1. Introduction
Among over the 370 genera belonging to the family Brassicaceae (Cruciferae), only the genus
Boechera shows asexual reproduction by seeds [
1,
2,
3,
4]. Apomixis is defined as asexual reproduction through seeds that results in progeny identical to the maternal plant. The harnessing of apomixis is widely considered as a key enabling technology for crop improvement because it allows the fixation of any heterozygous genotype, leading to simpler and faster breeding schemes [
5,
6,
7]. The
Boechera genus includes 110 sexual and apomictic species, widely distributed in North America. Plants from the
Boechera genus are represented by biannual and perennial herbs with a chromosome base number of
n = 7 [
8,
9].
Apomixis in the
Boechera genus is of special interest because it can occur at the diploid level, which is very rare [
1,
2,
3,
4,
5,
6,
7,
8]. Furthermore, the phylogenetic proximity of
Boechera to the model plant
Arabidopsis thaliana is attractive for potential functional studies. Although the genus
Boechera includes both sexual and apomictic species and accessions that are of variable ploidy and geographical origin, search for homologous sequences are feasible across the genus [
10]. The sexual accessions of
Boechera are self-compatible and largely self-pollinating [
11], unlike the sexual ancestors of most other apomicts, which are typically self-incompatible and cross-pollinating [
12]. This inbreeding causes low heterozygosity in sexual
Boechera species. Apomictic
Boechera accessions have likely arisen through independent hybridization events [
13]. Their hybridogenic origin is supported by the aberrant structure of their chromosomes, as they are often observed as a consequence of hybridization, leading to alloploidy, aneuploidy, the replacement of homeologous chromosomes, and aberrant chromosomes [
13,
14].
Certain apomictic
Boechera accessions are hypothesized to have arisen through hybridization between sexual
Boechera stricta and
Boechera retrofracta (
Figure 1).
Boechera retrofracta was previously included within
Boechera holboellii (sensu lato) [
15]. Up to now only the genome sequence of
B. stricta was available [
16], while the genome of
B. retrofracta has not been assembled yet.
In this paper, we present the assembly and annotation of the B. retrofracta genome. The availability of the B. retrofracta genome sequence together with the previously assembled B. stricta genome will greatly help in the assembly and annotation of related apomictic hybrid species and provide the basis to investigate the peculiarities of hybridization events, chromosomal organization, the stability of apomictic genomes, and the genetic factors underlying apomixis. The performed assembly and annotation allowed us to analyze of the APOLLO (APOmixis-Linked LOcus) genes, that are associated with apomixis in Boechera.
3. Results
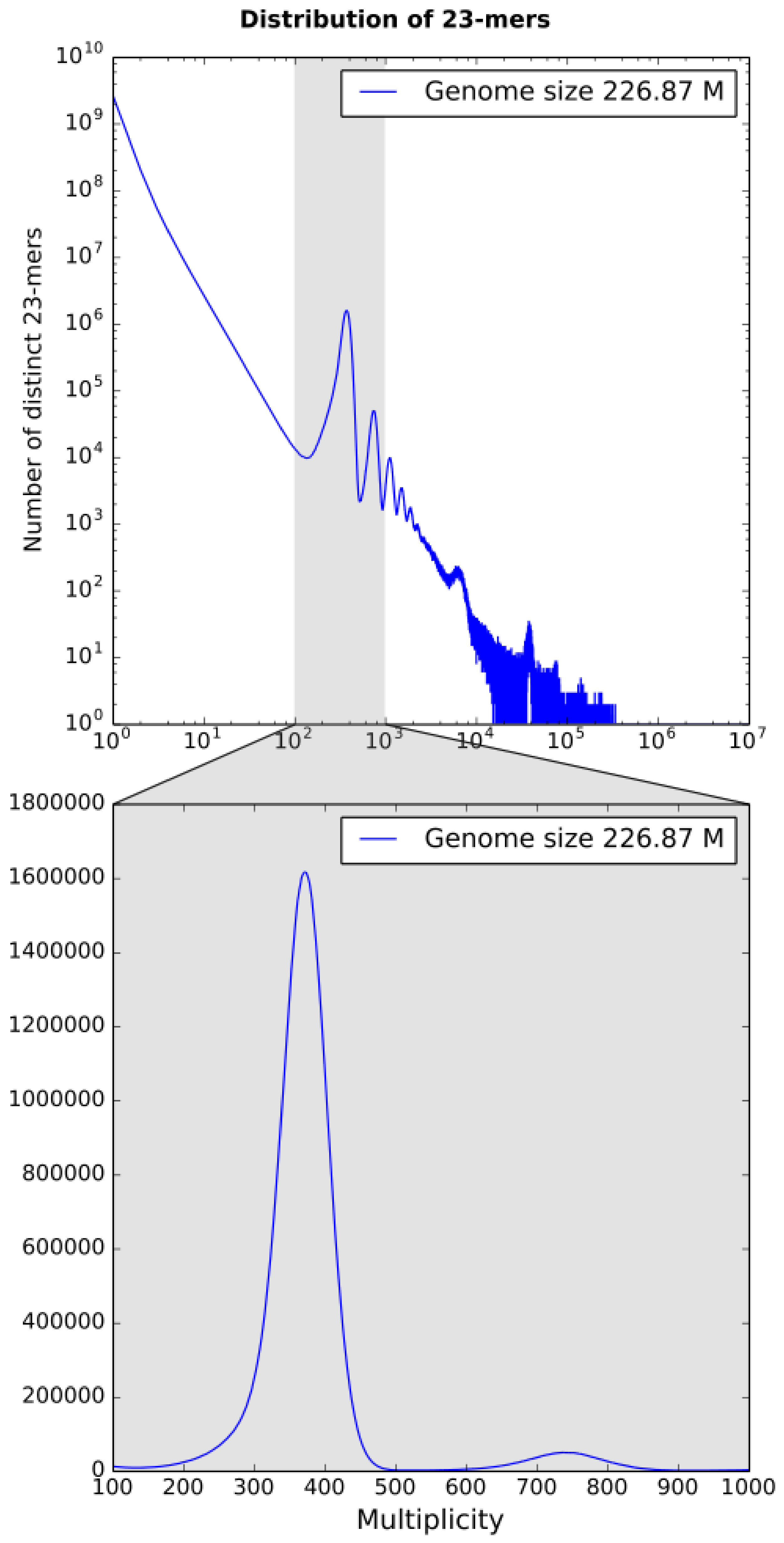
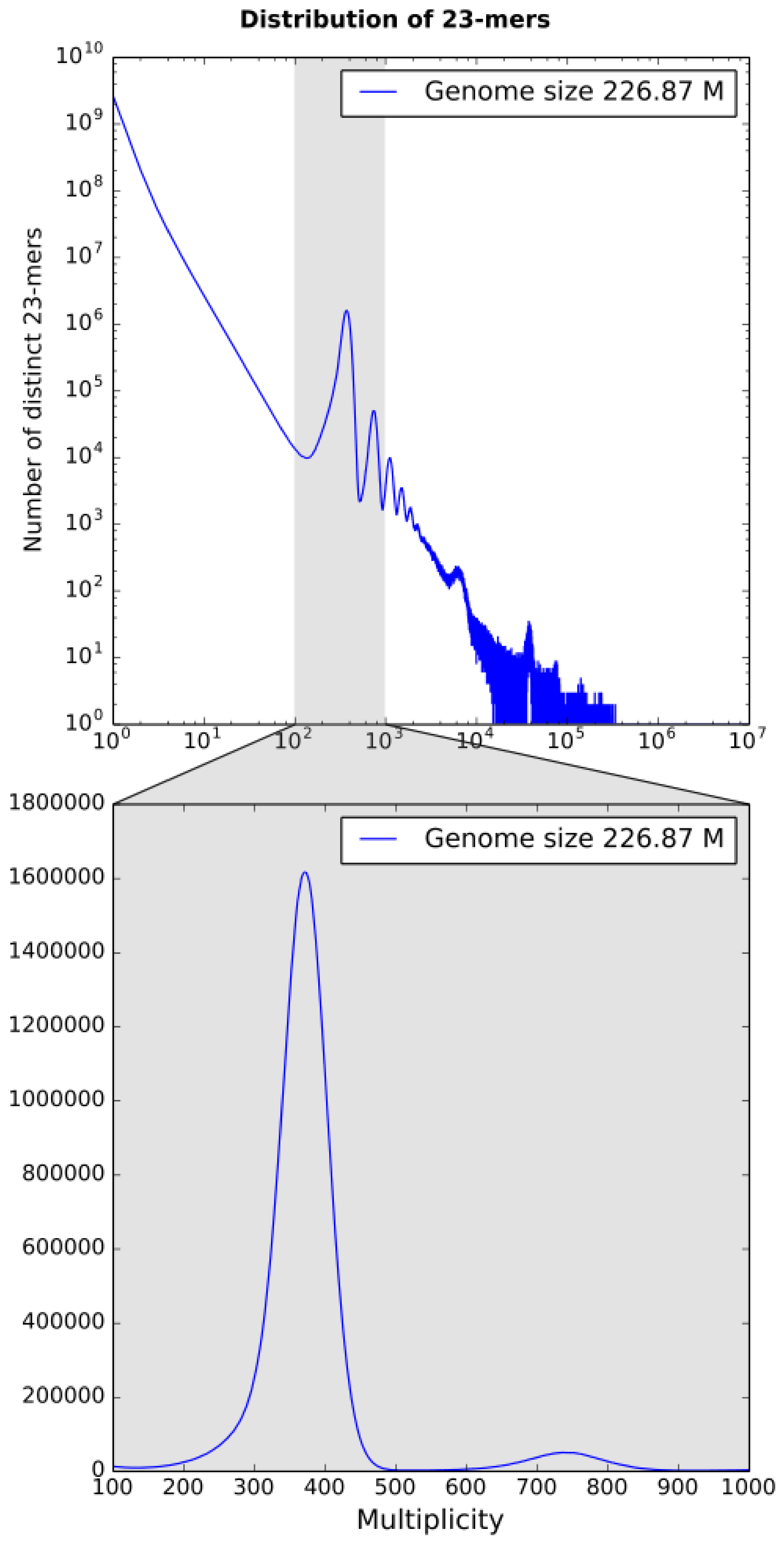
3.1. k-mer Based Statistics
k-mer spectrum built by KrATER [
23] is shown in
Figure 2. The 23-mer distribution has a peak of erroneous 23-mers at 1× coverage corresponding to sequencing errors and one major peak at 371× coverage corresponding to diploid 23-mers (shared between homologous chromosomes), but no significant peak related to heterozygous genome positions was detected (
Figure 2). However, we detected several small additional peaks at double (737×) and triple (1120×) depth, which are probably related to duplications and triplications, respectively.
The genome size of
B. retrofracta was estimated to be close to 227 Mbp, which is close to the previous estimations of a minimal genome size of 200 Mbp in the
Boechera genus [
24].
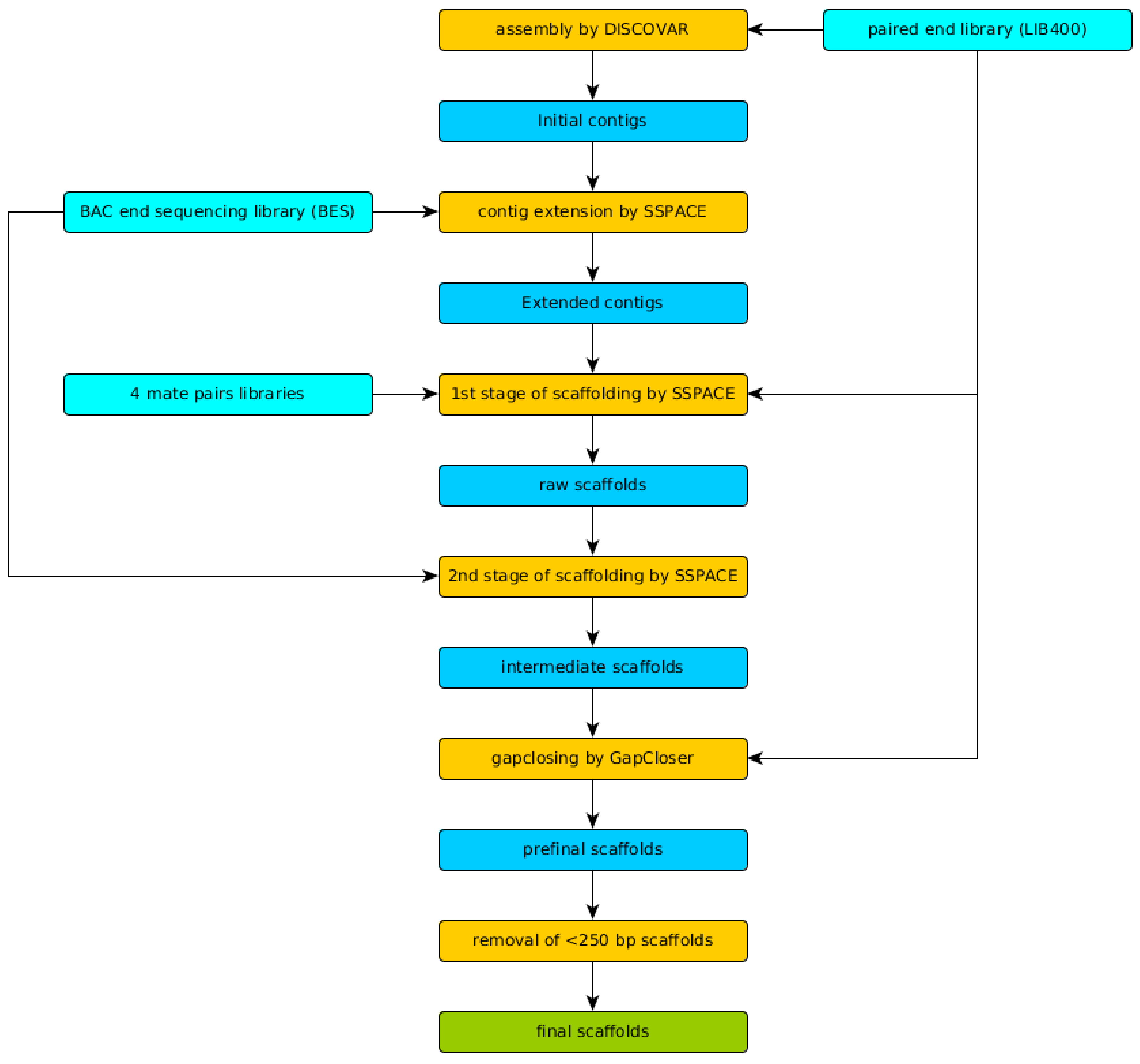
3.2. Genome Assembly and Evaluation
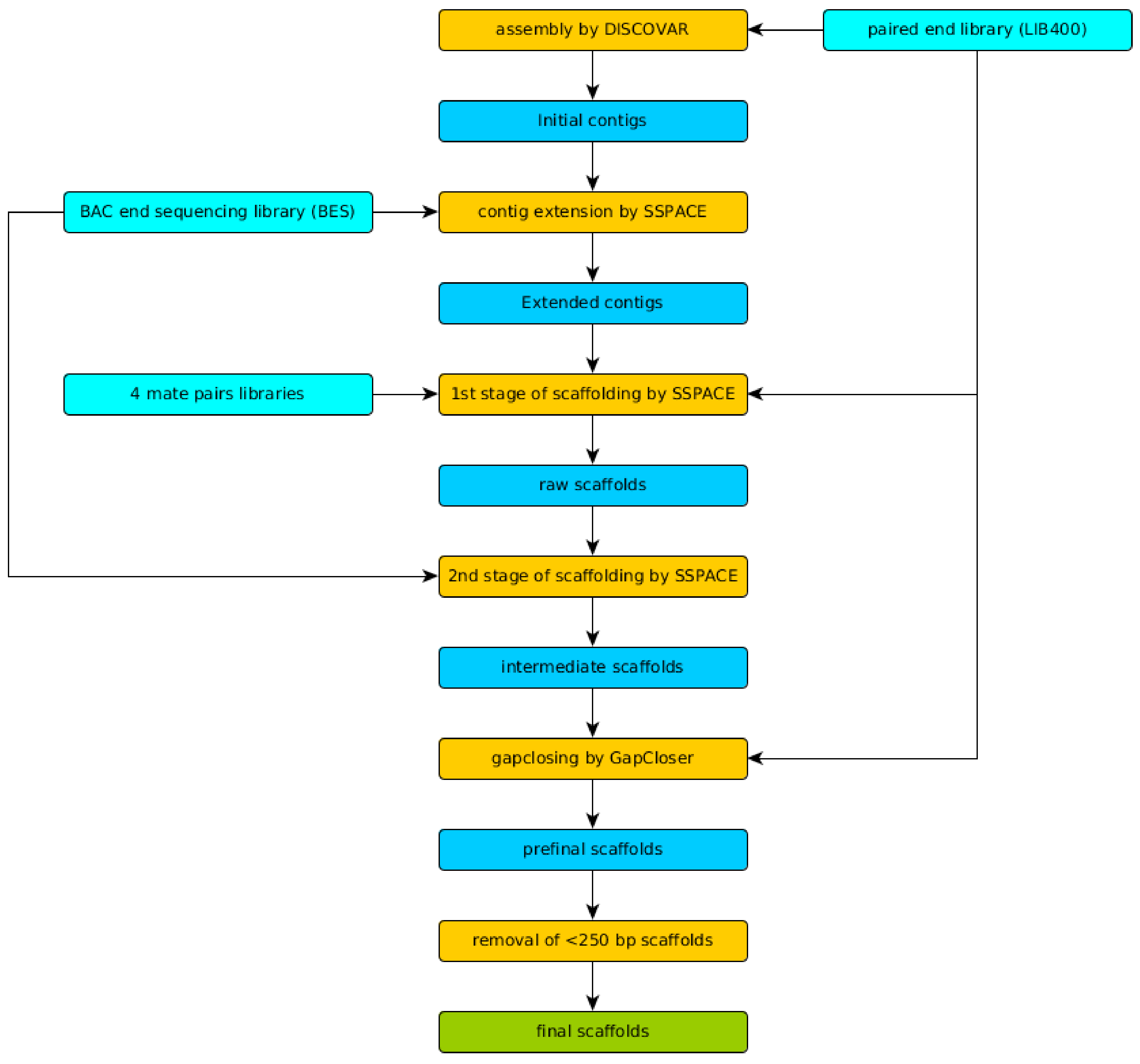
We have achieved N50 of 2,297,899 bp, L50 of 25, and a total assembly length of 222.25 Mbp for the final scaffolds, which is very close to our 23-mer based estimation. Detailed statistics including N50 and total assembly values for every stage of the assembly pipeline are listed in
Table 1 and
Table 2. It is important to note that the final assembly (
Table 1, column final scaffolds) has smaller size than previous intermediate assemblies due to the last filtration step. All scaffolds shorter than 250 bp (a read length of LIB400) were treated as artifacts of assembly and were removed. However, size of final assembly (222.25 Mbp) is closer to estimated genome size (226.87 Mbp) than the size of intermediate assemblies.
Evaluation of the assembly completeness was performed using CEGMA [
28] and BUSCO [
29]. In the assembled genome 242 (97.58%) complete core eukaryotic genes (CEGs) were identified. Out of 1440 BUSCO genes from the Embryophyta, set only 12 (0.8%) genes were not found, 6 were fragmented, 36 (2.5%) were duplicated and 1422 (98.8%) were complete. This high fraction of complete BUSCO genes suggests high completeness of the assembly and its integrity at least in gene-coding regions.
3.3. Repeats Annotation
In total approximately 85 Mbp (38.13%) of the assembly were masked. The detailed description of the annotated repeat types is listed in
Table 3. It is important to note that a large number (10.96% of the assembly size) of interspersed repeats was not classified. The results are shown in
Table 4.
3.4. Variant Calling and Genotyping
In the genome 3341 SNPs and 1317 indels were detected. Among these, 103 (3.08%) SNPs and 97 (7.37%) indels were homozygous and, therefore, most likely artifacts of alignment or assembly or SNP calling. Mean heterozygous SNP and indel densities in non-masked regions (138 Mbp in total) are 0.0235 SNP and 0.0089 indel per Kbp, respectively, suggesting a very low heterozygosity of the B. retrofracta genome.
3.5. Prediction of Protein-Coding Genes and Non-Coding RNAs
In total 27,048 genes with 28,269 transcripts were predicted. tRNA and rRNA genes predicted by tRNAscan-SE and Barrnap are given in
Table 5 and
Table 6 respectively.
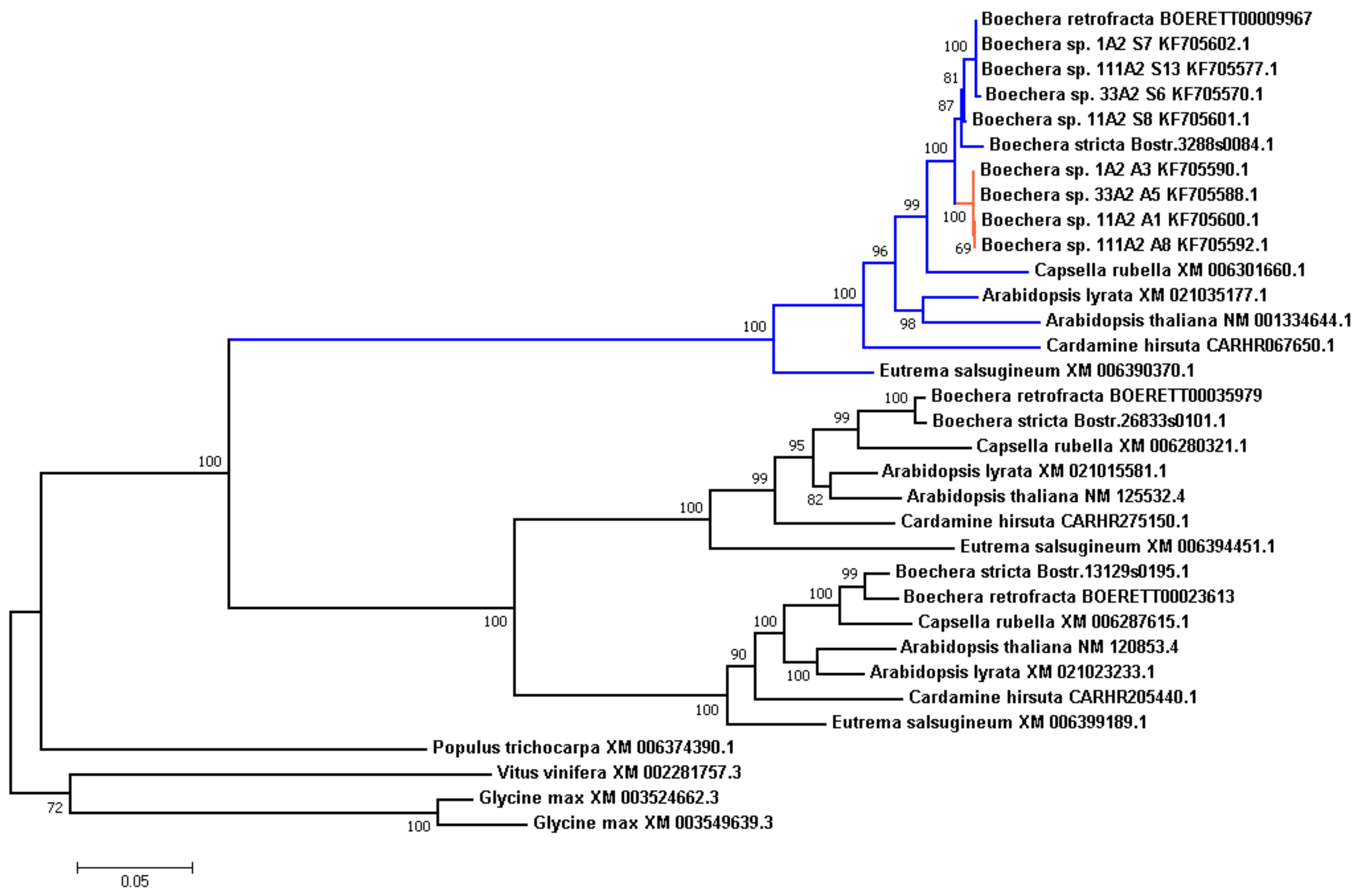
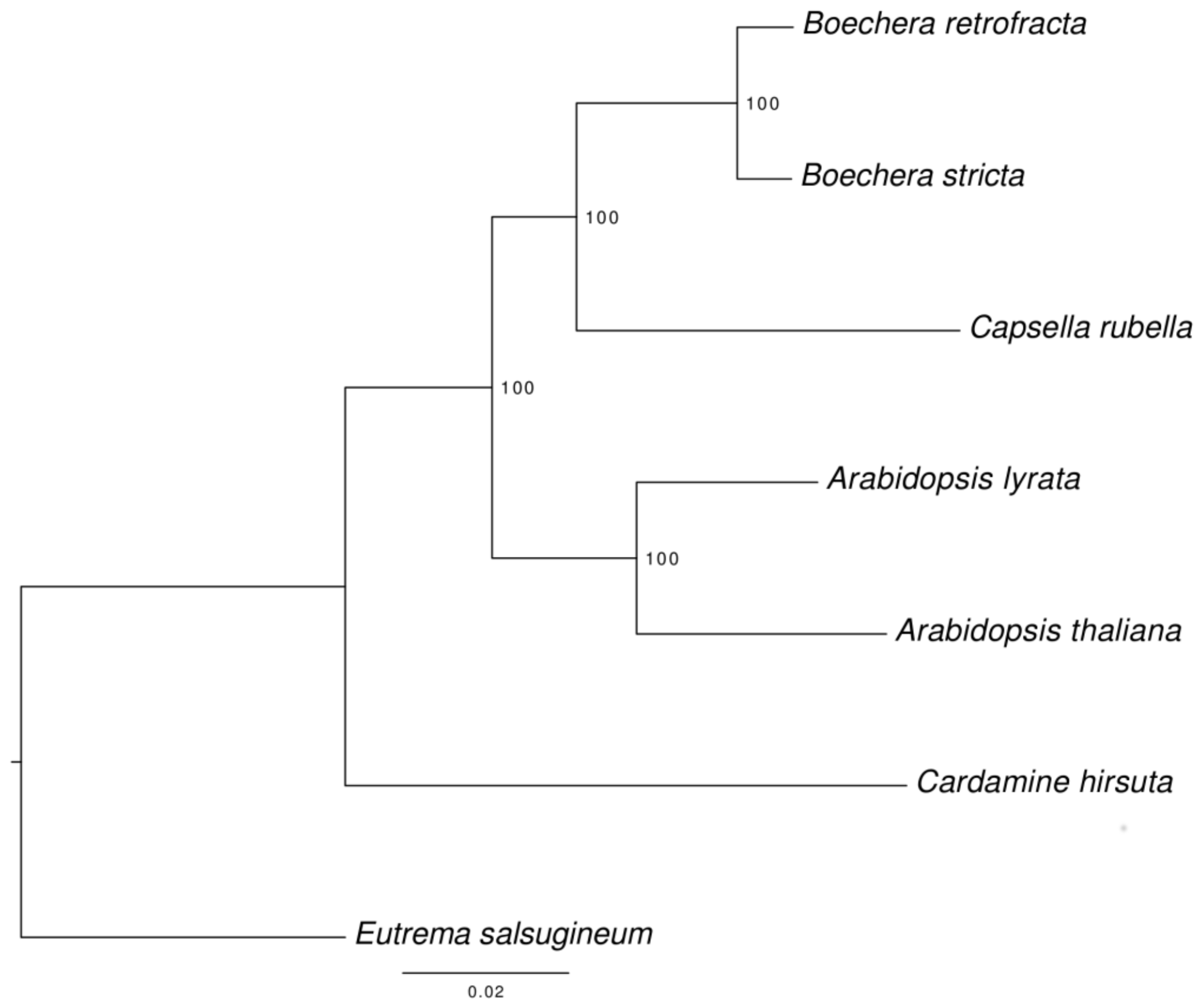
3.6. Species Tree Reconstruction
In course of the assignment of proteins to orthologous groups 8959 single-copy orthologs were identified among the seven species (B. retrofracta, B. stricta, A. thaliana, A. lyrata, C. rubella, C. hirsuta, and E. salsugineum).
The corresponding phylogenetic tree was rooted with
E. salsugineum as an outgroup (
Figure 3). All nodes have a high support and no topology discordance was found with the tree reconstructed previously by Huang et al [
52].
3.7. Analysis of Evolution of the APOLLO Locus
Results from Corral et al. [
56] suggest that
APOLLO (aspartate glutamate aspartate aspartate histidine exonuclease) is one of the important apomixis-related genes in
Boechera. It was shown that the
APOLLO locus has several alleles with apomixis-associated polymorphisms. All studied apomictic plants carry at least one of the “apoalleles”, while both copies in sexual genotypes were “sexalleles”.
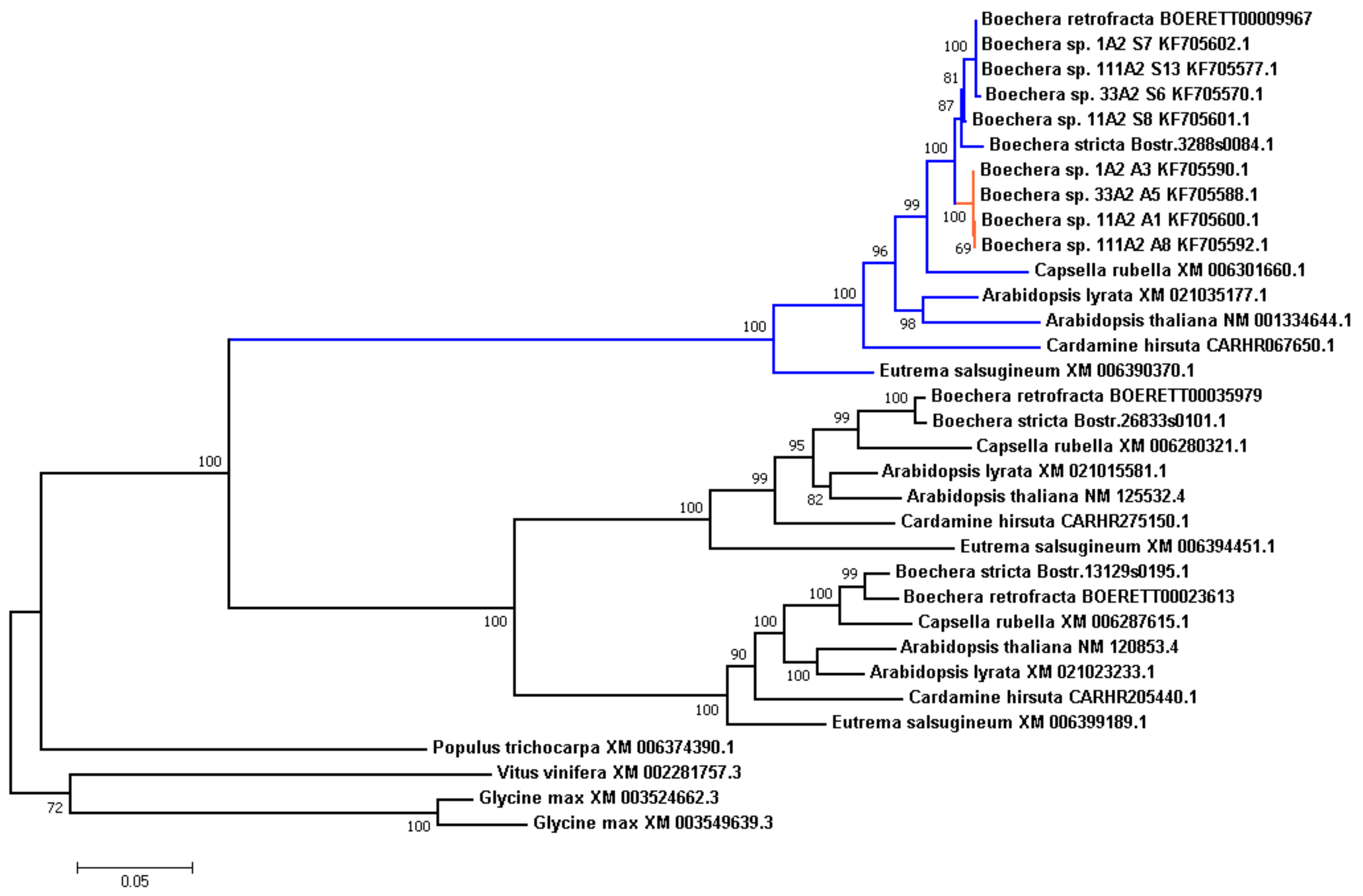
In this study we decided to take a closer look to this locus in our assembly and other Brassicaceae species in this study. Along with an exact copy of the
APOLLO locus, we also found two other, more distant copies, which may indicate past duplication events. We searched for these orthologs in other species, and reconstructed phylogenetic tree (
Figure 4). All Brassicaceae genomes in the study also carried these three copies, related to the clusters of orthologous genes ENOG410BURN (
APOLLO locus), ENOG410BUTR, and ENOG410C333 in the EggNOG database.
We observed that branches in the tree were grouped by genes rather than by species, suggesting that the triplication event took place before the separation of the Brassicaceae species in this study. It is worth noting that in Populus trichocarpa genome there is only one copy of these locus, which gives an upper-bound time estimate of the series of duplication events.
We also examined
APOLLO alleles (both apo- and sex-alleles) described in
Boechera ssp. by Corral et al. [
56]. We can see that these alleles arise after the separation of the
Boechera genus, and compose two separate clades. Given the fact that
B.retrofracta and
B.stricta are the sexual species, it was not surprising that in both cases all corresponding polymorphic sites were in the “sexallele”-state, and clustered with sex-alleles.
We calculated the Ka/Ks ratio for the internal branches in this tree and found that branch leading to apo-alleles is under positive selection (Ka/Ks 1.4646, the branch is shown in red in
Figure 4), which is typical for paralogues that are required to serve a novel function.
The
APOLLO gene was initially described in
A. thaliana as an exonuclease, protein NEN3, Q9CA74 in Uniprot database [
42], probably involved in enucleation of sieve elements, whereas two other copies were described as NEN1 (Q9FLR0) and NEN2 (Q0V842). Given that, we may suggest an evolutionary scenario where, after the series of duplications, one of the NEN protein copies in the common ancestor of
Boechera spp. might have acquired alter regulation, and might induce development of the apomictic reproduction from the ancestral “sexual” state, following by separation of the apomictic lineages.
That could explain the phenomena of the diploid apomictic Boechera, emerged as a result of duplication events rather than polyploidy.
3.8. Whole-Genome Comparison
As an example of whole-genome comparison a Circos plot was built for
B. retrofracta and
B. stricta (
Appendix C). Since both assemblies are performed on a scaffold level, it is difficult to highlight any large genome rearrangements. However, this plot is a visual way to represent the scatteredness of both assemblies.
4. Discussion
In this study we present a de novo assembly and annotation of the genome of
Boechera retrofracta, a perennial flowering plant belonging to Brassicaceae family that is native to North America. The genome of
B. retrofracta demonstrated a very low level of heterozygosity compare to the genomes of apomictic accessions [
2,
8,
9,
10,
11,
12,
13,
14,
15,
16]. Notably, repeats in the genome of
B. retrofracta occupied almost 40% of the genome space. Nearly half of them were long terminal repeats (LTRs) (18.27%). The genome size was found to be 227 Mb, nearly two-fold larger than the
Arabidopsis thaliana genome (
Table 7). At the same time the amount of protein-coding genes in the genome of
B. retrofracta is slightly less then in the
B. stricta and
A. thaliana genomes and much less than that in the
A. lyrata genome (
Table 1). Despite the largest genome size, the number of predicted transcripts in
B. retrofracta is the smallest among the four Brassicaceae species compared (
Table 1). The presence of a slightly greater number of genes in
B. stricta compared with
B. retrofracta, despite a smaller genome size, may be associated with aneuploidy of the chromosomal fragments, or genome rearrangements occurred as a result of interhybridization, which is characteristic of many
Boechera species and accessions.
As an example of how the genome of the sexual species
B. retrofracta could be used to study evolution and origin of apomixis, we performed an evolutionary analysis of the three alleles of the
APOLLO (APOmixis-Linked LOcus) gene (apo- and sex-alleles) described by Corral et al [
56]. We examined this gene in more detail in our assembly and in other Brassicaseae species. Along with the described copy of
APOLLO, we also found two other, more distant copies, which evidently arose by two sequential gene duplications (triplication). The
APOLLO phylogenetic tree may indicate that triplication event occurred before the separation of Brassicaceae species under study (
Figure 4). We also analyzed the
APOLLO alleles described in
Boechera ssp. It was clear that these alleles arose after separation of the
Boechera genus. In sexual
B. retrofracta and
B. stricta polymorphic sites corresponded to the “sexallele”-state and clustered with sex-alleles of the other species.
These results are compatible with an evolutionary scenario where, after the series of duplications, one of the NEN exonuclease protein (ancestor of APOLLO) copies in the common ancestor of Boechera spp. experiencing relaxed selection might be deregulated, promoting development of the apomictic reproduction from the ancestral “sexual” state, following by separation of the apomictic lineages. This model of evolution of APOLLO alleles might explain the phenomenon of apomictic development in Boechera in the diploid condition, emerged as a result of duplication events rather than polyploidy.
In conclusion, increasing number of sequenced genomes from the economically important Brassicaceae family will facilitate future genetic, genomic, evolutionary, and domestication studies in this family. B. retrofracta is thought to be an ancestor of certain hybrids including apomictic species, for example Boechera divaricarpa. Consequently, the genome assembly presented in this report may help with the challenging genome assembly of highly heterozygous hybrid Boechera species that are apomictic. Thus, the B. retrofracta genome reported here will provide a basis to decipher the hybridogenesis events that led to the formation of apomictic Boechera accessions.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}