N-Acetylcysteine Serves as Substrate of 3-Mercaptopyruvate Sulfurtransferase and Stimulates Sulfide Metabolism in Colon Cancer Cells

 , ,
, ,  , ,
, , 

Abstract
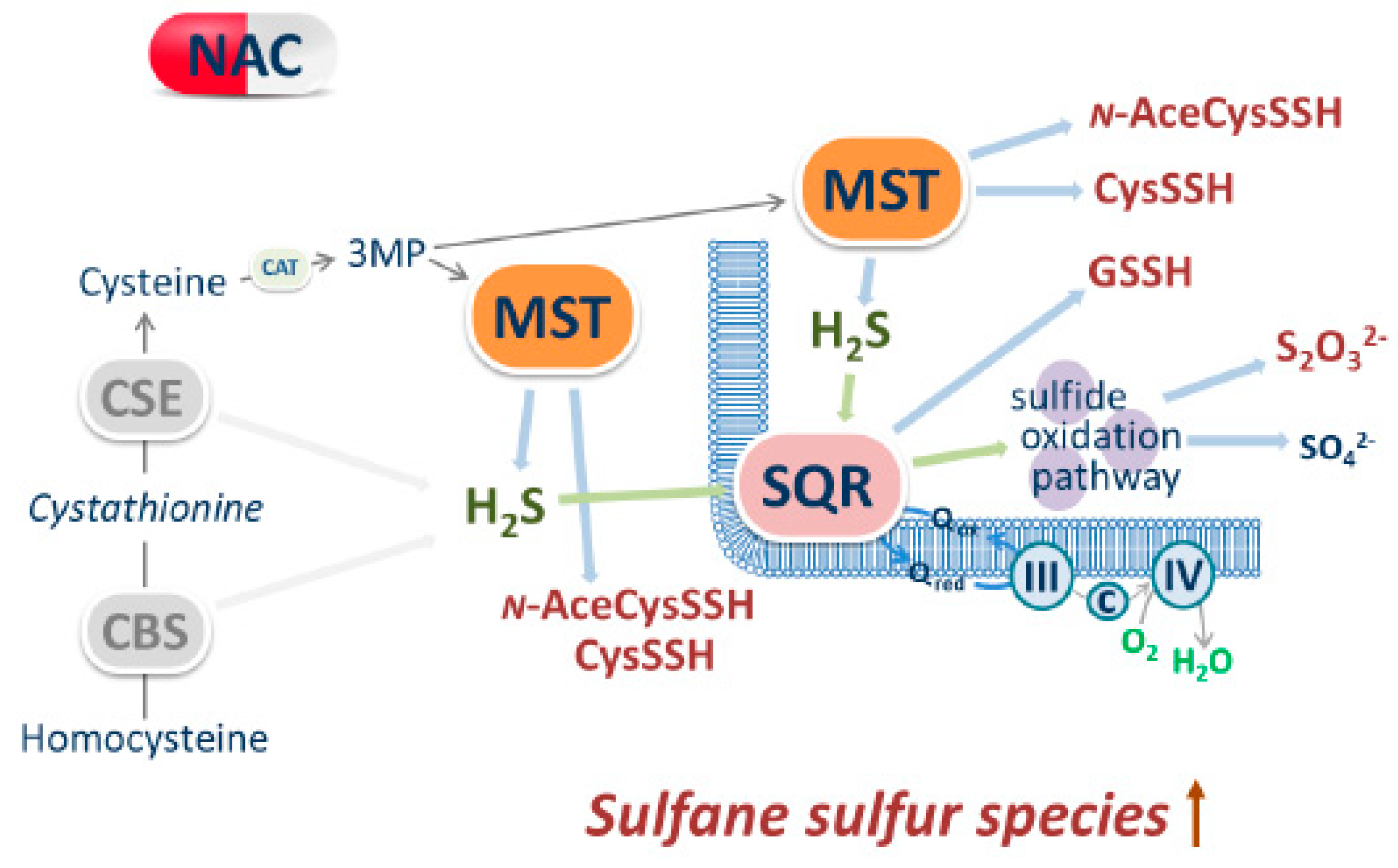
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Sulfide Stock Solutions
2.3. Cell Culture and Isolation of Mitochondria
2.4. Human MST Expression and Purification
2.5. SQR Activity Assay in Mitochondrial Preparations
2.6. MST Activity Assay in Cell Lysates
2.7. Recombinant Human MST Activity Assay
2.8. Evaluation of Mitochondrial Content by the Citrate Synthase Assay
2.9. Immunoblotting Assays
2.10. NAC Quantification by Reverse Phase High Performance Liquid Chromatography (RP-HPLC)
3. Results
3.1. Effect of NAC on Expression Levels of H2S Metabolism Enzymes
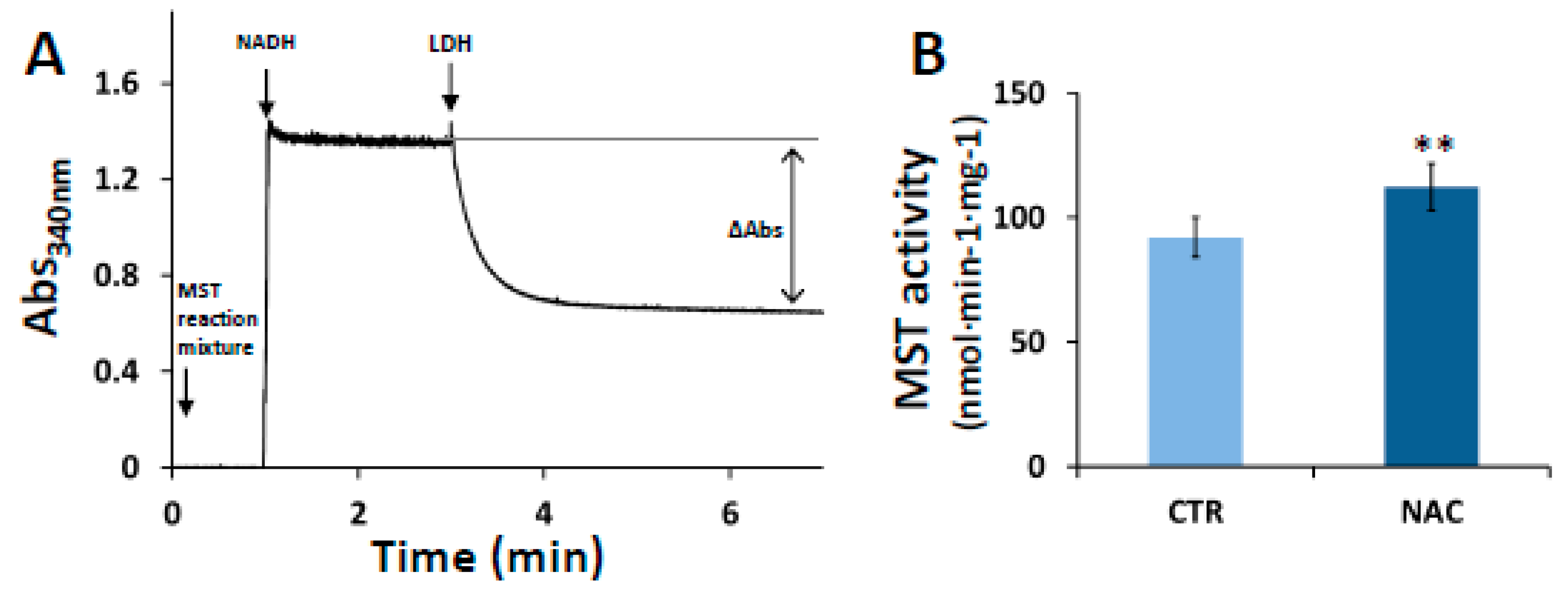
3.2. Effect of NAC on MST Activity
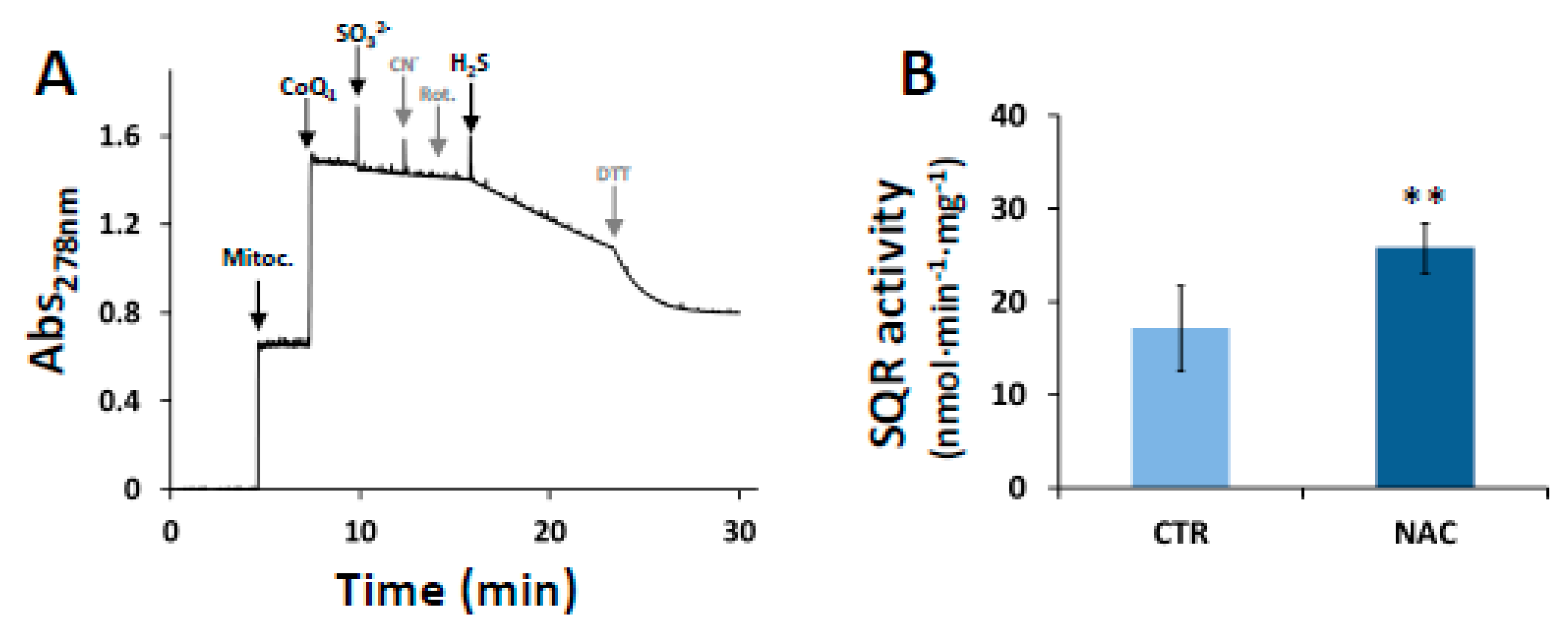
3.3. Effect of NAC on SQR Activity
3.4. Effect of NAC on Mitochondrial Mass
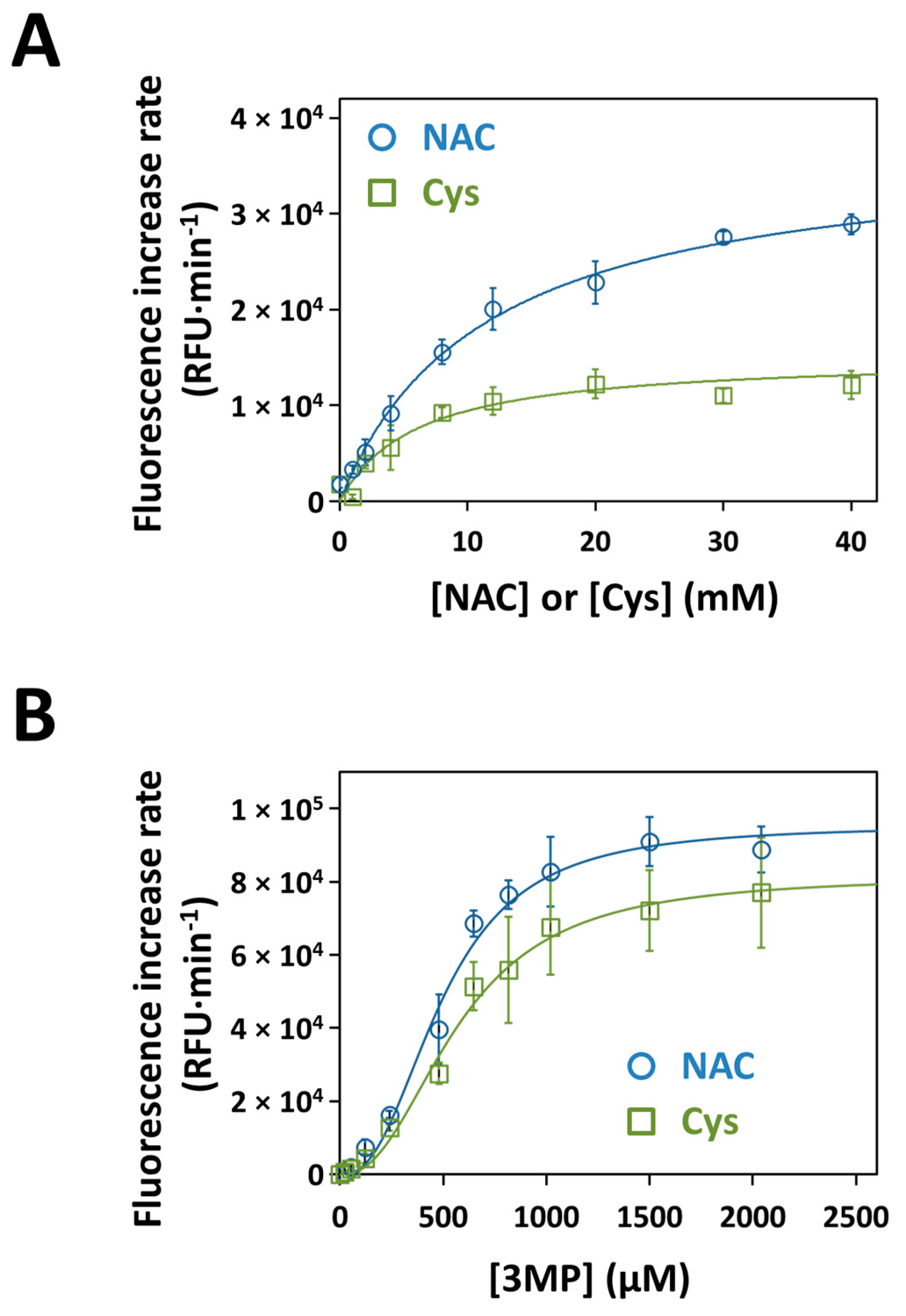
3.5. Kinetics of H2S Production by Recombinant Human MST with NAC as a Substrate
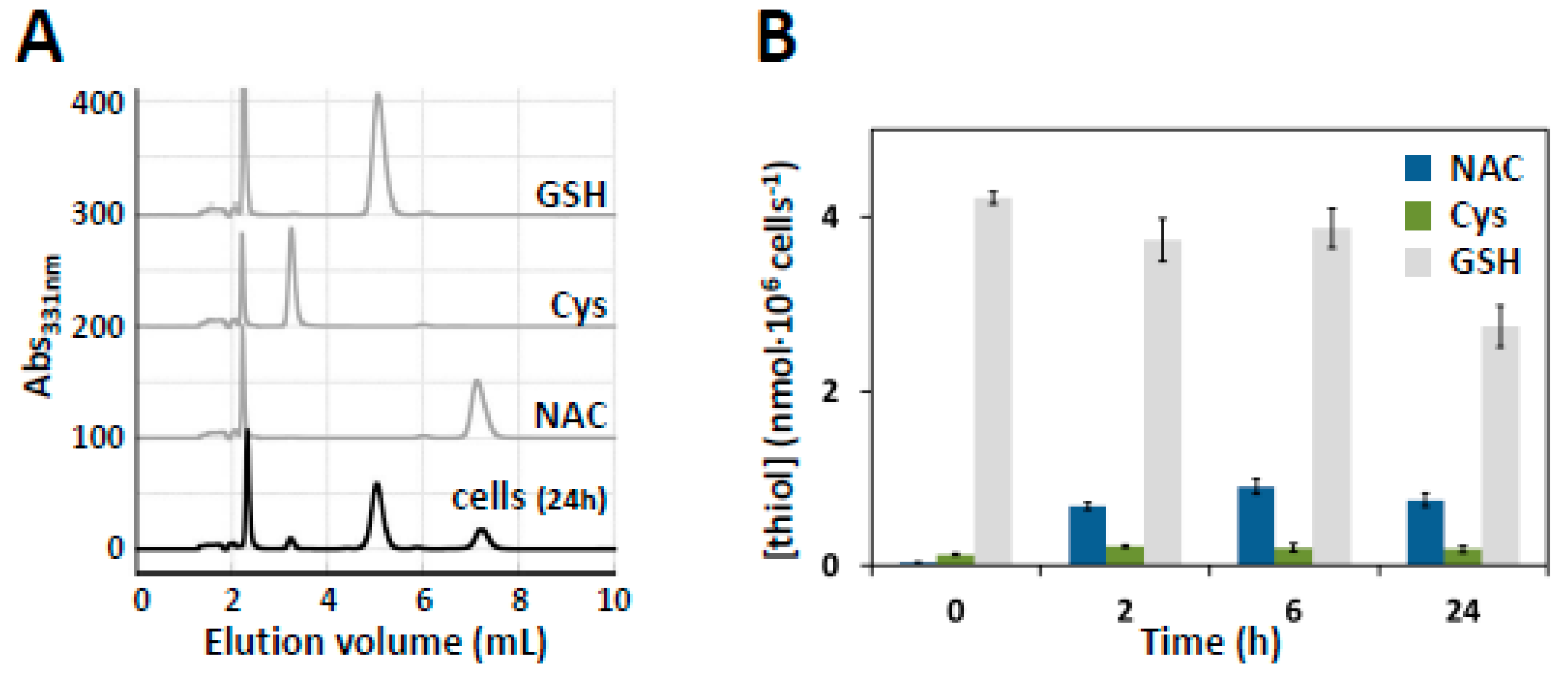
3.6. HPLC Quantification of Intracellular NAC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Wang, R. Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell. Longev. 2018, 2018, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P. Mechanistic Chemical Perspective of Hydrogen Sulfide Signaling. Methods Enzymol. 2015, 554, 3–29. [Google Scholar] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Nishimura, A.; Morita, M.; Matsunaga, T.; Hamid, H.A.; Akaike, T. Redox Signaling Regulated by Cysteine Persulfide and Protein Polysulfidation. Molecules 2016, 21, 1721. [Google Scholar] [CrossRef]
- Toohey, J.I. Sulfur signaling: Is the agent sulfide or sulfane? Anal. Biochem. 2011, 413, 1–7. [Google Scholar] [CrossRef]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.-I.; Kimura, H. Polysulfides are possible H2S -derived signaling molecules in rat brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Kumagai, Y. Sulfane Sulfur in Toxicology: A Novel Defense System Against Electrophilic Stress. Toxicol. Sci. 2019, 170, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Möller, M.N.; Alvarez, B. Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 2017, 617, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Dias, C.; Morello, J.; Correia, M.J.; Coelho, N.R.; Antunes, A.M.; Macedo, M.P.; Monteiro, E.C.; Soto, K.; Pereira, S.A. Mercapturate Pathway in the Tubulocentric Perspective of Diabetic Kidney Disease. Nephron 2019, 1–7. [Google Scholar] [CrossRef]
- Veiga-Da-Cunha, M.; Tyteca, D.; Stroobant, V.; Courtoy, P.J.; Opperdoes, F.R.; Van Schaftingen, E. Molecular Identification of NAT8 as the Enzyme That Acetylates Cysteine S-Conjugates to Mercapturic Acids. J. Biol. Chem. 2010, 285, 18888–18898. [Google Scholar] [CrossRef] [Green Version]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid. Redox Signal. 2012, 17, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and Kinetic Analysis of H2S Production by Human Mercaptopyruvate Sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013. [Google Scholar] [CrossRef]
- Fräsdorf, B.; Radon, C.; Leimkühler, S. Characterization and Interaction Studies of Two Isoforms of the Dual Localized 3-Mercaptopyruvate Sulfurtransferase TUM1 from Humans. J. Biol. Chem. 2014, 289, 34543–34556. [Google Scholar] [CrossRef] [Green Version]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Ogasawara, Y.; Kimura, H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 2011, 439, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Human Sulfide:Quinone Oxidoreductase Catalyzes the First Step in Hydrogen Sulfide Metabolism and Produces a Sulfane Sulfur Metabolite. Biochemistry 2012, 51, 6804–6815. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Yadav, P.K.; Ballou, D.P.; Banerjee, R. Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J. Biol. Chem. 2015, 290, 25072–25080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Role of human sulfide: Quinone oxidoreductase in H2S metabolism. Methods Enzymol. 2015, 554, 255–270. [Google Scholar] [PubMed]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the Human Mitochondrial Hydrogen Sulfide Oxidation Pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.B.; Malagrinò, F.; Arese, M.; Forte, E.; Sarti, P.; Giuffrè, A. Bioenergetic relevance of hydrogen sulfide and the interplay between gasotransmitters at human cystathionine beta-synthase. BBA Bioenergy 2016, 1857, 1127–1138. [Google Scholar] [CrossRef]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. BBA Bioenergy 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [Green Version]
- Blachier, F.; Davila, A.-M.; Mimoun, S.; Benetti, P.-H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.-J.; Sakamoto, N.; Fearon, E.R.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Druzhyna, N.; Szczesny, B.; Olah, G.; Módis, K.; Asimakopoulou, A.; Pavlidou, A.; Szoleczky, P.; Gerö, D.; Yanagi, K.; Törö, G.; et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine beta-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol. Res. 2016, 113, 18–37. [Google Scholar]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid. Redox Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef]
- Zuhra, K.; Sousa, P.M.F.; Paulini, G.; Lemos, A.R.; Kalme, Z.; Bisenieks, I.; Bisenieks, E.; Vigante, B.; Duburs, G.; Bandeiras, T.M.; et al. Screening Pyridine Derivatives against Human Hydrogen Sulfide-synthesizing Enzymes by Orthogonal Methods. Sci. Rep. 2019, 9, 684. [Google Scholar] [CrossRef]
- Cao, X.; Xie, Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.-S. A review of hydrogen sulfide synthesis, metabolism, and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Szabo, C. Hydrogen sulfide and cancer. Handb. Exp. Pharmacol. 2015, 230, 233–241. [Google Scholar]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S -producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef]
- Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabó, C.; Módis, K. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar]
- Breza, J.; Soltysova, A.; Hudecova, S.; Penesova, A.; Szadvari, I.; Babula, P.; Chovancova, B.; Lencesova, L.; Pös, O.; Breza, J.; et al. Endogenous H2S producing enzymes are involved in apoptosis induction in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 591. [Google Scholar] [CrossRef]
- Jurkowska, H.; Placha, W.; Nagahara, N.; Wróbel, M. The expression and activity of cystathionine-gamma-lyase and 3-mercaptopyruvate sulfurtransferase in human neoplastic cell lines. Amino Acids 2011, 41, 151–158. [Google Scholar] [CrossRef]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)—hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2018, 104083. [Google Scholar] [CrossRef]
- Malagrinò, F.; Zuhra, K.; Mascolo, L.; Mastronicola, D.; Vicente, J.B.; Forte, E.; Giuffrè, A. Hydrogen Sulfide Oxidation: Adaptive Changes in Mitochondria of SW480 Colorectal Cancer Cells upon Exposure to Hypoxia. Oxid. Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Giampazolias, E.; Tait, S.W. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Castaldo, S.A.; Freitas, J.T.; Conchinha, N.V.; Madureira, P.A. The tumorigenic roles of the cellular REDOX regulatory systems. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in Randomized Trials of Antioxidant Supplements for Primary and Secondary Prevention: Systematic Review and Meta-analysis. JAMA 2007, 297, 842. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef]
- Papaioannou, D.; Cooper, K.L.; Carroll, C.; Hind, D.; Squires, H.; Tappenden, P.; Logan, R.F. Antioxidants in the chemoprevention of colorectal cancer and colorectal adenomas in the general population: A systematic review and meta-analysis. Colorectal Dis. 2011, 13, 1085–1099. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N -acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Park, E.J.; Min, K.; Yoo, Y.H.; Kim, Y.-S.; Kwon, T.K. β-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 2014, 5, e1230. [Google Scholar] [CrossRef]
- Wang, F.; Liu, S.; Shen, Y.; Zhuang, R.; Xi, J.; Fang, H.; Pan, X.; Sun, J.; Cai, Z. Protective effects of N-acetylcysteine on cisplatin-induced oxidative stress and DNA damage in HepG2 cells. Exp. Ther. Med. 2014, 8, 1939–1945. [Google Scholar] [CrossRef]
- Fu, Y.; Yang, G.; Zhu, F.; Peng, C.; Li, W.; Li, H.; Kim, H.-G.; Bode, A.M.; Dong, Z. Antioxidants decrease the apoptotic effect of 5-Fu in colon cancer by regulating Src-dependent caspase-7 phosphorylation. Cell Death Dis. 2014, 5, e983. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.-K.; Ming, S.-H.; Nam, O.-C.; Armstrong, J.-S.; Whiteman, M. Hydrogen sulfide protects colon cancer cells from chemopreventative agent beta-phenylethyl isothiocyanate induced apoptosis. World J. Gastroenterol. 2005, 11, 3990–3997. [Google Scholar] [CrossRef]
- Raftos, J.E.; Whillier, S.; Chapman, B.E.; Kuchel, P.W. Kinetics of uptake and deacetylation of N-acetylcysteine by human erythrocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1698–1706. [Google Scholar] [CrossRef]
- Patriarca, S.; Furfaro, A.L.; Domenicotti, C.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N. Supplementation with N-acetylcysteine and taurine failed to restore glutathione content in liver of streptozotocin-induced diabetics rats but protected from oxidative stress. BBA Mol. Basis Dis. 2005, 1741, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef]
- Nagy, P.; Pálinkás, Z.; Nagy, A.; Budai, B.; Toth, I.; Vasas, A. Chemical aspects of hydrogen sulfide measurements in physiological samples. BBA Gen. Subj. 2014, 1840, 876–891. [Google Scholar] [CrossRef]
- Nashef, A.S.; Osuga, D.T.; Feeney, R.E. Determination of hydrogen sulfide with 5,5′-dithiobis-(2-nitrobenzoic acid), N-ethylmaleimide, and parachloromercuribenzoate. Anal. Biochem. 1977, 79, 394–405. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Ferrín, G.; Pérez-Martos, A.; Fernández-Silva, P.; Zeviani, M.; Enriquez, J.A. Isolation of mitochondria for biogenetical studies: An update. Mitochondrion 2010, 10, 253–262. [Google Scholar] [CrossRef]
- Jones, A.J.; Hirst, J. A spectrophotometric coupled enzyme assay to measure the activity of succinate dehydrogenase. Anal. Biochem. 2013, 442, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Theissen, U.; Martin, W.F. Sulfide: Quinone oxidoreductase (SQR) from the lugworm Arenicola marina shows cyanide- and thioredoxin-dependent activity. FEBS J. 2008, 275, 1131–1139. [Google Scholar] [CrossRef]
- Valentine, W.N.; Frankenfeld, J.K. 3-Mercaptopyruvate sulfurtransferase (EC 2.8.1.2): A simple assay adapted to human blood cells. Clin. Chim. Acta 1974, 51, 205–210. [Google Scholar] [CrossRef]
- Thorson, M.K.; Majtan, T.; Kraus, J.P.; Barros, A.M. Identification of cystathionine beta-synthase inhibitors using a hydrogen sulfide selective probe. Angew. Chem. Int. Ed. Engl. 2013, 52, 4641–4644. [Google Scholar] [CrossRef]
- Srere, P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. Methods Enzymol. 1969, 13, 3–11. [Google Scholar]
- Rivero-Gutierrez, B.; Anzola, A.; Martínez-Augustín, O.; De Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Brundu, S.; Nencioni, L.; Celestino, I.; Coluccio, P.; Palamara, A.T.; Magnani, M.; Fraternale, A. Validation of a Reversed-Phase High Performance Liquid Chromatography Method for the Simultaneous Analysis of Cysteine and Reduced Glutathione in Mouse Organs. Oxid. Med. Cell. Longev. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, J.; Bae, J.-S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.C.; Serpa, J. Glutathione in Ovarian Cancer: A Double-Edged Sword. Int. J. Mol. Sci. 2018, 19, 1882. [Google Scholar] [CrossRef]
- Kimura, Y.; Koike, S.; Shibuya, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci. Rep. 2017, 7, 10459. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Chowdhuri, S.R.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine beta-synthase in human breast Cancer. Free Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef] [Green Version]
- Iciek, M.; Wlodek, L. Biosynthesis and biological properties of compounds containing highly reactive, reduced sulfane sulfur. Pol. J. Pharmacol. 2001, 53, 215–225. [Google Scholar]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, H.; Wróbel, M. Inhibition of Human Neuroblastoma Cell Proliferation by N-acetyl-L-cysteine as a Result of Increased Sulfane Sulfur Level. Anticancer Res. 2018, 38, 5109–5113. [Google Scholar] [CrossRef]
- Jurkowska, H.; Wrobel, M. N-acetyl-L-cysteine as a source of sulfane sulfur in astrocytoma and astrocyte cultures: Correlations with cell proliferation. Amino Acids 2008, 34, 231–237. [Google Scholar] [CrossRef]
- Uttamsingh, V.; Keller, D.A.; Anders, M.W.; Keller, D. Acylase I-Catalyzed Deacetylation of N-Acetyl-l-cysteine and S-Alkyl-N-acetyl-l-cysteines. Chem. Res. Toxicol. 1998, 11, 800–809. [Google Scholar] [CrossRef]
- Yamauchi, A.; Ueda, N.; Hanafusa, S.; Yamashita, E.; Kihara, M.; Naito, S. Tissue distribution of and species differences in deacetylation of N-acetyl-L-cysteine and immunohistochemical localization of acylase I in the primate kidney. J. Pharm. Pharmacol. 2002, 54, 205–212. [Google Scholar] [CrossRef]
- Nakanishi, T.; Akabane, E.R.; Nanami, M.; Kiyobayashi, Y.; Moriguchi, R.; Hasuike, Y.; Otaki, Y.; Miyagawa, K.; Itahana, R.; Izumi, M. Comparison of Cytotoxicity of Cysteine and Homocysteine for Renal Epithelial Cells. Nephron Exp. Nephrol. 2005, 100, e11–e20. [Google Scholar] [CrossRef]
- Nishiuch, Y.; Sasaki, M.; Nakayasu, M.; Oikawa, A. Cytotoxicity of cysteine in culture media. In Vitro Cell. Dev. Biol. Anim. 1976, 12, 635–638. [Google Scholar] [CrossRef]
- Baker, D.H. Comparative species utilization and toxicity of sulfur amino acids. J. Nutr. 2006, 136, 1670S–1675S. [Google Scholar] [CrossRef]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J.; et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Ziwanovic, J.; Alvarez, B.; Banerjee, R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KM (A) | Vmax (A) | Vmax/KM (A) | KM (3MP) | Vmax (3MP) | Vmax/KM (3MP) | |
|---|---|---|---|---|---|---|
| mM | RFU·min−1 | RFU·min−1·mM−1 | mM | RFU·min−1 | RFU·min−1·mM−1 | |
| Cys | 6.0 ± 1.0 | 15,136 ± 839 | 2523 | 0.57 ± 0.04 | 81,560 ± 3972 | 143,088 |
| NAC | 11.4 ± 1.0 | 37,178 ± 1442 | 3261 | 0.49 ± 0.02 | 95,396 ± 2487 | 194,686 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuhra, K.; Tomé, C.S.; Masi, L.; Giardina, G.; Paulini, G.; Malagrinò, F.; Forte, E.; Vicente, J.B.; Giuffrè, A. N-Acetylcysteine Serves as Substrate of 3-Mercaptopyruvate Sulfurtransferase and Stimulates Sulfide Metabolism in Colon Cancer Cells. Cells 2019, 8, 828. https://doi.org/10.3390/cells8080828
Zuhra K, Tomé CS, Masi L, Giardina G, Paulini G, Malagrinò F, Forte E, Vicente JB, Giuffrè A. N-Acetylcysteine Serves as Substrate of 3-Mercaptopyruvate Sulfurtransferase and Stimulates Sulfide Metabolism in Colon Cancer Cells. Cells. 2019; 8(8):828. https://doi.org/10.3390/cells8080828
Chicago/Turabian StyleZuhra, Karim, Catarina S. Tomé, Letizia Masi, Giorgio Giardina, Giulia Paulini, Francesca Malagrinò, Elena Forte, João B. Vicente, and Alessandro Giuffrè. 2019. "N-Acetylcysteine Serves as Substrate of 3-Mercaptopyruvate Sulfurtransferase and Stimulates Sulfide Metabolism in Colon Cancer Cells" Cells 8, no. 8: 828. https://doi.org/10.3390/cells8080828