,150–405,050 | 405,003–405,033 | 404,650–404,733 | 404,165–404,173 | ||
| N. castellii | NC_016499.1 | pos | 381,827–383,194 [64] | 382,404–382,432 [19] | 382,506–382,519 [64] | 382,647–382,710 [64] | 382,994–383,155 [64] | 383,176–383,184 [64] |
| N. dairenensis | NC_016479.1 | neg | 1,519,837–1,521,377 | 1,520,648–1,520,678 | 1,520,550–1,520,562 | 1,520,303–1,520,369 | 1,519,864–1,520,027 | 1,519,849–1,519,857 |
| C. castellii | CAPW01000002.1 | neg | 272,769–274,000 | 273,158–273,179 | 272,992–273,085 | 272,781–272,789 | ||
| N. bacillisporus | CAPX01000073.1 | pos | 1230–2215 | 2197–2204 | ||||
| C. glabrata | NC_006032.2 | neg | 419,194–421,150 [19] | 421,007–421,081 [19] | 420,914–420,932 [19] | 420,657–420,852 [19] | 419,206–419,214 [19] | |
| C. bracarensis | CAPU01000044.1 | pos | 2586–4361 | 2836–2854 | 4342–4350 | |||
| N. delphensis | CAPT01000167.1 | neg | 254,761–256,469 | 256,151–256,169 | 254,773–254,781 | |||
| C. nivariensis | CAPV01000033.1 | pos | 87,530–89,215 | 87,780–87,798 | 89,196–89,204 | |||
| S. uvarum | NOWY01000011.1 | pos | 45,720–46,940 | 45,996–46,050 | 46,193–46,203 | 46,301–46,377 | 46,703–46,848 | 46,921–46,929 |
| S. eubayanus | NC_030979.1 | pos | 476,134–47,7336 | 476,392–476,446 | 476,588–476,598 | 476,694–476,770 | 477,100–477,240 | 477,317–477,325 |
| S. arboricola | NC_026172.1 | pos | 287,410–288,645 | 287,705–287,739 | 287,888–287,898 | 288,019–288,096 | 288,417–288,558 | 288,626–288,634 |
| S. kudriavzevii | AY639012.1 | pos | 1–1215 [17] | 284–320 [17] | 424–434 | 585–662 | 981–1128 [9] | 1201–1209 |
| S. mikatae | AABZ01000048.1 | neg | 18,591–19,809 [17] | 19,497–19,532 [17] | 19,349–19,356 | 19,156–19,232 | 18,687–18,833 [9] | 18,603–18,611 |
| S. paradoxus | CP020294.1 | pos | 307,733–308,897 [17] | 308,010–308,045 [17] | 308,154–308,161 | 308,281–308,353 | 308,660–308,803 [9] | 308,878–308,886 |
| S. cerevisiae | NC_001134.8 | pos | 307,597–308,757 [17,18] | 307,880–307,914 [68] | 308,057–308,064 [64] | 308,185–308,256 [32] | 308,563–308,682 [9] | 308,737–308,746 [64] |
| S. pastorianus | AZCJ01000004.1 | neg | 478,773–479,970 [17] | 479,664–479,718 [17] | 479,512–479,520 | 479,340–479,417 | 478,866–479,012 [9] | 478,785–478,793 |
| S. cer. x S. kud. | AGVY01000004.1 | pos | 284,183–285,344 | 284,465–284,501 | 284,645–284,655 | 284,772–284,843 | 285,150–285,269 | 285,325–285,333 |
| S. bayanus | AACG02000058.1 | pos | 58,142–59,362 [17] | 58,418–58,472 [17] | 58,613–58,620 | 58,723–58,799 | 59,125–59,270 [9] | 59,343–59,351 |
| S. sp. ‘boulardii’ | CM003558.1 | pos | 287,536–288,696 | 287,818–287,854 | 287,998–288,008 | 288,124–288,195 | 288,502–288,621 | 288,677–288,685 |
| S. sp. M14 | MVPU01000005.1 | neg | 473,800–474,997 | 474,691–474,745 | 474,537–474,547 | 474,368–474,444 | 473,894–474,038 | 473,812–473,820 |
| S. cariocanus | AY639010.1 | pos | 1–1163 [17] | 278–313 [17] | 424–434 | 549–621 | 928–1072 [9] | 1147–1155 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldl, M.; Thiel, B.C.; Ochsenreiter, R.; Holzenleiter, A.; De Araujo Oliveira, J.V.; Walter, M.E.M.T.; Wolfinger, M.T.; Stadler, P.F. TERribly Difficult: Searching for Telomerase RNAs in Saccharomycetes. Genes 2018, 9, 372. https://doi.org/10.3390/genes9080372
Waldl M, Thiel BC, Ochsenreiter R, Holzenleiter A, De Araujo Oliveira JV, Walter MEMT, Wolfinger MT, Stadler PF. TERribly Difficult: Searching for Telomerase RNAs in Saccharomycetes. Genes. 2018; 9(8):372. https://doi.org/10.3390/genes9080372
Chicago/Turabian StyleWaldl, Maria, Bernhard C. Thiel, Roman Ochsenreiter, Alexander Holzenleiter, João Victor De Araujo Oliveira, Maria Emília M. T. Walter, Michael T. Wolfinger, and Peter F. Stadler. 2018. "TERribly Difficult: Searching for Telomerase RNAs in Saccharomycetes" Genes 9, no. 8: 372. https://doi.org/10.3390/genes9080372