IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Ethics
2.2. Blood Samples
2.3. Cytokine Receptor Analysis
2.4. STAT-pY Analysis
2.5. Genotyping
2.6. Fluorescence-Activated Cell Sorting of T, B, and NK Cells
2.7. mRNA Analysis of T, B, and NK Cells
2.8. Pathway-Associated wGRS
2.9. Statistical Analysis
3. Results
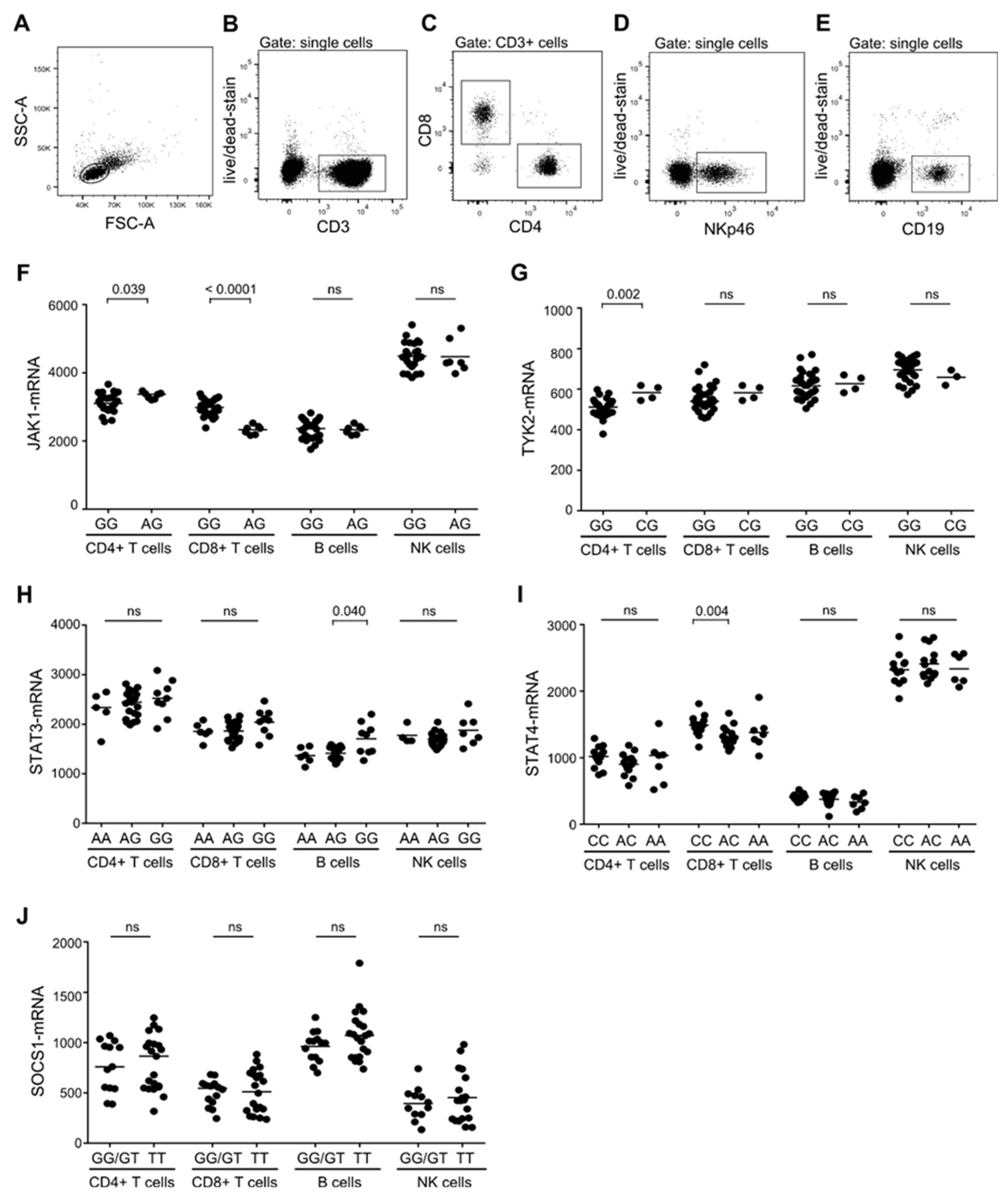
3.1. Association Between MS-Risk Alleles and Expression Level of Molecules in the IL-6, IL-12, and IL-23 Induced STAT-Pathway
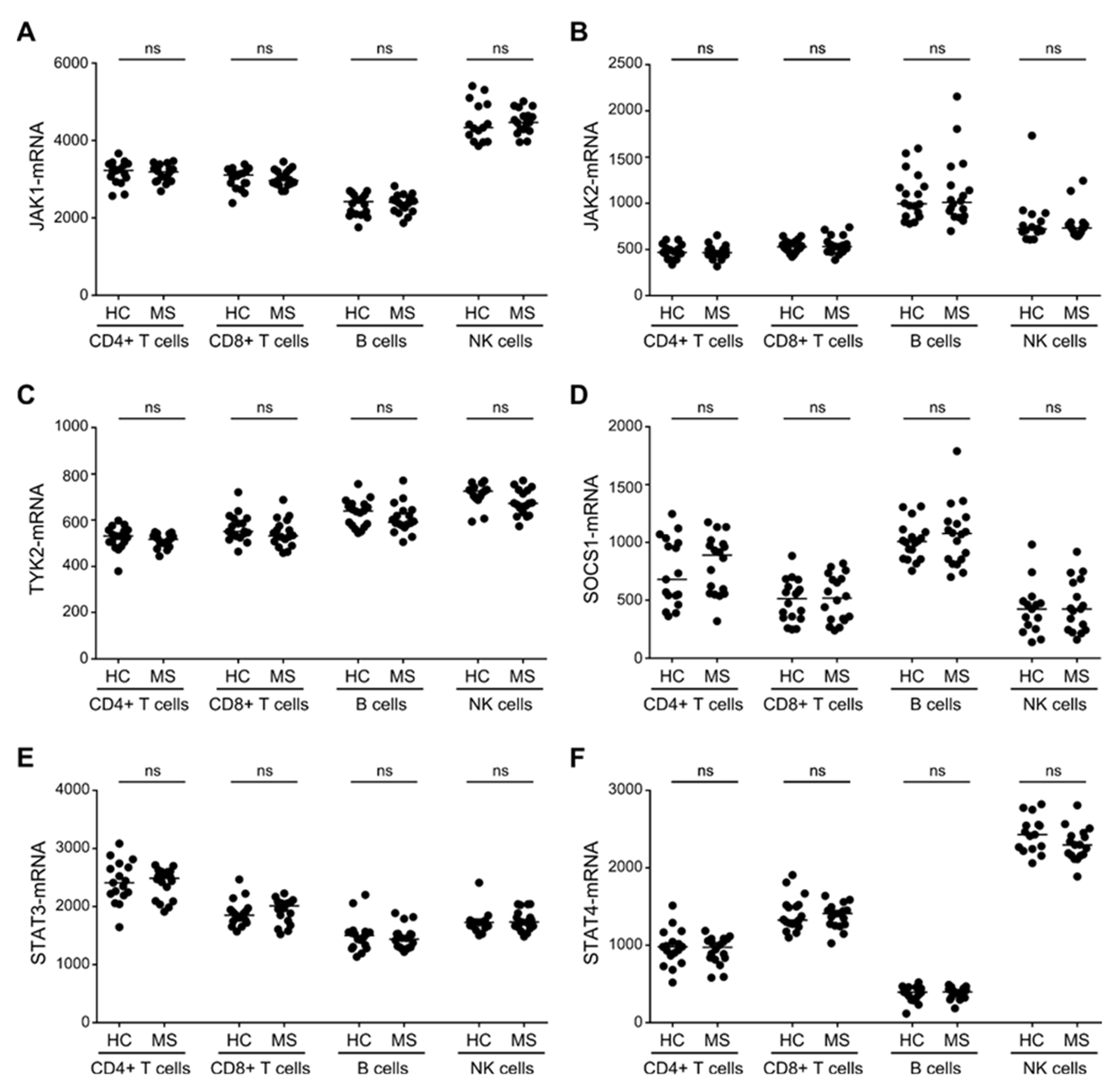
3.2. No Difference in the Expression of the IL-6R, IL-12R, and IL-23R in T, B, and NK Cells Between Patients with MS and Healthy Controls
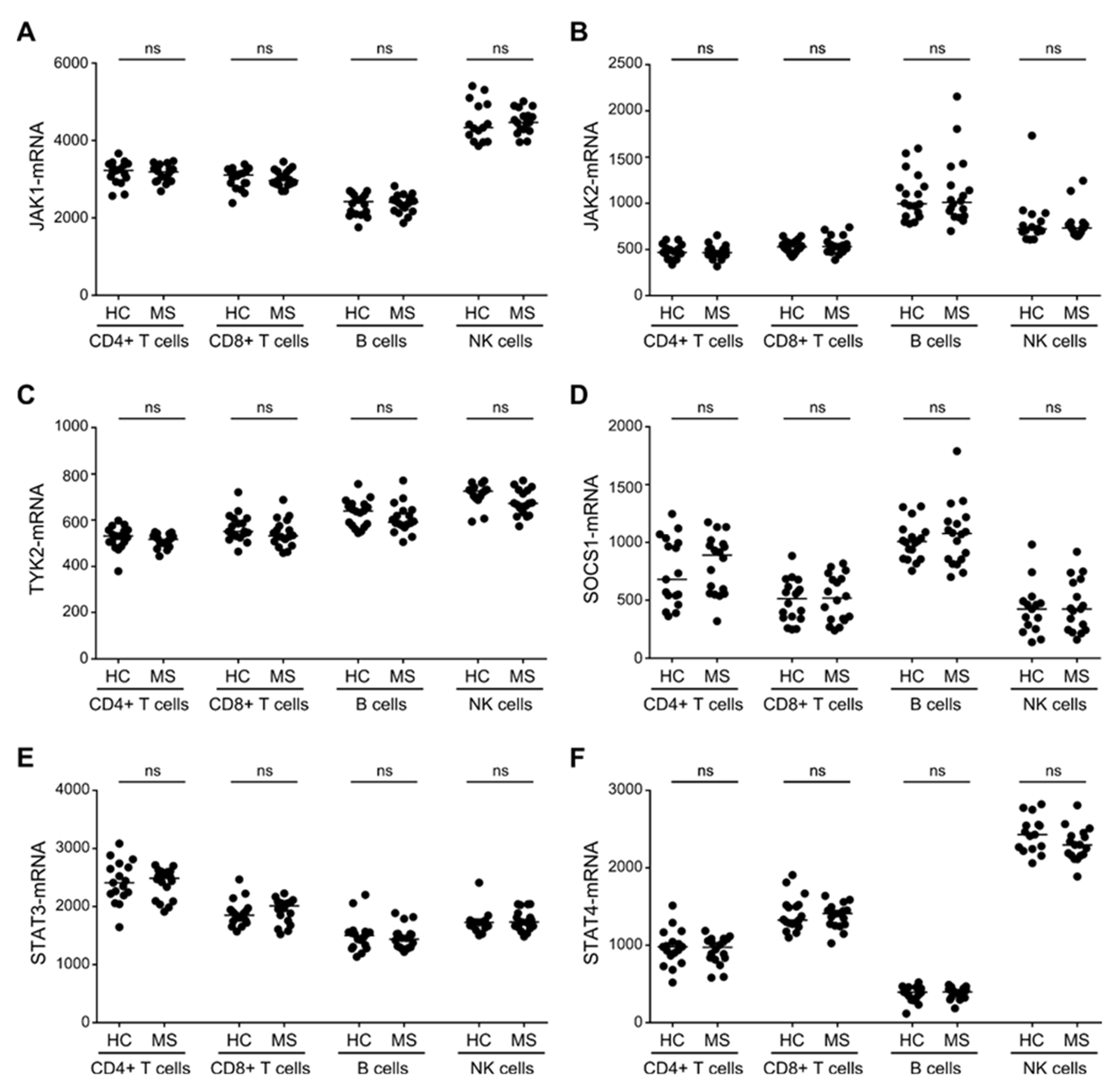
3.3. Similar Expression Levels of STAT3/4-Pathway Molecules in T, B and NK Cells between Patients with MS and Healthy Controls
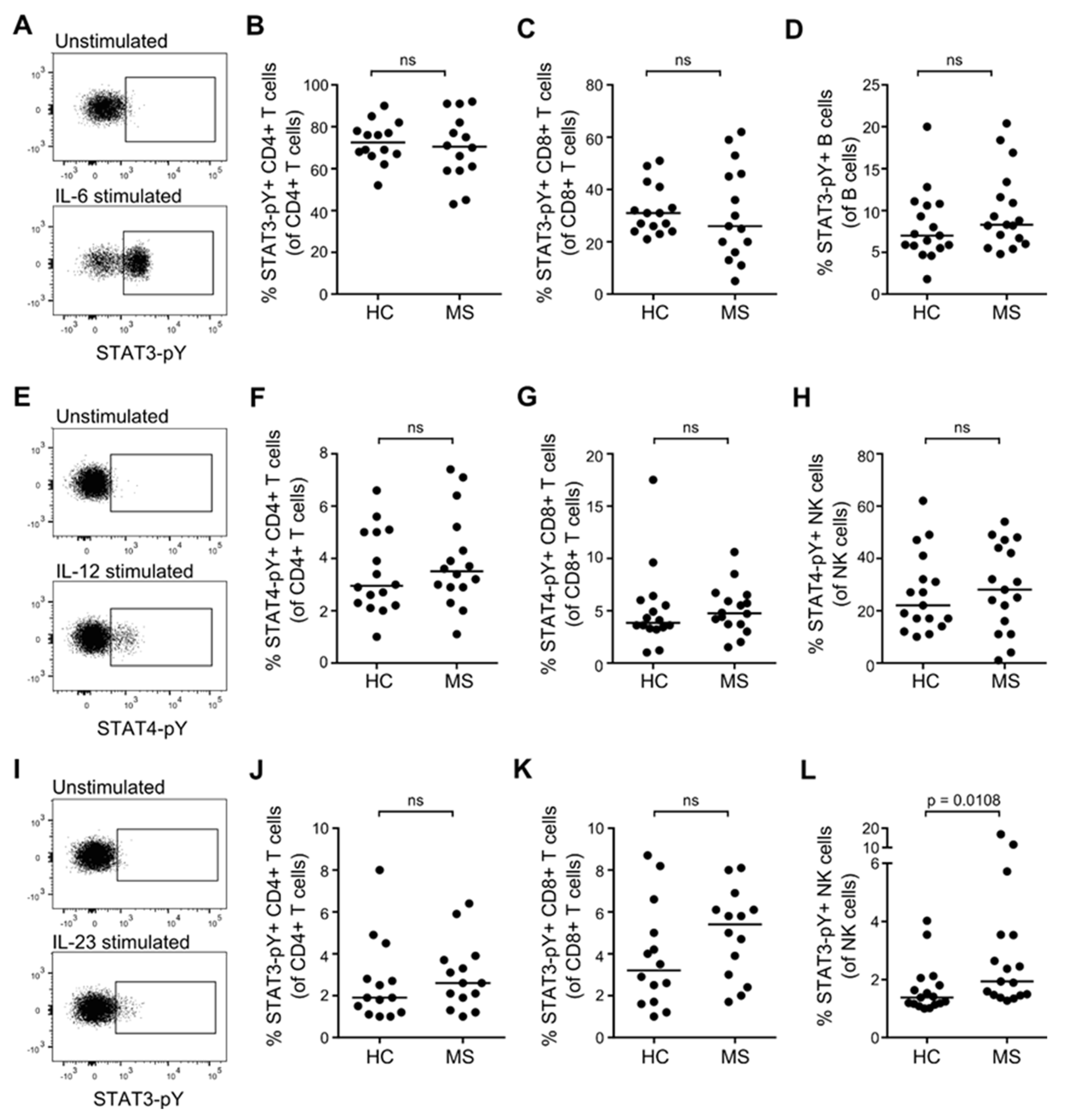
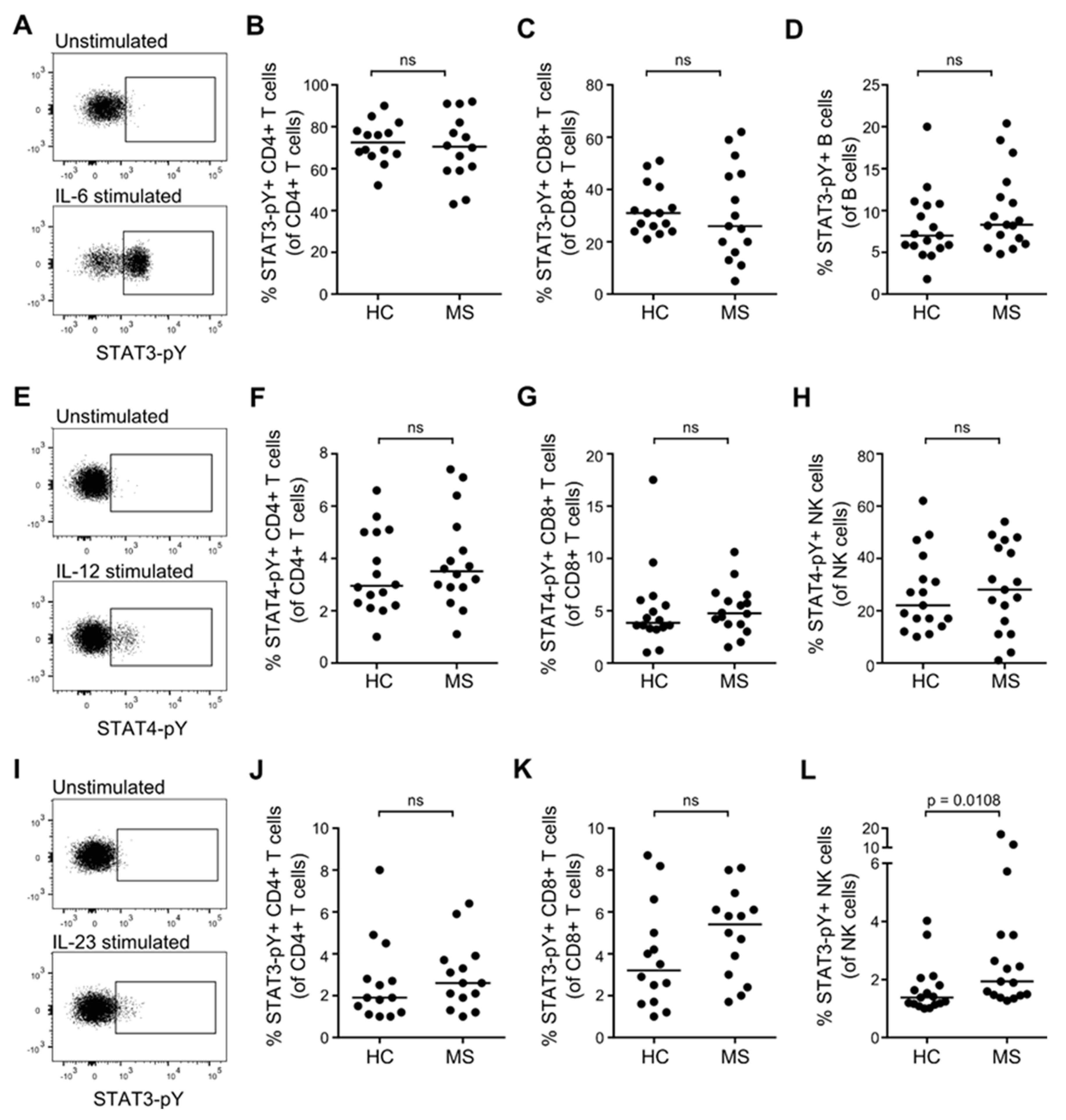
3.4. STAT Activation Induced by IL-6, IL-12, and IL-23 in Resting T, B and NK Cells from Patients with MS and Healthy Controls
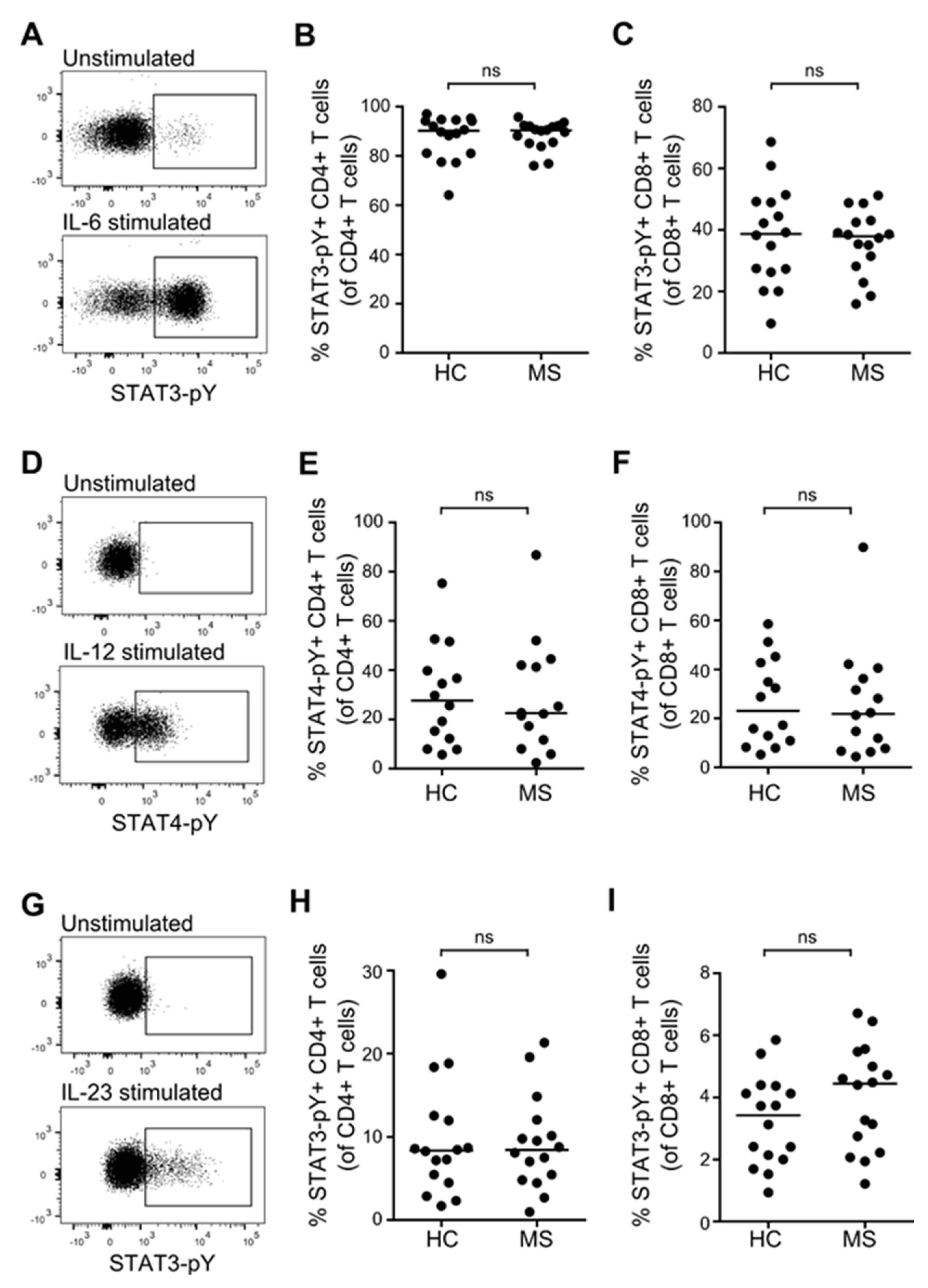
3.5. STAT Activation Induced by IL-6, IL-12, and IL-23 in Primed T Cells from Patients with MS and Healthy Controls
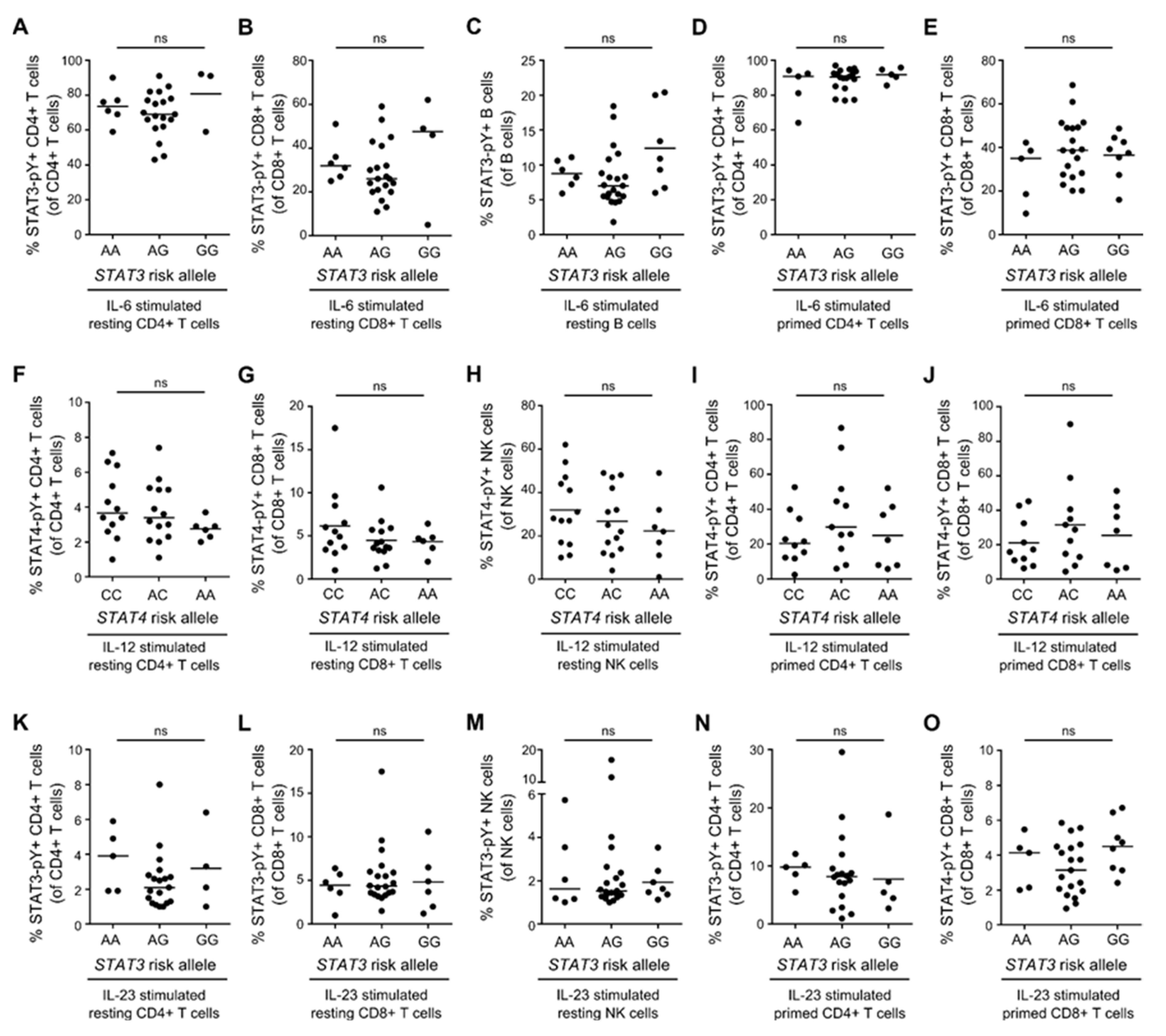
3.6. STAT3/STAT4 MS-Risk Alleles Are Not Associated with the Level of STAT3-pY/STAT4-pY
3.7. Association between IL-6, IL-12, and IL-23 Responsiveness of Primed CD4+ and CD8+ T cells
3.8. IL-6, IL-12, and IL-23 STAT-Pathway wGRS and Responsiveness of Primed T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [Green Version]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [Green Version]
- The International Multiple Sclerosis Consortium. The Multiple Sclerosis Genomic Map: Role of peripheral immune cells and resident microglia in susceptibility. BioRxiv 2018, 143933. [CrossRef]
- Lee, P.W.; Smith, A.J.; Yang, Y.; Selhorst, A.J.; Liu, Y.; Racke, M.K.; Lovett-Racke, A.E. IL-23R-activated STAT3/STAT4 is essential for Th1/Th17-mediated CNS autoimmunity. JCI Insight 2017, 2, e91663. [Google Scholar] [CrossRef]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.I.; Compston, A.; Edan, G.; Goodkin, D.; Hartung, H.P.; Lublin, F.D.; McFarland, H.F.; Paty, D.W.; Polman, C.H.; Reingold, S.C.; et al. Recommended diagnostic criteria for multiple sclerosis: Guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann. Neurol. 2001, 50, 121–127. [Google Scholar] [CrossRef]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2018. [Google Scholar] [CrossRef]
- Hagberg, N.; Joelsson, M.; Leonard, D.; Reid, S.; Eloranta, M.L.; Mo, L.; Nilsson, M.K.; Syvänen, A.C.; Bryceson, Y.T.; Rönnblom, L.; et al. The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-gamma production in T cells from patients with SLE. Ann. Rheum. Dis. 2018, 77, 1070–1077. [Google Scholar] [CrossRef]
- Zundler, S.; Neurath, M.F. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015, 26, 559–568. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef]
- Couturier, N.; Bucciarelli, F.; Nurtdinov, R.N.; Debouverie, M.; Lebrun-Frenay, C.; Defer, G.; Moreau, T.; Confavreux, C.; Vukusic, S.; Cournu-Rebeix, I.; et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain: A J. Neurol. 2011, 134, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 2008, 181, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, L.Y.; Ivarsson, K.; Fryknäs, M.; Rickardson, L.; Tobin, G.; Ekman, S.; Larsson, R.; Gullberg, U.; Nilsson, K.; Öberg, F.; et al. Stat1 activation attenuates IL-6 induced Stat3 activity but does not alter apoptosis sensitivity in multiple myeloma. BMC Cancer 2012, 12, 318. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Padberg, F.; Feneberg, W.; Schmidt, S.; Schwarz, M.J.; Körschenhausen, D.; Greenberg, B.D.; Nolde, T.; Müller, N.; Trapmann, H.; König, N.; et al. CSF and serum levels of soluble interleukin-6 receptors (sIL-6R and sgp130), but not of interleukin-6 are altered in multiple sclerosis. J. Neuroimmunol. 1999, 99, 218–223. [Google Scholar] [CrossRef]
- O’Gorman, C.; Lucas, R.; Taylor, B. Environmental risk factors for multiple sclerosis: A review with a focus on molecular mechanisms. Int. J. Mol. Sci. 2012, 13, 11718–11752. [Google Scholar] [CrossRef]
- Chen, Y.C.; Yang, X.; Miao, L.; Liu, Z.G.; Li, W.; Zhao, Z.X.; Sun, X.J.; Jiang, G.X.; Chen, S.D.; Cheng, Q. Serum level of interleukin-6 in Chinese patients with multiple sclerosis. J. Neuroimmunol. 2012, 249, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Holdbrooks, A.T.; De Sarno, P.; Rowse, A.L.; Yanagisawa, L.L.; McFarland, B.C.; Harrington, L.E.; Raman, C.; Sabbaj, S.; Benveniste, E.N.; et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J. Immunol. 2014, 192, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gibson, S.A.; Benveniste, E.N.; Qin, H. Opportunities for Translation from the Bench: Therapeutic Intervention of the JAK/STAT Pathway in Neuroinflammatory Diseases. Crit. Rev. Immunol. 2015, 35, 505–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet. Neurol. 2008, 7, 796–804. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Essen, M.R.; Søndergaard, H.B.; Petersen, E.R.S.; Sellebjerg, F. IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis. Cells 2019, 8, 285. https://doi.org/10.3390/cells8030285
von Essen MR, Søndergaard HB, Petersen ERS, Sellebjerg F. IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis. Cells. 2019; 8(3):285. https://doi.org/10.3390/cells8030285
Chicago/Turabian Stylevon Essen, Marina R., Helle B. Søndergaard, Eva R.S. Petersen, and Finn Sellebjerg. 2019. "IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis" Cells 8, no. 3: 285. https://doi.org/10.3390/cells8030285