Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box

 , , and
, , and 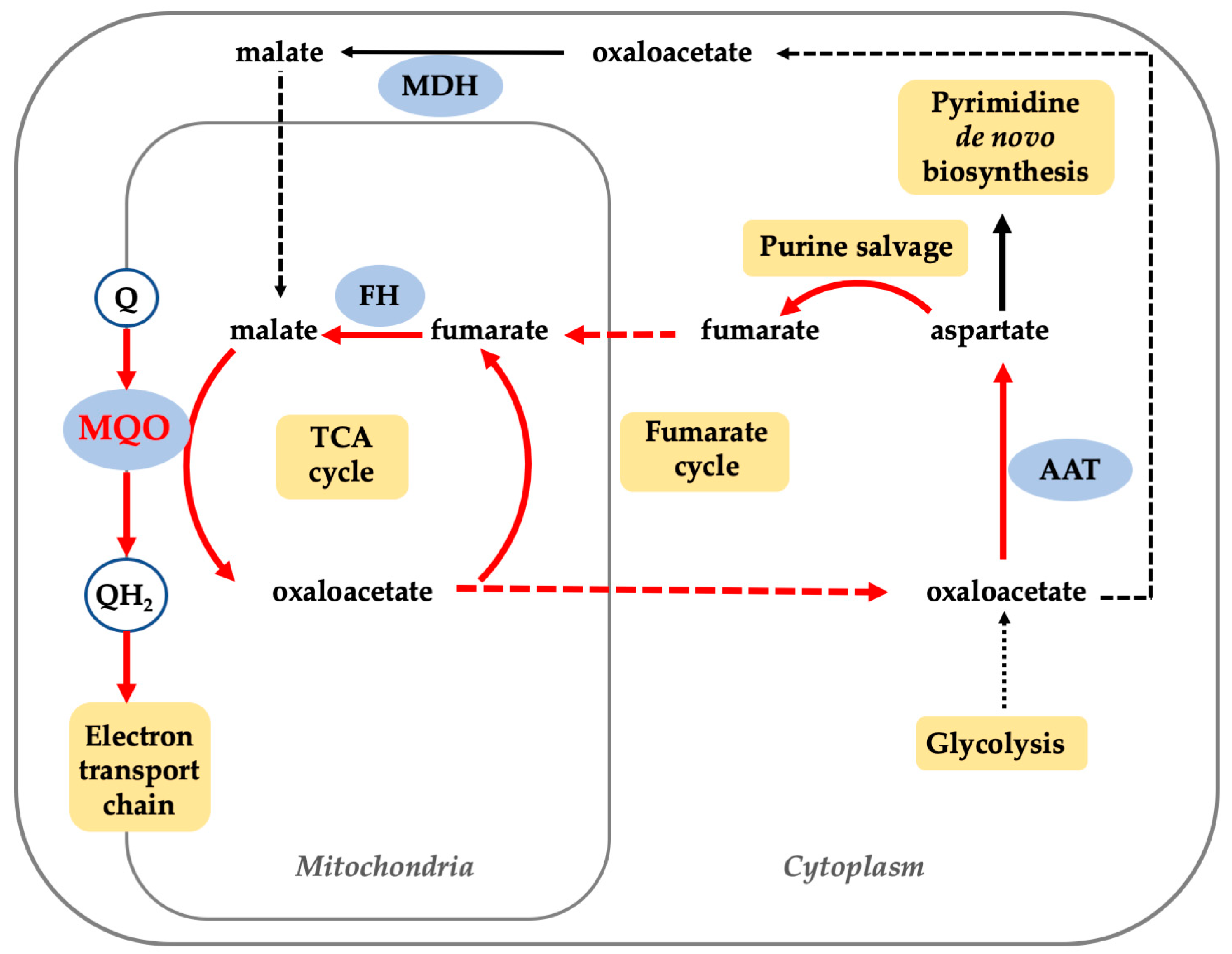{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. PfMQO Expression and Purification
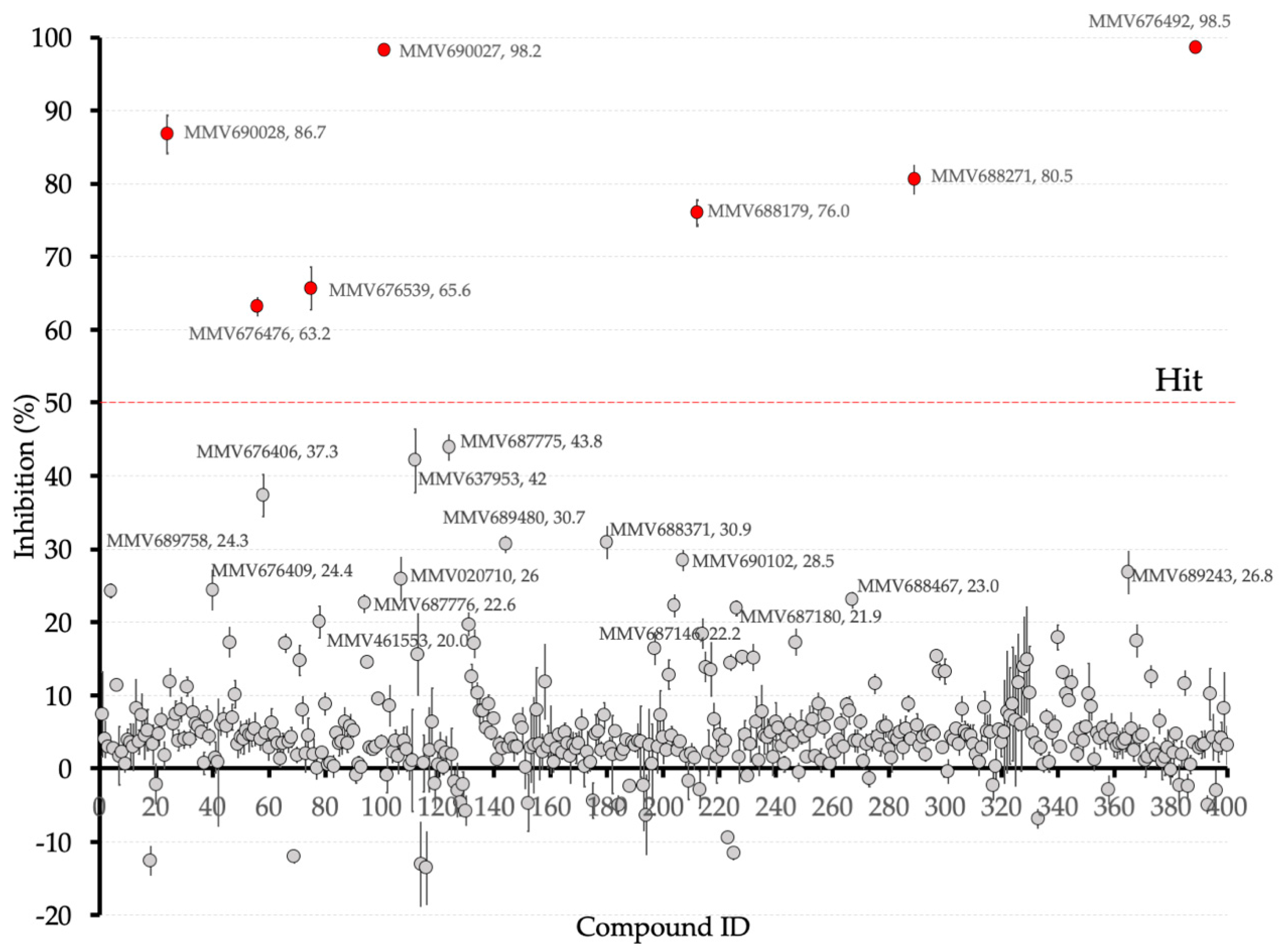
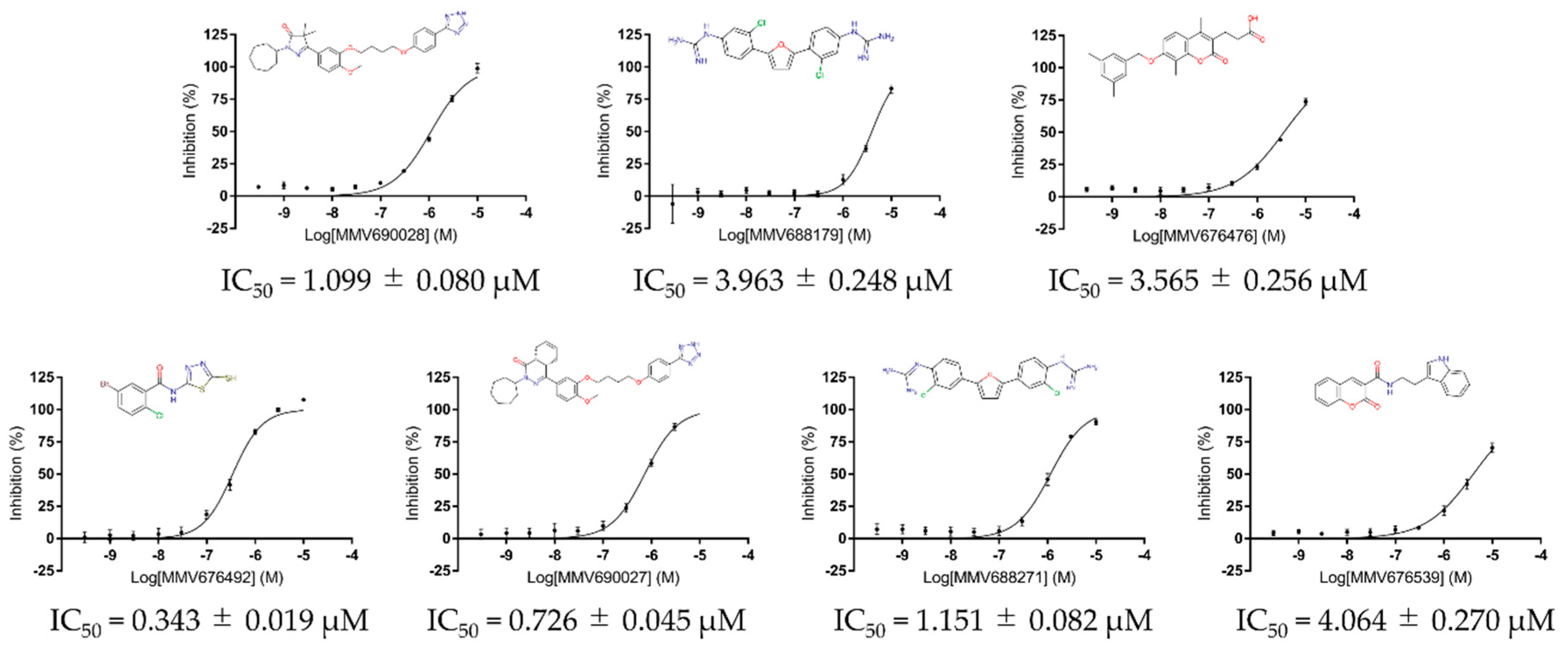
2.2. Screening of the Pathogen Box against Recombinant PfMQO
2.3. Plasmodium falciparum Culture
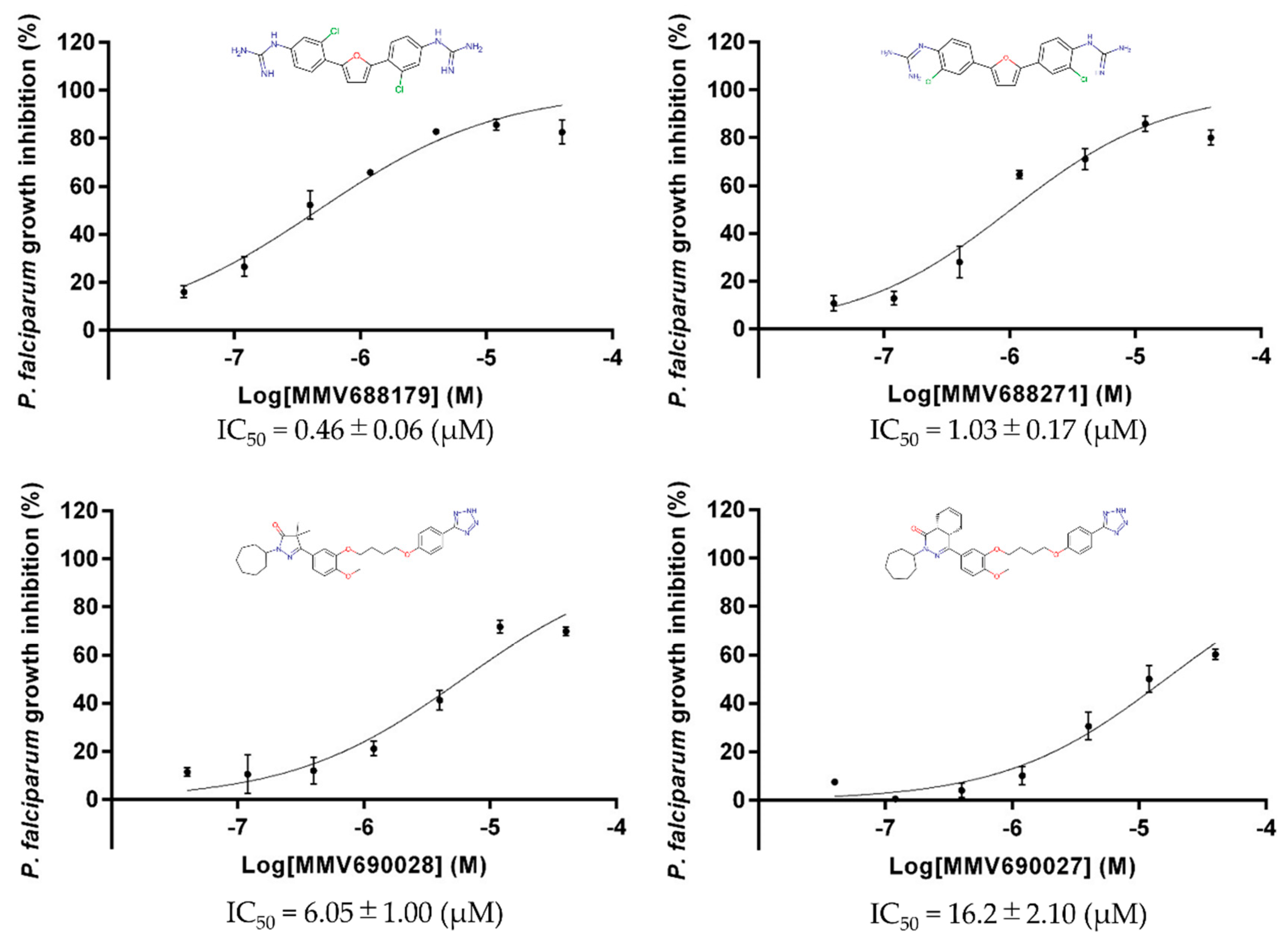
2.4. Plasmodium falciparum Growth Inhibition Assay
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO|Malaria. Available online: http://www.who.int/malaria/en/ (accessed on 22 July 2018).
- Thu, A.M.; Phyo, A.P.; Landier, J.; Parker, D.M.; Nosten, F.H. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 2017, 284, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Gamo, F.-J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.-L.; Vanderwall, D.E.; Green, D.V.S.; Kumar, V.; Hasan, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Chua, H.H.; Cobbold, S.A.; Nijagal, B.; Macrae, J.I.; Dickerman, B.K.; Gilson, P.R.; Ralph, S.A.; McConville, M.J. Metabolomics-based screening of the Malaria Box reveals both novel and established mechanisms of action. Antimicrob. Agents Chemother. 2016, 60, 6650–6663. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, D.; Brinker, A.; McNamara, C.; Henson, K.; Kato, N.; Kuhen, K.; Nagle, A.; Adrián, F.; Matzen, J.T.; Anderson, P.; et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. USA 2008, 105, 9059–9064. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, R. Introduction to the Special Issue on Malaria. FEBS J. 2017, 284, 2550–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Achieng, A.O.; Rawat, M.; Ogutu, B.; Guyah, B.; Michael Ong’echa, J.; J Perkins, D.; Kempaiah, P. Antimalarials: Molecular Drug Targets and Mechanism of Action. Curr. Top. Med. Chem. 2017, 17, 2114–2128. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, C.D.; Siregar, J.E.; Mollard, V.; Vega-Rodríguez, J.; Syafruddin, D.; Matsuoka, H.; Matsuzaki, M.; Toyama, T.; Sturm, A.; Cozijnsen, A.; et al. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016, 352, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Paton, D.G.; Childs, L.M.; Itoe, M.A.; Holmdahl, I.E.; Buckee, C.O.; Catteruccia, F. Exposing Anopheles mosquitoes to antimalarials blocks Plasmodium parasite transmission. Nature 2019, 567, 239–243. [Google Scholar] [CrossRef]
- Ke, H.; Lewis, I.A.; Morrisey, J.M.; McLean, K.J.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Jacobs-Lorena, M.; Llinás, M.; Vaidya, A.B. Genetic Investigation of Tricarboxylic Acid Metabolism during the Plasmodium falciparum Life Cycle. Cell Rep. 2015, 11, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagini, G.A.; Viriyavejakul, P.; O’Neill, P.M.; Bray, P.G.; Ward, S.A. Functional Characterization and Target Validation of Alternative Complex I of Plasmodium falciparum Mitochondria. Antimicrob. Agents Chemother. 2006, 50, 1841–1851. [Google Scholar] [CrossRef]
- Mi-Ichi, F. Parasite Mitochondria as a Target of Chemotherapy: Inhibitory Effect of Licochalcone A on the Plasmodium falciparum Respiratory Chain. Ann. N. Y. Acad. Sci. 2005, 1056, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Hoelz, L.V.; Calil, F.A.; Nonato, M.C.; Pinheiro, L.C.; Boechat, N. Plasmodium falciparum dihydroorotate dehydrogenase: A drug target against malaria. Future Med. Chem. 2018, 10, 1853–1874. [Google Scholar] [CrossRef] [PubMed]
- Hartuti, E.D.; Inaoka, D.K.; Komatsuya, K.; Miyazaki, Y.; Miller, R.J.; Wang, X.; Sadikin, M.; Prabandari, E.E.; Waluyo, D.; Kuroda, M.; et al. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim. Biophys. Acta 2018, 1859, 191–200. [Google Scholar] [CrossRef]
- Boysen, K.E.; Matuschewski, K. Arrested Oocyst Maturation in Plasmodium Parasites Lacking Type II NADH:Ubiquinone Dehydrogenase. J. Biol. Chem. 2011, 286, 32661–32671. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.; Ganesan, S.M.; Dass, S.; Morrisey, J.M.; Pou, S.; Nilsen, A.; Riscoe, M.K.; Mather, M.W.; Vaidya, A.B. Mitochondrial type II NADH dehydrogenase of Plasmodium falciparum (PfNDH2) is dispensable in the asexual blood stages. PLoS ONE 2019, 14, e0214023. [Google Scholar] [CrossRef]
- Tanaka, T.Q.; Hirai, M.; Watanabe, Y.; Kita, K. Toward understanding the role of mitochondrial complex II in the intraerythrocytic stages of Plasmodium falciparum: Gene targeting of the Fp subunit. Parasitol. Int. 2012, 61, 726–728. [Google Scholar] [CrossRef]
- Hino, A.; Hirai, M.; Tanaka, T.Q.; Watanabe, Y.; Matsuoka, H.; Kita, K. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem. (Tokyo) 2012, 152, 259–268. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.-C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Shears, M.J.; Botté, C.Y.; McFadden, G.I. Fatty acid metabolism in the Plasmodium apicoplast: Drugs, doubts and knockouts. Mol. Biochem. Parasitol. 2015, 199, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Niikura, M.; Komatsuya, K.; Inoue, S.-I.; Matsuda, R.; Asahi, H.; Inaoka, D.K.; Kita, K.; Kobayashi, F. Suppression of experimental cerebral malaria by disruption of malate:quinone oxidoreductase. Malar. J. 2017, 16, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downie, M.J.; Kirk, K.; Mamoun, C.B. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryot. Cell 2008, 7, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Bulusu, V.; Jayaraman, V.; Balaram, H. Metabolic fate of fumarate, a side product of purine salvage pathway in the intraerythrocytic stages of Plasmodium falciparum. J. Biol. Chem. 2011, 286, 9236–9245. [Google Scholar] [CrossRef]
- Fleige, T.; Pfaff, N.; Gross, U.; Bohne, W. Localisation of gluconeogenesis and tricarboxylic acid (TCA)-cycle enzymes and first functional analysis of the TCA cycle in Toxoplasma gondii. Int. J. Parasitol. 2008, 38, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Mogi, T.; Kita, K. Diversity in mitochondrial metabolic pathways in parasitic protists Plasmodium and Cryptosporidium. Parasitol. Int. 2010, 59, 305–312. [Google Scholar] [CrossRef]
- Danne, J.C.; Gornik, S.G.; MacRae, J.I.; McConville, M.J.; Waller, R.F. Alveolate Mitochondrial Metabolic Evolution: Dinoflagellates Force Reassessment of the Role of Parasitism as a Driver of Change in Apicomplexans. Mol. Biol. Evol. 2013, 30, 123–139. [Google Scholar] [CrossRef]
- Mogi, T.; Murase, Y.; Mori, M.; Shiomi, K.; Ōmura, S.; Paranagama, M.P.; Kita, K. Polymyxin B Identified as an Inhibitor of Alternative NADH Dehydrogenase and Malate: Quinone Oxidoreductase from the Gram-positive Bacterium Mycobacterium smegmatis. J. Biochem. (Tokyo) 2009, 146, 491–499. [Google Scholar] [CrossRef]
- Kather, B.; Stingl, K.; van der Rest, M.E.; Altendorf, K.; Molenaar, D. Another Unusual Type of Citric Acid Cycle Enzyme inHelicobacter pylori: The Malate: Quinone Oxidoreductase. J. Bacteriol. 2000, 182, 3204–3209. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Chai, S.C.; Goktug, A.N.; Chen, T. Assay Validation in High Throughput Screening—From Concept to Application. Drug Discov. Dev. Mol. Med. 2015. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Nwaka, S.; Besson, D.; Ramirez, B.; Maes, L.; Matheeussen, A.; Bickle, Q.; Mansour, N.R.; Yousif, F.; Townson, S.; Gokool, S.; et al. Integrated Dataset of Screening Hits against Multiple Neglected Disease Pathogens. PLoS Negl. Trop. Dis. 2011, 5, e1412. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.L. Is Open Science the Future of Drug Development? Yale J. Biol. Med. 2017, 90, 147–151. [Google Scholar] [PubMed]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, T.; Lopez-Ribot, J.L. Screening the “Pathogen Box” for the Identification of Candida albicans Biofilm Inhibitors. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Kronstad, J.W. Discovery of a Novel Antifungal Agent in the Pathogen Box. mSphere 2017, 2, e00120-17. [Google Scholar] [CrossRef] [PubMed]
- Preston, S.; Jiao, Y.; Jabbar, A.; McGee, S.L.; Laleu, B.; Willis, P.; Wells, T.N.C.; Gasser, R.B. Screening of the ‘Pathogen Box’ identifies an approved pesticide with major anthelmintic activity against the barber’s pole worm. Int. J. Parasitol. Drugs Drug Res. 2016, 6, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Pasche, V.; Laleu, B.; Keiser, J. Early Antischistosomal Leads Identified from in Vitro and in Vivo Screening of the Medicines for Malaria Venture Pathogen Box. ACS Infect. Dis. 2018, 5, 102–110. [Google Scholar] [CrossRef]
- Spalenka, J.; Escotte-Binet, S.; Bakiri, A.; Hubert, J.; Renault, J.-H.; Velard, F.; Duchateau, S.; Aubert, D.; Huguenin, A.; Villena, I. Discovery of New Inhibitors of Toxoplasma gondii via the Pathogen Box. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Hennessey, K.M.; Rogiers, I.C.; Shih, H.-W.; Hulverson, M.A.; Choi, R.; McCloskey, M.C.; Whitman, G.R.; Barrett, L.K.; Merritt, E.A.; Paredez, A.R.; et al. Screening of the Pathogen Box for inhibitors with dual efficacy against Giardia lamblia and Cryptosporidium parvum. PLoS Negl. Trop. Dis. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Paquet, T.; Manach, C.L.; Cabrera, D.G.; Younis, Y.; Henrich, P.P.; Abraham, T.S.; Lee, M.C.S.; Basak, R.; Ghidelli-Disse, S.; Lafuente-Monasterio, M.J.; et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Aguado, A.; Laleu, B.; Balmer, V.; Ritler, D.; Hemphill, A. In vitro screening of the open source Pathogen Box identifies novel compounds with profound activities against Neospora caninum. Int. J. Parasitol. 2017, 47, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Sykes, M.L.; Jones, A.J.; Shelper, T.B.; Simpson, M.; Lang, R.; Poulsen, S.-A.; Sleebs, B.E.; Avery, V.M. Screening the MMV Pathogen Box across multiple pathogens reclassifies starting points for open source drug discovery. Antimicrob. Agents Chemother. 2017. [Google Scholar] [CrossRef]
- Stephens, C.E.; Brun, R.; Salem, M.M.; Werbovetz, K.A.; Tanious, F.; Wilson, W.D.; Boykin, D.W. The activity of diguanidino and ‘reversed’ diamidino 2,5-diarylfurans versus Trypanosoma cruzi and Leishmania donovani. Bioorg. Med. Chem. Lett. 2003, 13, 2065–2069. [Google Scholar] [CrossRef]
- Ramos-Molina, B.; Lick, A.N.; Blanco, E.H.; Posada-Salgado, J.A.; Martinez-Mayorga, K.; Johnson, A.T.; Jiao, G.-S.; Lindberg, I. Identification of potent and compartment-selective small molecule furin inhibitors using cell-based assays. Biochem. Pharmacol. 2015, 96, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Orrling, K.M.; Jansen, C.; Vu, X.L.; Balmer, V.; Bregy, P.; Shanmugham, A.; England, P.; Bailey, D.; Cos, P.; Maes, L.; et al. Catechol Pyrazolinones as Trypanocidals: Fragment-Based Design, Synthesis, and Pharmacological Evaluation of Nanomolar Inhibitors of Trypanosomal Phosphodiesterase B1. J. Med. Chem. 2012, 55, 8745–8756. [Google Scholar] [CrossRef]
- Jansen, C.; Wang, H.; Kooistra, A.J.; de Graaf, C.; Orrling, K.M.; Tenor, H.; Seebeck, T.; Bailey, D.; de Esch, I.J.P.; Ke, H.; et al. Discovery of Novel Trypanosoma brucei Phosphodiesterase B1 Inhibitors by Virtual Screening against the Unliganded TbrPDEB1 Crystal Structure. J. Med. Chem. 2013, 56, 2087–2096. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Miyazaki, Y.; Inaoka, D.K.; Hartuti, E.D.; Watanabe, Y.-I.; Shiba, T.; Harada, S.; Saimoto, H.; Burrows, J.N.; Benito, F.J.G.; et al. Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box. Genes 2019, 10, 471. https://doi.org/10.3390/genes10060471
Wang X, Miyazaki Y, Inaoka DK, Hartuti ED, Watanabe Y-I, Shiba T, Harada S, Saimoto H, Burrows JN, Benito FJG, et al. Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box. Genes. 2019; 10(6):471. https://doi.org/10.3390/genes10060471
Chicago/Turabian StyleWang, Xinying, Yukiko Miyazaki, Daniel Ken Inaoka, Endah Dwi Hartuti, Yoh-Ichi Watanabe, Tomoo Shiba, Shigeharu Harada, Hiroyuki Saimoto, Jeremy Nicholas Burrows, Francisco Javier Gamo Benito, and et al. 2019. "Identification of Plasmodium falciparum Mitochondrial Malate: Quinone Oxidoreductase Inhibitors from the Pathogen Box" Genes 10, no. 6: 471. https://doi.org/10.3390/genes10060471