Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB)

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Cohort
2.3. Clinical Examination of Subjects
3. Genetic analysis
3.1. Whole Exosome Sequencing (WES)
3.2. Variant Filtering and Analysis
4. Result
4.1. Patients and Clinical Characteristics
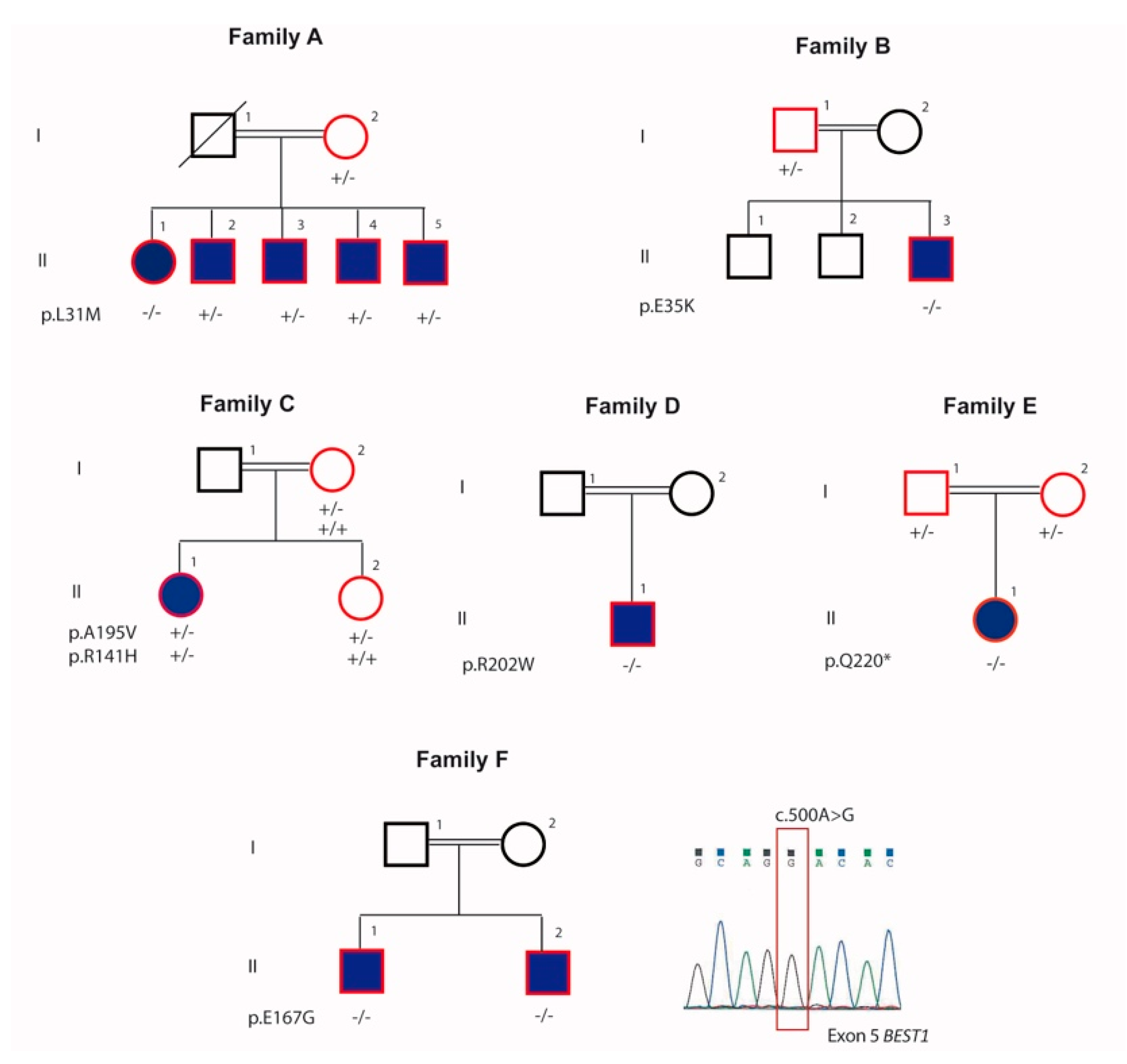
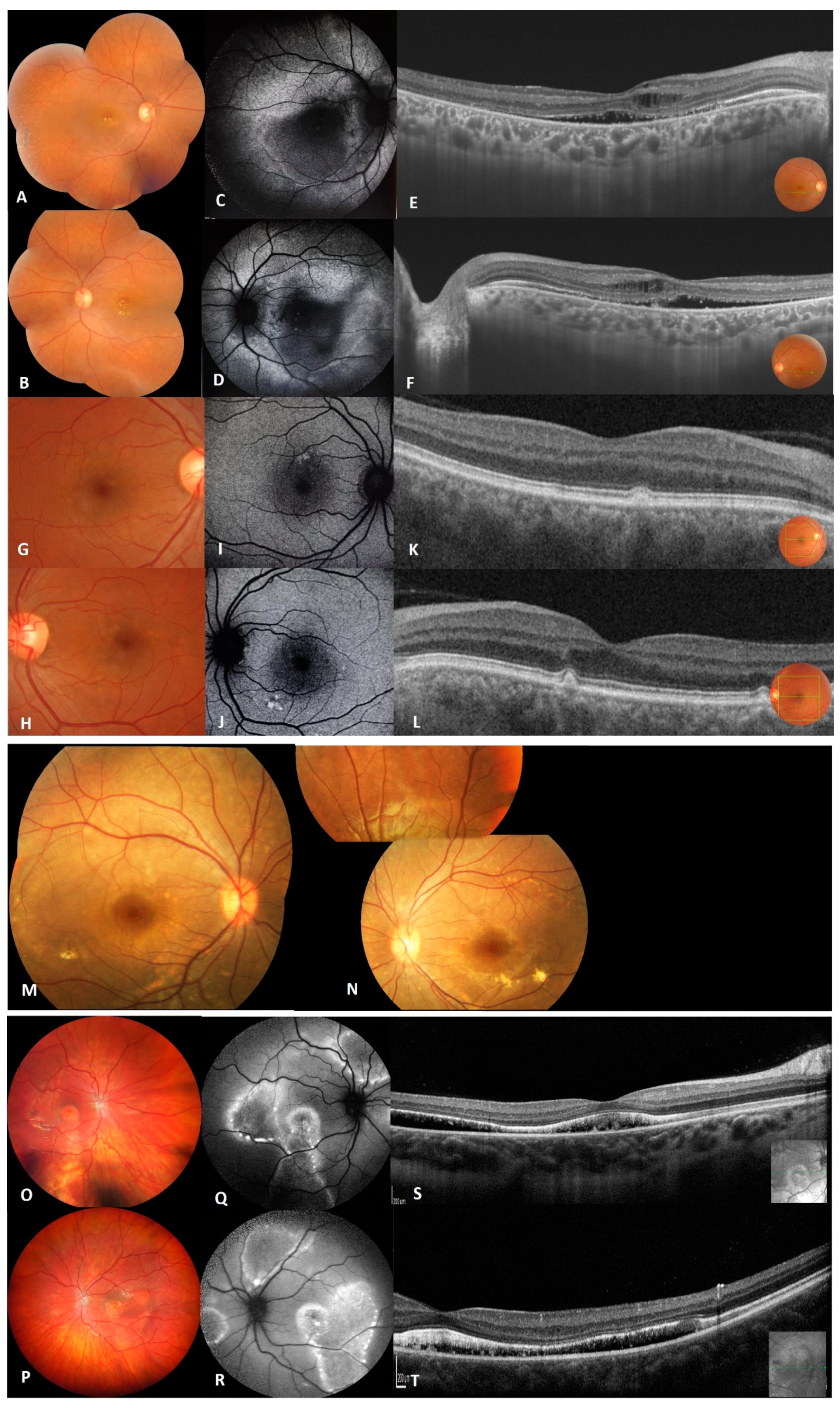
4.1.1. Family A
4.1.2. Family B
4.1.3. Family C
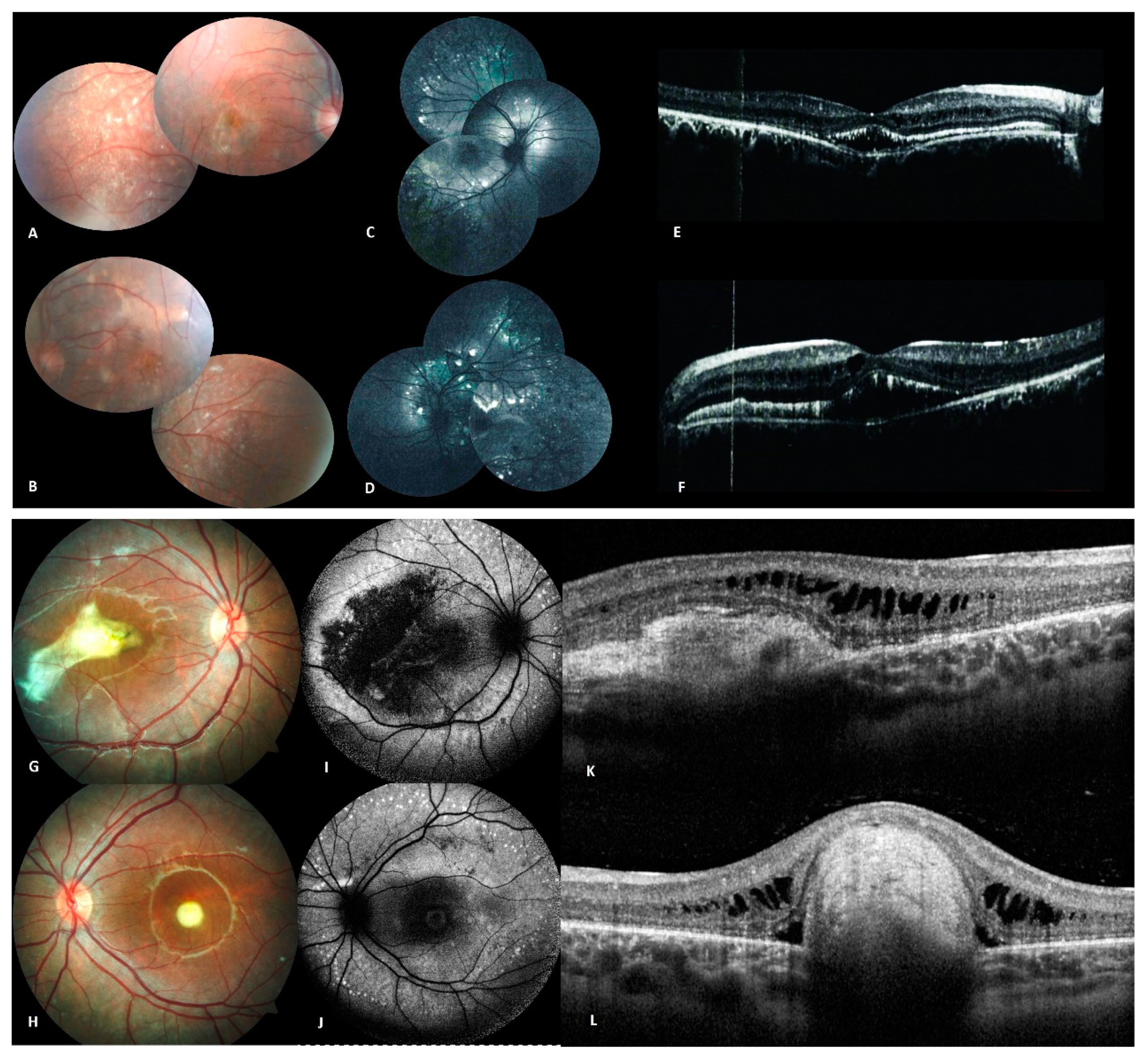
4.1.4. Family D
4.1.5. Family E
4.1.6. Family F
4.2. Exome Sequencing and Causal Variants Identification
5. Discussion
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Schatz, P.; Klar, J.; Andreasson, S.; Ponjavic, V.; Dahl, N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006, 27, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pasquay, C.; Wang, L.F.; Lorenz, B.; Preising, M.N. Bestrophin 1-Phenotypes and Functional Aspects in Bestrophinopathies. Ophthalmic Genet. 2015, 36, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Gerth, C.; Zawadzki, R.J.; Werner, J.S.; Héon, E. Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (ARB). Doc. Ophthalmol. 2009, 118, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, S.; Preising, M.N.; Ciccolella, N.; Causio, F.; Lorenz, B.; Fischetto, R. Autosomal recessive bestrophinopathy: New observations on the retinal phenotype–clinical and molecular report of an Italian family. Ophthalmologica 2011, 225, 228–235. [Google Scholar] [CrossRef]
- Pomares, E.; Bures-Jelstrup, A.; Ruiz-Nogales, S.; Corcóstegui, B.; González-Duarte, R.; Navarro, R. Nonsense-mediated decay as the molecular cause for autosomal recessive bestrophinopathy in two unrelated families. Investig. Ophthalmol. Vis. Sci. 2012, 53, 532–537. [Google Scholar] [CrossRef]
- Boon, C.J.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef]
- Chibani, Z.; Abid, I.Z.; Molbaek, A.; Söderkvist, P.; Feki, J.; Hmani-Aifa, M. Novel BEST1 gene mutations associated with two different forms of macular dystrophy in Tunisian families. Clin. Exp. Ophthalmol. 2019. [Google Scholar] [CrossRef]
- Tian, L.; Sun, T.; Xu, K.; Zhang, X.; Peng, X.; Li, Y. Screening of BEST1 gene in a Chinese cohort with Best vitelliform macular dystrophy or autosomal recessive bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3366–3375. [Google Scholar] [CrossRef]
- Kinnick, T.R.; Mullins, R.F.; Dev, S.; Leys, M.; Mackey, D.A.; Kay, C.N.; Lam, B.L.; Fishman, G.A.; Traboulsi, E.; Iezzi, R.; et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 2011, 31, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.J.; Qi, Y.H.; Hu, F.Y.; Wang, D.D.; Xu, P.; Guo, J.L.; Li, J.K.; Zhang, Y.J.; Li, W.; Chen, F.; et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br. J. Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Introini, U.; Casalino, G.; Khan, K.N.; Eandi, C.; Alovisi, C.; Michaelides, M.; Bandello, F. Clinical Course of Autosomal Recessive Bestrophinopathy Complicated by Choroidal Neovascularization. Ophthalmic Surg. Lasers Imaging Retina 2018, 49, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lin, M.; Guo, X.; Xiao, X.; Li, J.; Hu, H.; Xiao, H.; Xu, X.; Zhong, Y.; Long, S.; et al. Novel BEST1 mutations and special clinical characteristics of autosomal recessive bestrophinopathy in Chinese patients. Acta. Ophthalmol. 2019, 97, 247–259. [Google Scholar] [CrossRef]
- Gao, T.; Tian, C.; Hu, Q.; Liu, Z.; Zou, J.; Huang, L.; Zhao, M. Clinical and mutation analysis of patients with Best vitelliform macular dystrophy or autosomal recessive bestrophinopathy in Chinese population. Biomed. Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Kalevar, A.; Chen, J.J.; McDonald, H.R.; Fu, A.D. Autosomal recessive bestrophinopathy: Multimodal imaging update. Retin. Cases Brief Rep. 2018, 12, 51–54. [Google Scholar] [CrossRef]
- Nakanishi, A.; Ueno, S.; Hayashi, T.; Katagiri, S.; Kominami, T.; Ito, Y.; Gekka, T.; Masuda, Y.; Tsuneoka, H.; Shinoda, K.; et al. Clinical and genetic findings of autosomal recessive bestrophinopathy in Japanese cohort. Am. J. Ophthalmol. 2016, 168, 86–94. [Google Scholar] [CrossRef]
- Kubota, D.; Gocho, K.; Akeo, K.; Kikuchi, S.; Sugahara, M.; Matsumoto, C.S.; Shinoda, K.; Mizota, A.; Yamaki, K.; Takahashi, H.; et al. Detailed analysis of family with autosomal recessive bestrophinopathy associated with new BEST1 mutation. Doc. Ophthalmol. 2016, 132, 233–243. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Ohkuma, Y.; Sekiryu, T.; Takeuchi, T.; Gekka, T.; Kondo, M.; Iwata, T.; Tsuneoka, H. Mutation analysis of BEST1 in Japanese patients with Best’s vitelliform macular dystrophy. Br. J. Ophthalmol. 2015, 99, 1577–1582. [Google Scholar] [CrossRef]
- Tian, R.; Yang, G.; Wang, J.; Chen, Y. Screening for BEST1 gene mutations in Chinese patients with bestrophinopathy. Mol. Vis. 2014, 20, 1594–1604. [Google Scholar]
- Boon, C.J.; van den Born, L.I.; Visser, L.; Keunen, J.E.; Bergen, A.A.; Booij, J.C.; Riemslag, F.C.; Florijn, R.J.; van Schooneveld, M.J. Autosomal recessive bestrophinopathy: Differential diagnosis and treatment options. Ophthalmology 2013, 120, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.E.; Millar, I.D.; Burgess-Mullan, R.; Maher, G.J.; Urquhart, J.E.; Brown, P.D.; Black, G.C.; Manson, F.D. Functional Characterization of Bestrophin-1 Missense Mutations Associated with Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3730–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Sex | CDVA | OCT features | ERG | EOG | Mutations | REF | |
|---|---|---|---|---|---|---|---|---|
| Family A | [9] | |||||||
| Proband (II.1) | 44 | F | 0.32 RE 0.2 LE | cystoid intra-retinal and serous subretinal fluid | Moderate reduced response in both scotopic and photopic conditions | Absent light peak | p.(L31M);(L31M) | |
| Mother (I.2) | 75 | F | 0.4 BE | Very small sub-foveal deposits | Not done | Not done | p.(L31M);(=) | |
| Sibling (II.2) | 50 | M | 0.8 BE | Very small focal subretinal macular deposits | Normal | Absent light peak | p.(L31M);(=) | |
| Sibling (II.3) | 48 | M | 1.0 BE | Normal | Normal | Absent light peak | p.(L31M);(=) | |
| Sibling (II.4) | 49 | M | 1.0 BE | Normal | Normal | Absent light peak | p.(L31M);(=) | |
| Sibling (II.5) | 53 | M | 1.0 BE | Normal | Normal | Absent light peak | p.(L31M);(=) | |
| Family B | [10] | |||||||
| Proband (II.3) | 7 | M | 0.4 BE | Not done | Not done | Not done | p.(E35K);(E35K) | |
| Father (I.1) | 49 | M | Not done | Not done | Not done | Not done | p.(E35K);(=) | |
| Family C | [11] | |||||||
| Proband (II.1) | 17 | F | RE: 0.16 LE:0.25 | multiple yellowish, vitelliform deposits in the macula and along the vessel arcades exhibiting a pseudohypopyon appearance serous subretinal fluid | ERG performed on the proband confirmed reduced photopic and scotopic responses (OS) and responses still in normal range (OD), whereas the mfERG was reduced centrally and paracentrally | Reduced Arden ratio | p.(R141H);(A195V) | |
| Mother (I.2) | 55 | F | 1.0, OU | Normal findings | Not done | Not done | p.(A195V);(=) | |
| Sibling (II.2) | 18 | F | 1.0, OU | Normal findings | Not done | Not done | p.(A195V);(=) | |
| Family D | [12] | |||||||
| Proband (II.1) | 28 | M | RE: 0.36 LE: 0.8 | hyperreflective subretinal deposits, as well as RPE detachment of retina affecting the macula | Reduced scotopic and photopic responses | Reduced Arden ratio | p.(R202W);(R202W) | |
| Family E | [13] | |||||||
| Proband (II.1) | 13 | F | RE: 0.1, LE: 0.32 | Subretinal depositis and subretinal fluid | normal | Reduced Arden ratio | p.(Q220*);(Q220*) | |
| Father (I.1) | 46 | M | Not done | Not done | Not done | Not done | p.(Q220*);(=) | |
| Mother (I.2) | 45 | F | Not done | Not done | Not done | Not done | p.(Q220*);(=) | |
| Family F | Our study | |||||||
| Proband (II.1) | 35 | M | 0.8 with +2 ddc | Schisis subfoveal important | Reduced scotopic and photopic responses | Pathologic | p.(E167G);(E167G) | |
| Sibling (II.2) | 29 | M | 0.16 with +6 RE, 0.16 with +6 LE | Schisis, subretinal subfoveal fluid | Within normal limits | Not done | p.(E167G);(E167G) | |
| Our Study | Literature | Mutations | ACMG/AMG * Classification | |
|---|---|---|---|---|
| Family A | Bilateral shallow anterior chamber macular vitelliform lesions with yellow flecks and dots, cystoid intra-retinal and serous subretinal fluid. Abnormal fundus in one heterozygous carrier. Reduction in the light rise in EOG in homozygous and all heterozygous carriers. | Normal intraocular pressure and normal anterior ocular segments in both eyes. Axial length was reduced, inflammatory vitreous cells in both eyes. Multifocal macular and extramacular involvement with yellowish deposits in the central macula for both eyes and extending to the midretinal periphery in the left eye [9]. | p.(L31M);(L31M) | PM2 |
| Family B | Normal interior segment, yellowish vitelliform deposits located in the posterior pole, pre-auricular tag at the left ear. | Diffuse yellowish lesion with tiny yellow white spots scattered in the macula and near the inferior temporal vascular arcade, cystoid macula edema [10]. | p.(E35K);(E35K) | PM2 |
| Family C | Multiple yellow vitelliform deposits in the macula and along the vessel arcades exhibiting a pseudohypopyon appearance, yellow deposits on the optic disc, serous subretinal fluid. | RPE thinning and pigment mottling in the right eye and some subretinal fibrosis in the left eye. Numerous very fine deposits anterior to the temporal vascular arcades [11]. | p.(R141H);(A195V) | PS3/PS3 |
| Family D | Multiple yellow intraretinal deposits in the posterior pole and along the vessel arcades, hyperreflective subretinal deposits as well as RPE detachment affecting the macula. | Multiple small, round, yellow lesions caused by vitelliform material deposits throughout the posterior pole corresponding to focal hyperreflective lesions with subretinal and intraretinal fluid [12]. | p.(R202W);(R202W) | PS3 |
| Family E | Macular yellow vitelliform lesions and extramacular lesions along the temporal vascular arcades. | ARB complicated by choroidal neovascularization (CNV) [13]. | p.(Q220*);(Q220*) | PM2 |
| Family F | Bilateral subfoveal schisis as well as subretinal detachment, RPE alterations, hyperautofluroescence delimited with hyperfluorescent ring. | - | p.(E167G);(E167G) | PM2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habibi, I.; Falfoul, Y.; Todorova, M.G.; Wyrsch, S.; Vaclavik, V.; Helfenstein, M.; Turki, A.; El Matri, K.; El Matri, L.; Schorderet, D.F. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes 2019, 10, 953. https://doi.org/10.3390/genes10120953
Habibi I, Falfoul Y, Todorova MG, Wyrsch S, Vaclavik V, Helfenstein M, Turki A, El Matri K, El Matri L, Schorderet DF. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes. 2019; 10(12):953. https://doi.org/10.3390/genes10120953
Chicago/Turabian StyleHabibi, Imen, Yosra Falfoul, Margarita G. Todorova, Stefan Wyrsch, Veronika Vaclavik, Maria Helfenstein, Ahmed Turki, Khaled El Matri, Leila El Matri, and Daniel F. Schorderet. 2019. "Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB)" Genes 10, no. 12: 953. https://doi.org/10.3390/genes10120953