The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases


1
State Key Laboratory of Silkworm Genome Biology, Southwest University, Beibei, Chongqing 400716, China
2
Engineering Research Center for Cancer Biomedical and Translational Medicine, Southwest University, Beibei, Chongqing 400716, China
3
Chongqing Engineering and Technology Research Center for Silk Biomaterials and Regenerative Medicine, Southwest University, Beibei, Chongqing 400716, China
*
Author to whom correspondence should be addressed.
Cells 2018, 7(12), 274; https://doi.org/10.3390/cells7120274
Submission received: 16 November 2018
/
Revised: 7 December 2018
/
Accepted: 14 December 2018
/
Published: 17 December 2018
(This article belongs to the Special Issue Mitochondrial Biology in Health and Disease)
{kind=link}
{kind=link}
Abstract
:Mitochondria are dynamic cellular organelles that consistently migrate, fuse, and divide to modulate their number, size, and shape. In addition, they produce ATP, reactive oxygen species, and also have a biological role in antioxidant activities and Ca2+ buffering. Mitochondria are thought to play a crucial biological role in most neurodegenerative disorders. Neurons, being high-energy-demanding cells, are closely related to the maintenance, dynamics, and functions of mitochondria. Thus, impairment of mitochondrial activities is associated with neurodegenerative diseases, pointing to the significance of mitochondrial functions in normal cell physiology. In recent years, considerable progress has been made in our knowledge of mitochondrial functions, which has raised interest in defining the involvement of mitochondrial dysfunction in neurodegenerative diseases. Here, we summarize the existing knowledge of the mitochondrial function in reactive oxygen species generation and its involvement in the development of neurodegenerative diseases.
1. Introduction
Mitochondria are important cellular organelles that control various vital physiological processes in the bodies of organisms. Being a major consumer of oxygen, mitochondria utilize approximately 98% of the total amount of inhaled oxygen. Mitochondria efficiently generate energy that is required for almost all types of cellular activities; this energy is also used to contract both voluntary and involuntary muscles [1,2]. Additionally, the produced energy is used to sustain ionic gradients across the plasma membrane, which are essential for the excitability of excitable cells, permitting the accumulation of secreted material into vesicles, and allowing for the vesicle fusion and cycling essential for the secretion of neurotransmitters [3]. Thus, a mitochondrial functional disorder causes different disorders, such as alterations in tissue functions that may manifest as disease, as well as major defects in tissue function that may lead to handicap or death. In fact, mitochondria are necessary in the bodies of animals not just to provide energy, but also to maintain other physiological activities of cells, such as cellular calcium signaling, and any defect in this process might lead to dysfunction and disease [4,5].
The respiratory chain in mitochondria is a greatly efficient system. It catalyzes alternating one-electron oxidation reduction reactions that predisposes each carrier of an electron to side reactions with oxygen. It has been shown that mitochondria are the major intracellular source of reactive oxygen species under normal physiological conditions [6,7]. According to estimates, 1–2% of total daily oxygen consumption goes to reactive oxygen species production, and a woman with an average weight of 60 kg generates 160–320 mmol of free radicals each day from cellular respiration, while a man of 80 kg produces almost 215–430 mmol per day [8].
Neurodegenerative diseases are a group of heterogeneous disorders with discrete clinical symptoms and genetic etiologies. The diseases are characterized by the progressive loss of physiologically or anatomically associated neuronal systems. Parkinson’s disease and Huntington’s disease are typical examples of neurodegenerative diseases [9]. In spite of this heterogeneity, mitochondrial dysfunction is considered to be a unifying basic mechanism involved in different types of neuronal degeneration. Mitochondrial dysfunction has widespread detrimental consequences for cellular functions; for instance, it leads to impaired energy generation, impaired cellular calcium buffering, the activation of phospholipases and proteases, nitric oxide synthase, and the production of reactive oxygen species [10]. Thus, they play a crucial role in ageing, and can directly interact with variety of specific proteins that are thought to be involved in genetic forms of neurodegenerative disorders [11,12]. Furthermore, there are several lines of evidence that suggest that mitochondrial dysfunction is linked with neurodegenerative disorders [10]. However, our understanding is limited regarding the different mechanisms by which mitochondrial dysfunction influences physiological functions. Here, our main focus is to review recent data on the role of mitochondria in reactive oxygen species and its deleterious impact on the progression of neurodegenerative diseases.
2. The Structure and Functions of Mitochondria
A mitochondrion is a double-membraned, semi-autonomous cellular organelle that is separated from the cytoplasm of a cell by the mitochondrial membranes. The outer mitochondrial membrane is spongy in nature, which allows for free cross-movement of small, uncharged molecules and ions through the porin [13,14]. However, larger molecules, particularly proteins, have to be transported inside the mitochondrion via special translocases [15]. The inner membrane is highly impermeant and forms the main barrier between the mitochondrial matrix and the cytosol. The space between the outer and inner mitochondrial membranes is called the intermembrane space and harbors a variety of different proteins, which seem to be involved in different physiological functions of the cell. The inner membrane has at least three morphologically discrete subregions, including cristae, cristae junctions, and boundary membranes. These structures alter their shape in response to the cell’s metabolic needs and stress [16,17].
The structure of cristae appears to differ greatly between different tissues, and the functional importance of these variations in the structure of cristae remains largely mysterious [4]. Different groups of researchers have attempted to generate detailed three-dimensional reconstructions of cristae and to model the bioenergetic consequences of altering the shape of their structure [18,19,20]. Some researchers have correlated the complex structure of cristae with their energy demands; for instance, Perkins et al. [18] compared the structure of cristae in the mitochondrion of rod and cone cells, and observed greater cristae connectivity, narrower crista junctions, and almost 3-fold more surface area for cristae membranes in cone cells compared to that in rods. Further, they suggested that, as cones and rods utilize a different bioenergetic signature, the aerobic ATP demand and production is higher in cones than in rods. Hence, cones use two different strategies to enhance their aerobic ATP production: increasing the number of mitochondria and increasing the surface area of cristae membranes. However, it must be noted that we still have little exact information regarding the functional importance of the complex features of, and structural variations in, mitochondria.
Mitochondria regulate various biological processes in a cell for its survival and precise functioning. They functionally control the production of energy, the electron transport chain, cell signaling, apoptosis, and programmed cell death, while their functional disorder has serious health consequences. Considering the involvement of mitochondria in the key processes of cells as well as their health impacts, researchers all over the world are attempting to explore the role of mitochondrial dysfunction in the pathogenesis of different diseases [3,21,22].
3. Mitochondrial Sites for the Generation of Free Radicals
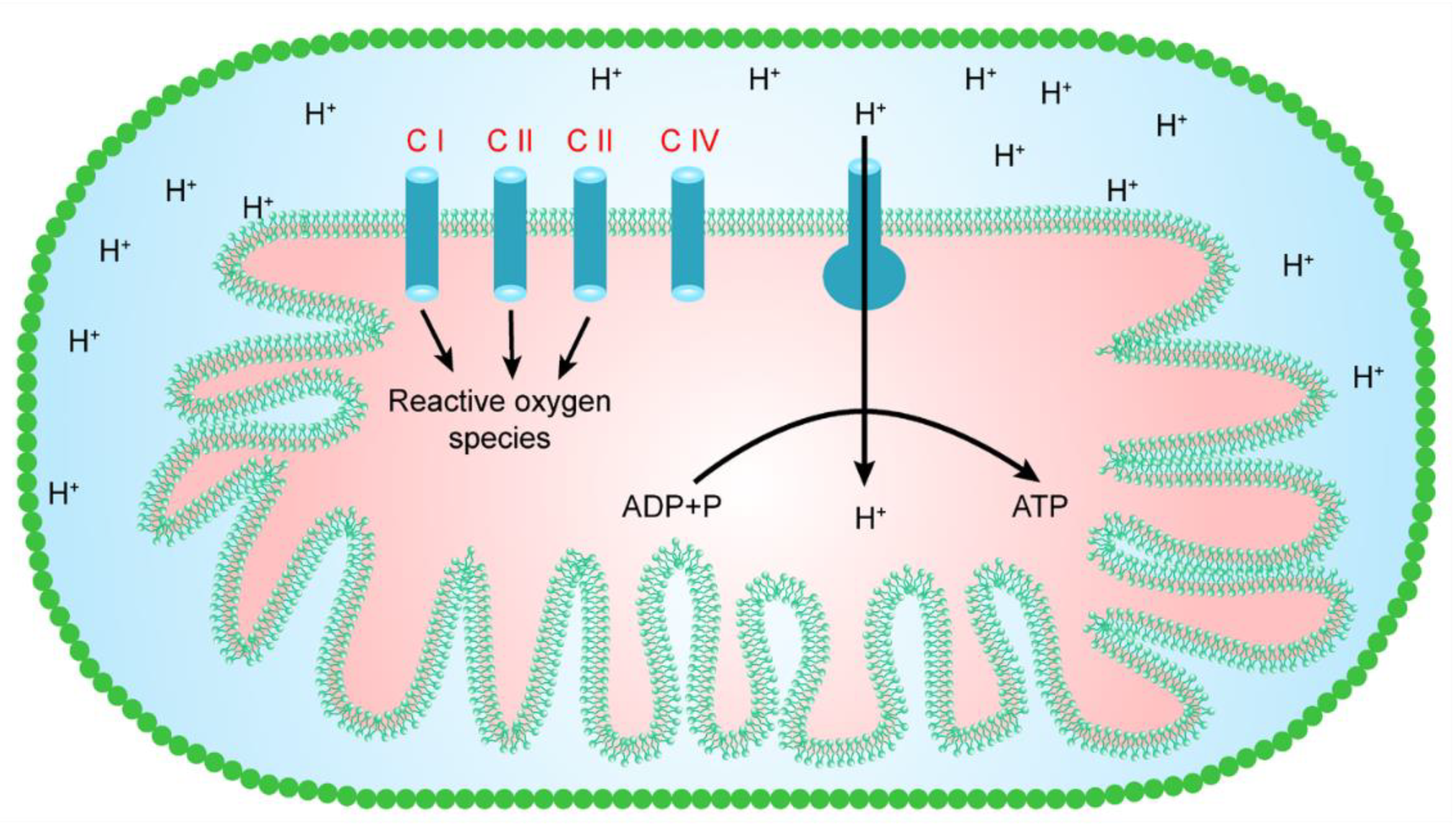
Reactive oxygen species are produced in various cellular compartments. However, mitochondria are a major contributor to reactive oxygen species, as they generate almost 90% of the total number of cellular reactive oxygen species [23]. Hence, mitochondria seem to represent one of the key sources of reactive oxygen species production in the majority of cell types. However, mitochondria in different cells/tissues may clearly differ in their capacity to generate free radicals using various substrates, and this capacity of the mitochondria may depend on the composition of a membrane, the age of an organism, and the species of animal. In a mitochondrion, at least eight sites are known to be involved in the production of reactive oxygen species. However, as shown in Figure 1, mitochondrial complexes I, II, and III mainly lie within the respiratory chain, and are thought to make a major contribution to the generation of reactive oxygen species [24,25]. Oxidative phosphorylation is the main process that produces unpaired electrons. These unpaired electrons interact with O2, resulting in the production of greatly reactive free radicals (superoxide ions). The superoxide ions are converted into other reactive oxygen species, such as H2O2 and hydroxyl ions (-OH) [4].
4. Complex I and the Generation of Free Radicals
Mitochondrial complex I, also known as NADH CoQ reductase, catalyzes the electron transfer from NADH to ubiquinone, which is accompanied by the movement of protons from the mitochondrial matrix to the intermembrane space [26,27]. During the 20th century, it was shown that mitochondrial complex I is involved in the generation of reactive oxygen species [24]. Additionally, it is believed that mitochondrial complex I is a key source of reactive oxygen species and is a major contributor to cellular oxidative stress. Hence, since then, an enormous literature has been developed on the mechanism by which reactive oxygen species are produced from complex I. For instance, the transfer of an electron at complex I may cause the production of reactive oxygen species, whereas rotenone, a complex I inhibitor, seems to suppress the generation of reactive oxygen species, presumably by preventing the electron’s passage further into the distal end where reactive oxygen species are produced [4,28]. Cadenas et al. [8] suggested that water-soluble co-enzyme Q homologs are utilized as electron acceptors in isolated complex-I-induced H2O2 formation; the production rate of H2O2 was partially prevented by rotenone, suggesting that water-soluble quinones react with oxygen when reduced at sites both downstream and upstream of the rotenone block. The one electron donor to oxygen in complex I is a non-physiological hydrophilic site [29,30] that reduces many quinones to their corresponding semiquinone forms, which are unstable and reduce oxygen to a superoxide. This mechanism is shared by several quinones, including anthracyclines [31], and the CoQ analog idebenone [32]. The hydrophilic, rotenone-insensitive site can apparently reduce oxygen to reactive oxygen species in the absence of intermediate acceptors [33]. Furthermore, many studies have reported that mitochondrial complex I is a major source of reactive oxygen species production in mitochondria [34,35] and localizes the oxygen-reducing site between the ferricyanide and the quinone reduction sites [36].
A recent study proposed two sites in a complex I reactive oxygen production model using the mitochondria of rat skeletal muscle. One site is in equilibrium with the NAD pool, seemingly the flavin of the FMN moiety, and the other site is dependent not only on the NAD redox state, but also on the proton motive force ∆p and the reduction state of the Q pool, seemingly a semiquinone in the Q-binding site [37]. Concurrently, Pryde and Hirst [5] devised a single mechanism of reactive oxygen species production by complex I under all conditions (during both NADH oxidation and reverse electron transfer) using bovine heart sub-mitochondrial particles. Generally, NADH-induced reactive oxygen species generation is prevented by complex I flavin-site inhibitors, but not by inhibitors of ubiquinone reduction and it is independent of the proton motive force (∆p). A reverse electron transfer by complex I in sub-mitochondrial particles, driven by the oxidation of succinate and the ∆p generated by ATP hydrolysis, reduces the flavin, leading to NAD+ and O2 reduction. However, similar to forward electron transport, reverse electron-transfer-stimulated reactive oxygen species generation is prevented by ubiquinone reduction and flavin site inhibitors. The potential dependence of NADH-stimulated reactive oxygen species generation (set by the NAD+ potential) matches with that of reverse electron-transfer-stimulated reactive oxygen species generation (set by the succinate potential and ∆p), and they both match the potential dependence of the flavin. Hence, the study suggested that NADH- and reverse electron-transfer-stimulated reactive oxygen species are generated by the flavin.
While mitochondrial complex I has been shown to be a key source of reactive oxygen species, and also a remarkable contributor to cellular oxidative stress in mitochondria, its ability to produce reactive oxygen species may vary under different oxidation–reduction pressure conditions [38]. In particular, a study on bovine heart mitochondria showed that the concentration and ratio of NAD+ and NADH regulate the production rate of reactive oxygen species [39]. For instance, at a lower NADH level, rotenone seemed to prevent the generation of reactive oxygen species in coupled sub-mitochondrial particles from the heart of beef. So, this phenomenon excludes a crucial function of complex I in reactive oxygen species under a lower reduction state of the iron–sulfur clusters of complex I. However, at a higher NADH level, rotenone induced reactive oxygen species generation. Recently, it was discovered that, with the exception of the concentration and ratio of NAD+ and NADH, many other factors also control the production of reactive oxygen species. For example, in reverse electron transport, the generation of reactive oxygen species is influenced by changes in the concentration of O2, the magnitude of the proton motive force, and the redox states of the co-enzyme Q (CoQ) and NADH pools [35]. Such a condition simulates the state of reduction of complex I when the respiratory chain is impaired.
Collectively, mitochondrial complex I generates large amounts of free radicals using two biological mechanisms: (1) a high ratio of NAD+/NADH leads to a reduction of the FMN site on complex I; and (2) electron transport to the CoQ pool coupled with a high protonmotive force ∆p leads to reverse electron transport. Although the site at which free radicals are generated during reverse electron transport has not been identified, the rate at which free radicals are generated under reverse electron transport appears to be the highest that can occur in mitochondria [37,39]. Furthermore, the mechanisms underlying the generation of free radicals that have been developed by the sub-mitochondrial particles, the isolated enzyme, and the coupled membrane vesicles need to be tested on mitochondrial complex I.
5. Complex II and the Generation of Free Radicals
The question of whether complex II, which is also known as succinate dehydrogenase, is a major source of reactive oxygen species remained controversial for many decades. Previous studies on isolated mitochondria and cells showed that complex II was not recognized as a remarkable contributor to reactive oxygen species until it experienced mutation [6,40]. Recently, the generation of reactive oxygen species from complex II of mitochondria has gained extensive scientific interest. Since then, many studies executed on sub-mitochondrial particles and intact isolated mitochondria have demonstrated that complex II, under specific conditions, and particularly when it is supplied with a high succinate concentration (almost 5 mM), produces a considerable amount of reactive oxygen species [41,42]. Interestingly, a recent study on skeletal muscle mitochondria of rats demonstrated that complex II produces reactive oxygen species in both the forward and reverse reactions as well as with a low and a high concentration of succinate [43,44]. Additionally, the authors suggested that complex II generates reactive oxygen species in the reverse reaction, for instance when electrons are provided from the reduced ubiquinone pool, and in the forward reaction, for instance when the electrons are supplied from succinate. Furthermore, they proposed a mechanism for the production of reactive oxygen species through complex II that entirely depends upon its possession of the carboxylate binding site and the enzyme reduction state; e.g., reactive oxygen species are produced when the binding site for carboxylate is not occupied and the flavin is reduced. Finally, it has been demonstrated that the complex-II-associated rates approach or exceed the maximum rates of complexes I and III, indicating that complex II may be an important contributor to the physiological and pathological generation of reactive oxygen species [24].
It has been shown that a functional loss of mitochondrial complex II can lead to succinate accumulation and enhanced reactive oxygen species production in cells [45]. This appears to particularly be the case for mutation in the subunits of complex II, and it is believed that it is associated with certain diseases. Mitochondrial complex II contains four subunits: SDHA, SDHB, SDHC, and SDHD. Mutation is mostly associated with SDHB, SDHC, and SDHC, while mutation in SDHA rarely occurs. A loss of SDHB, SDHC, and SDHD would allow for the acceptance of an electron, but not progression along the respiratory chain, and consequently may enhance the production of reactive oxygen species [46]. Ishii et al. [47] suggested that a mutation in the SDHC subunit of complex II in fibroblasts of a transgenic mouse and conditional transgenic mice stimulates dysfunction of the respiratory chain in mitochondria and enhances reactive oxygen species’ generation. Likewise, in a study on hamster fibroblasts, the authors observed that SDHD is also responsible for the production of reactive oxygen species [48]. Further, a mutation in SDHC of complex II triggered hypersensitivity that enhanced the concentration of oxygen in Caenorhabditis elegans, and stimulated the production of reactive oxygen species that was found in succinate sub-mitochondrial particles and fueled mitochondria from the mutant Caenorhabditis elegans [49,50].
To summarize, a growing amount of evidence suggests that mitochondrial complex II is an important modulator of reactive oxygen species generation under different physiological conditions. Hence, it can adapt different functions as a generator or regulator of reactive oxygen species depending on the supply of substrate, etc. [45,46]. Complex II’s biological functions need a rigorous analysis to identify the source of reactive oxygen species. This will be useful to provide a clear understanding of complex II’s functions in various pathophysiological conditions. Furthermore, the basic mechanisms by which mitochondrial complex II generates reactive oxygen species should be further studied in various model organisms.
6. Complex III and the Generation of Free Radicals
Complex III is an important multi-subunit, membrane-bound enzyme that is essential to respiratory energy transduction pathways in various organisms. Apart from energy production, it is known to contribute a considerable number of reactive oxygen species [51,52]. A remarkable advancement has been made in our understanding of complex III’s biological role in the production of reactive oxygen species. Various studies have described the mechanism by which complex III generates reactive oxygen species. According to these studies, it can produce reactive oxygen species by both a forward and a reverse electron transfer.
The first report regarding the involvement of complex III in reactive oxygen species generation surfaced in 1970s. The study explored the role of complex III in reactive oxygen species production using mitochondria from a rat’s heart. It showed that antimycin A, an oxidative phosphorylation inhibitor, cannot entirely prevent electron transport from ubiquinol to cytochrome c: the antimycin-A-insensitive reduction of cytochrome c is mediated by reactive oxygen species [53]. Another study proposed a mechanism for electron transport and reactive oxygen species generation from complex III, and suggested that two electrons from ubiquinol oxidation would be transported to the cytochrome c, but by using two pathways. One of the electrons would be transferred to cytochrome c through the chain reactions such as from quinol oxidation center to the iron–sulfur protein and finally to cytochrome c1, while the second one would be delivered to O2 for the production of reactive oxygen species. Additionally, preventing the quinone reduction at the quinone reduction (Qi) center using inhibitor molecules (e.g., antimycin A) remarkably enhanced quinol oxidation (Qo) center mediated reactive oxygen species generation [54]. Further, loss of iron–sulfur protein abolishes reactive oxygen species generation, whereas the loss of cytochrome b retains production of reactive oxygen species [55,56]. Many studies investigated the impact of particular Qi and Qo center inhibitors e.g., antimycin A, myxothiazol, on reactive oxygen species generation described complementary reactions in detail [57]. For example, declining electron transfer rate between the hemes b H and b L of cytochrome b, or eradicating the following oxidation of both the hemes by the Qi center inhibitor antimycin A, caused accumulation of electrons on cytochrome b [58]. This resulted in the accumulation of semiquinone radical at the Qo center and to the escape of electrons to O2 to produce reactive oxygen species [57]. If a semiquinone radical is formed at the Qo center during the normal turnover of a complex III, reactive oxygen species generation is likely to be relatively low to reduce electron leakage and energy wastage. However, reactive oxygen species generation at the Qo center possibly become remarkable under specific conditions, such as a highly reduced quinone pool and presence of inhibitor molecules (e.g., antimycin A). These abnormal conditions may occur only in extreme physiological conditions and in situation of complex III damage [59,60]. A recent study proposed a mechanism for generation of reactive oxygen species, when a complex III undergoes damage during disease resulted in the opening of permeability transition pores, consequently malate/succinate-fueled mediated generation of reactive oxygen species from complex III due to activation of malic enzyme through increases in matrix [Mg2+], [ADP] and [NAD+]. Further generation of reactive oxygen species in these physiological conditions is related to Mg2+ dependent NADH production by malic enzyme. For maximal production of reactive oxygen species, the production rate of NADH has to be almost equal or below that of NADH oxidation, as further increase in NADH elevate ubiquinol-related complex III reduction beyond the optimal range for reactive oxygen species generation [61].
The production of reactive oxygen species by reverse electron transfer pathway has gained attention in the current decade. Consequently, many studies were conducted to explore the mechanism and rate of reactive oxygen species generation by this pathway. Most of these studies suggested that partial oxidation of the quinone pool in a physiologically relevant condition remarkably enhances the rate of reactive oxygen species generation by antimycin A inhibited complex III [62,63]. Another study on sub-mitochondrial particles showed that complex III mediated reactive oxygen species generation is higher, when complex II activity is inhibited by its inhibitors (i.e., oxaloacetate or malonate), linking quinone pool redox state to reactive oxygen species generation by the Qo center of complex III. They inferred that produced reactive oxygen species was generated at the Qo center via reverse electron transfer from reduced heme b L of cytochrome b to O2 by semiquinol [64]. Afterwards, Quinlan et al. [42] argued that this effect is perhaps due to the redox state of hemes b H and b L of cytochrome b, which are highly sensitive to the membrane potential and redox state of quinone pool. Similarly, studies using bacterial model provided evidences that reactive oxygen species generation at the Qo center also used reverse electron transfer from reduced heme b L of cytochrome b, later on Sarewicz et al. [7] proposed a mechanism that shows transport of the Fe/S protein cluster from the Qo center enhanced reactive oxygen species production, while its stagnation reduced the production at Qo center.
Taken together, the exact mechanism for production of reactive oxygen species at the Qo center has still remained to elucidate and is currently a matter of debate. Furthermore, remarkable amount of reactive oxygen species from the Qo center have been measured under non-native conditions, for example, in the presence of antimycin A (inhibitor of the Qi center) [53,57]. Therefore, the basic question that remains to be explored is which physiological processes or factors promote production of reactive oxygen species from the Qo center in vivo.
7. Other Cellular Components and Free Radicals
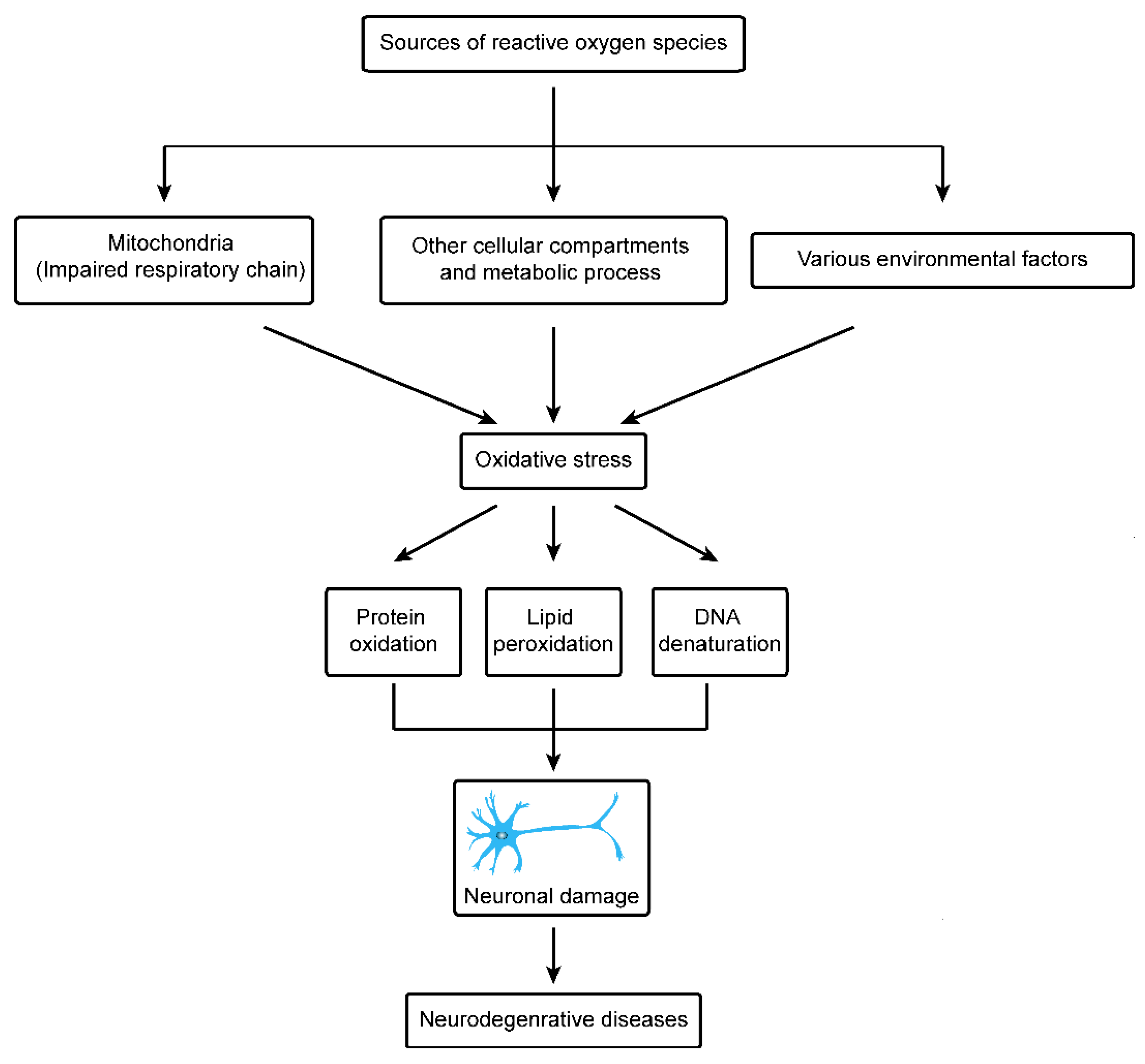
There is increasing evidence that besides mitochondria, cellular components and other factors are also involved in the production of reactive oxygen species. Peroxisome, endoplasmic reticulum phagocytic cells, neuro-inflammation, dopamine, genetic mutation, anti-oxidant depletion etc. may induce the reactive oxygen species in cells. Thus, contributing to degeneration of neurons by lipid peroxidation, DNA and protein oxidation [65,66].
8. Environmental Sources of Oxidative Stress and Neurodegenerative Disorder
Besides the endogenous factors, exogenous sources of reactive oxygen species also contribute in the pathogenesis of neurodegenerative diseases including Parkinson’s and Huntington’s disease etc. Multiple lines of evidence suggest the etiology of neurodegenerative disorders is multifactorial and comprised of an interaction between environmental factors and genetic predisposition. Migliorea and his coworkers [67] reviewed that the environmental exposure to air pollution, metals, herbicides, insecticides and diet are the major risk factors in pathogenesis of neurodegenerative diseases by stimulating oxidative stress. Hence, to understand the pathogenesis of neurodegenerative disorders exogenous factors are also equally important, however, they are beyond the scope of the present review.
9. Physiological Functions of Mitochondria in Neurodegenerative Disease
In recent years, important advancements have been made in the field of medical science to understand initiating factors and cure of neurodegenerative diseases, however still it is a major cause of concern in the health profession. Neurodegenerative diseases can be divided into Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, injury to the central nervous system by chronic low-grade hypoxia, motor neuron diseases, Wilson’s disease, Huntington’s disease and Freidreich’s ataxia [9]. Exact knowledge on the pathophysiological mechanisms is still insufficient and unclear in all of these neurodegenerative diseases. However, functional disorders of mitochondria have been reported to be a major cause in pathogenic process. A key challenge of modern neuroscience is to investigate and understand the level to which these variations in the functions of mitochondria depict the level (primary or secondary components) of the pathophysiological process, further to understand the fundamental biological pathways which lead to the initiation and development of neurodegenerative disease. A recent study suggested that mitochondria play critical functions in most neurodegenerative disorders. The mitochondria are mainly involved in ATP generation, reactive oxygen species generation, antioxidant activity and Ca2+ buffering. Neurons being high energy demanding cells are closely related to maintenance, dynamic activity, and functions of mitochondria. In most neurodegenerative diseases, mitochondrial dynamics and activities are impaired, causing low ATP production, high levels of reactive oxygen species, and apoptosis [68]. In this article our main focus is to review recent data regarding Huntington and Parkinson’s disease and the role of mitochondria in progression of these neurodegenerative diseases, particularly through generation of reactive oxygen species (Figure 2).
10. Mitochondria and Huntington’s Disease
Huntington’s disease is a detrimental neurodegenerative disorder, and people from the United States of America and Australia are highly affected by this disease, while a lower number of cases are also reported from China, Africa, Japan and other countries [69,70]. This disease is characterized by the irregular expansion of CAG trinucleotide repeats in exon 1 of the gene, which resides on the chromosome 4 and translates into an abnormal huntingtin protein. Healthy individuals usually contain 6 to 35 repeats of CAG, while this number is observed more than 36 repeats in patients [71]. Medically, Huntington’s disease is caused by dysfunction of motor neurons, psychiatric disturbances, and cognitive decay, consequently causing the patient to experience progressive muscle loss and brain dysfunction [72,73]. This disorder is commonly fatal within 15 to 20 years after disease onset.
Emerging evidence suggests that mitochondrial dysfunction is associated with the pathogenesis of Huntington’s disease. Panov et al. [74] observed mitochondria extracted from lymphoblasts of Huntington’s disease patients contain lower mitochondrial membrane potential compared with the control. Likewise, they found similar characteristics in mitochondria isolated from the brains of transgenic mice that transcribe mutant huntingtin protein. This physiological defect led the initiation of behavioral and pathological abnormalities. Based on these observations, they concluded that calcium abnormalities of the mitochondria arises at the onset of Huntington’s disease pathogenesis and probably is the direct influence of the huntingtin protein mutation on the tissues and organelle; however, it remains undiscovered exactly how the huntingtin mutant protein elicits its harmful effects. Mattson et al. [75] reviewed that mitochondrial-mediated oxidative stress, perturbed homeostasis of Ca2+, and apoptosis contribute to the onset of Huntington’s disease.
A recent study further highlighted this mechanism and demonstrated that the functional impairment of mitochondria and dysregulation of transcription are involved in Huntington’s disease pathogenesis. For example, Cui et al. [76] suggested that the mutant huntingtin protein causes perturbation of mitochondrial functions by preventing expression of PGC-1α, which controls different metabolic processes, including respiration and mitochondrial biogenesis. Crossbreeding of PGC-1α knockout mice with Huntington’s disease knock in mice lead to enhanced neurodegeneration of striatal neurons and motor abnormalities in mice with Huntington’s disease. Whereas, expression of PGC-1α partially reverses the toxic effects of mutant huntingtin in cultured striatal neurons.
11. Mitochondria and Parkinson’s Disease
Parkinson’s disease is one of the most predominant neurogenerative diseases. Clinically, it is characterized by apoptotic loss of dopaminergic neurons, resulting in subsequent and gradual loss of muscle control. This disease is diagnosed by resting tremors, rigidity of muscle, change in gait and speech, postural instability, depression, fatigue, anxiety, sleep disturbances, and decline in dementia and cognition. Premature death of patients occurs due to complications e.g., pneumonia, injuries etc. [77]. Parkinson’s disease is prevalent in the adult population, usually in people over 65 years. The male population is approximately 1.5–2 times more susceptible to this disease than the female population [78].
There are multiple lines of evidence that link functional disorders of mitochondrial complex I and dopaminergic neurons in Parkinson’s disease [79]. A most appealing model of this disease was discovered in the 20th century, which described how MPTP (1-methyl-phenyl 4-phenyl-1,2,3,6-tetrahydropyridine) of the mitochondria is responsible for Parkinson’s disease [80,81]. Later on, several researchers independently discovered that sporadic Parkinson’s disease patients have reduced complex I in different brain areas, peripheral cells and neural and extra neural tissues and in cells (cytoplasmic hybrid), which are derived from Parkinson’s disease patients [70,82,83]. Another study on hybrid cell lines showed that complex I deficiency is correlated with the increased reactive oxygen species generation [84]. Further, many studies based on model organs and human neuroblastoma cells showed that in some patients, the disorder may reflect exposure to the plant extracted insecticide rotenone (complex I inhibitor) [85,86]. Exposure to low doses of rotenone induce apoptosis in human neuroblastoma cells. While, rotenone inhibition stimulates aggregation and accumulation of ubiquitin, α-synuclein, gradual oxidative damage and caspase-dependent cell death, which is the central mechanism in Parkinson’s disease pathogenesis. The binding site for rotenone appears irrefutably defined as mitochondrial complex I. It seems injury of cells is not a simple role of metabolic machinery inefficiency, as similar levels of ATP suppression stimulated by other poisonous chemicals such as glycolysis inhibition by 2-deoxyglucose completely failed to cause approximately the same cell injury. Rather, cells protect themselves using their antioxidant protection system, suggesting that oxidative stress is the primary mechanism for cell injury. Mitochondrial complex I is particularly susceptible to modification caused by oxidative stress, and thereby is a potential producer of reactive oxygen species [85].
A recent study on cell lines, human brain tissues and mice suggest that mutations in α-synuclein is associated with the pathogenesis and progression of Parkinson’s disease. Further, point mutations in α-synuclein are responsible for its decreased association with mitochondria associated membranes, coincident with a lower degree of apposition of endoplasmic reticulum with mitochondria, a reduce in mitochondria associated membranes function and increase in fragmentation of mitochondria [86]. Mutations in Parkin and PINK1 also contribute in the progression of different types of Parkinson’s diseases. The Parkin gene is highly expressed in brain tissues including the substantia nigra, and contains 12 exons, of which five (exons 3–7) are generally deleted and cause the pathogenesis and progression of disease [87]. Likewise, PINK1 (PTEN-induced kinase 1) is a mitochondrially-located molecule and has a protective impact on a cell. A mutation in its kinase domain can make cells susceptible to oxidative stress, and is thereby involved in the progression of disease [88].
Besides the above-mentioned factors, many others have been reported to be involved in dysfunction of mitochondria associated membranes. However, these factors were identified in other brain associated disorders, which is beyond the scope of our review. In short, c-secretase activity itself is highly enriched in a sub-compartment of the endoplasmic reticulum that is biochemically and physically connected to mitochondria. Mutation in the catalytic components of c-secretase (Presenilin-1 and -2) increase mitochondria associated membranes functions and communication between endoplasmic reticulum and mitochondria, which is the prominent characteristic of the familial and sporadic forms of Alzheimer disease. These results will help to understand calcium deregulation, mitochondrial dysfunction and oxidative stress in this disease and will also help to explore the contribution of this mutant in other brain associated diseases [89]. Similarly, Sigma 1 receptor plays a crucial biological role in the protection of motor neurons and its mutation lead to degeneration of motor neurons and caused Amyotrophic lateral sclerosis. A recent study reported that mutation occurs in highly conserved amino acid reside in the sigma receptor of SIGMAR1 gene. The neuronal cells which express this mutant protein are less resistant to apoptosis stimulated by cellular stress [90]. Here, we garnered different contributing factors which are responsible for neurodegenerative diseases e.g., Alzheimer disease and that should be the focus of future studies to determine the biological role of these factors in brain associated diseases.
On the whole, cellular viability depends on the biological functions of mitochondria, and alterations in its biological functions can lead to cell functional abnormality and even cell death. Neuronal cells are especially susceptible to mitochondrial dysfunction due to their dependence for energy requirement on the mitochondrial metabolism. Dysfunction of mitochondrial respiration has been demonstrated to be involved in the pathogenesis of neurodegenerative diseases. However, currently the biological functions of mitochondria in neurodegenerative diseases appear to extend well beyond abnormalities in respiration. Although it remains to be explored whether alterations of mitochondria in neurodegenerative diseases constitute a primary or a secondary event, or are just part of a larger multifactorial pathogenic process.
12. Oxidative Stress and Parkinson’s Disease
The brain is the central organ in living organisms and is a major consumer of oxygen to provide uninterrupted supply of energy to approximately 86 billion neurons for their daily activities, it has been estimated it uses almost 20% of the basal oxygen (O2) from the total oxygen supplied to the human body [91,92]. This uptake of oxygen is utilized by mitochondria in respiratory chain to generate energy/ATP and reactive oxygen species are produced as by-product other than energy production, which is a major cause of DNA damage and one of the eminent characteristics of Parkinson’s disease [93,94].
Growing evidence suggests that oxidative metabolism has extensive functions in the Parkinson’s disease, as defective respiratory chain and mutation of mitochondrial DNA in the dopaminergic neurons are the most common features of Parkinson’s disease patients [95,96]. In the neurons of patients, metabolism of dopamine is greatly linked to oxidative stress and its biochemical degradation produces reactive oxygen species including hydrogen peroxide (H2O2) and other metabolic products. As the H2O2 is highly membrane permeable, these products enter into adjacent neurons where it has ability to interact with Fe2+ to hydroxyl radicals [97,98]. The remarkable elevation of iron in the redox active form in the dopaminergic neurons plays a key function in pathogenesis of Parkinson’s disease [99]. A recent study proposed a mechanism for iron accumulation in the brain neurons; functional disorder of mitochondria not only enhance reactive oxygen species generation but also decrease synthesis of iron–sulfur cluster and unorthodox activation of Iron Regulatory protein 1, a key regulator of iron homeostasis in a cell. This protein stimulates the accumulation of iron and hydroxyl radicals in a cell [100]. Iron ions can produce reactive oxygen species since ferrous ions (Fe2+) and ferric ions (Fe3+) can react with hydrogen peroxide and superoxide, respectively, in a chain reaction producing the hydroxyl free radical which together with oxidation of dopamine can induce neurotoxicity [101,102]. The iron accumulation in different areas of brain particularly in the dopaminergic neurons have been reported in the Parkinson’s disease, which suggest that iron mediated lipid peroxidation plays a major biological role in the development and progression of this disease [103,104]. For instance, the accumulation of byproducts of lipid peroxidation have been observed in the cerebral spinal fluid and serum of the patients [105]. A recent study suggested that the neurodegenerative process is increased, when neuro-melanin containing organelles accumulate high load of toxins and iron during aging. Additionally, the release of neuromelanin from degenerating neurons stimulates microglia and latter cause neurons death with further release of neuromelanin initiates a self-propelling mechanism of neuro-inflammation and neurodegeneration [106].
It has been shown with the onset of Parkinson’s disease metabolism of lipid peroxidation and proteins oxidation is increased in the dopaminergic neurons, and antioxidant level is also decreased. Additionally, thiobarbituric acid, malondialdehyde, and 4-hydroxy-2,3-nonenal level is also elevated in the substantia nigra of the patients [107,108]. Later, two-fold increase in protein oxidation in the substantia nigra was observed [109,110]. Furthermore, decrease in glutathione contents in human brain enhances the level of hydroxyl radicals in the patients [111]. Likewise, several experimental studies on human and model organisms suggest that glutathione peroxidase activities and glutathione contents are significantly decreased in the brain [112,113]. Multiple evidences, suggest that reduced activities of enzymatic and non-enzymatic antioxidants particularly in the dopaminergic neurons play crucial role in the progression and development of this disease [114,115,116]. Another study demonstrated that decrease in glutathione level and increase in oxidized glutathione contents is a common feature in the patients, whereas glutathione level is also decreased in the substantia nigra, due to loss of neurons [117].
13. Oxidative Stress and Huntington’s Disease
Besides major advancements in the field of molecular biology and medical sciences, the precise mechanism of neuronal death in Huntington’s disease remained mysterious. However, numerous reports suggest that oxidative stress play a major role in the development and progression of the disease [118,119]. In fact, susceptible brain neurons in the disease may not be able to cope the conditions of increasing reactive oxygen species. The higher level of reactive oxygen species may trigger intracellular cascades of oxidative stress through oxidizing DNA and protein, and inducing peroxidation of lipids in plasma membrane [120,121]. Mutant huntingtin proteins affect the mitochondrial activities by stimulating the opening of the mitochondrial permeability transition pore with the concomitant release of cytochrome c and the stimulation of the apoptotic mitochondrial pathway. This abnormal interaction also changes calcium buffering of mitochondria, further deteriorating mitochondrial disorder and enhancing reactive oxygen species production [122,123]. This mutant huntingtin protein interacts with Drp1, elevates GTPase Drp1 enzymatic activity, enhances abnormal dynamics of mitochondria and consequently, defective synaptic deficiencies and anterograde mitochondrial movement [124].
Although the oxidative damage has not been detected in the early stages of disease, yet it is assumed to have a crucial role in the progression of Huntington’s disease [125]. Many researchers observed a changed expression pattern and activity of nitric oxide synthase, antioxidants, and ascorbate in R6/2 HD and R6/1 transgenic mouse [126,127]. Reactive oxygen species generation is also enhanced in the striatum of transgenic mice [128], whereas, a recent study observed increase in lipid peroxidation in different mouse models [129]. Furthermore, decreased expression of antioxidants has also been detected in the model species, while increase of these antioxidant enzymes reduce the toxic effects of mutant huntingtin proteins in cultured neurons [130].
In the late stages of disease, level of oxidative stress plays a crucial role in the progression of Huntington’s disease. Mitochondrial dysfunction and impaired respiratory chain have been thought to involve in the reactive oxygen species mediated Huntington’s disease pathogenesis [131,132]. Brain tissues of the patients show a higher level of dysfunction in the components of oxidative phosphorylation [128]. Elevated oxidative stress markers and depletion of antioxidants in brain and peripheral tissues have also been observed in the patients [133]. Increase in reactive oxygen species in the patients causes the denaturation of other biomolecules, for instance, it has been proposed that reactive oxygen species mediated mitochondrial dysfunction, links with defective glucose metabolism in Huntington’s disease patients [134,135]. Another study suggested that the increase in reactive oxygen species level enhance lipid peroxidation and damage the cellular membranes, further, the higher lipid peroxidation is associated with lower glutathione content in Huntington’s diseases patients compared with the healthy subject [132,136]. A recent study on mice model organism proposed that oxidative stress greatly denature the DNA, and this denaturation accumulates at CAG repeats in a length dependent manner. Similarly, the rate of DNA damage is increased under the relevant physiological condition, for example reactive oxygen species mediated decrease in protein level and depletion of major base excision repair enzymes [137]. Later on, comparative study of striatal cells derived from Huntington’s diseases knock-in mice expressing mutant hunting versus wild cells indicated that higher level of reactive oxygen species greatly damages non-enzymatic antioxidants along with the other proteins [138]. Additionally, reactive oxygen species mediated abnormal metabolism of tryptophan further worsen the ongoing brain functional disorder [139,140,141].
Altogether, the mechanisms implicated in the pathogenesis and progression of Huntington’s and Parkinson’s disease have not been fully understood. However, there is increasing evidence that oxidative stress is one of the key events in the pathogenesis of these diseases. Apart from oxidative stress, failure of antioxidant enzymes in neurodegenerative disease patients plays a crucial role in neuro-degeneration. In the human brain, reactive oxygen species are mainly produced by mitochondrial dysfunction, dopamine metabolism and inflammation of neurons. Thus, the different protective mechanisms implicated in the modulation of these biological processes are an interesting area of research focus in current years. Moreover, the study of Huntington’s and Parkinson’s disease related proteins in combination with experimental research using model organism has yielded substantial insights into the molecular pathways of neuro-degeneration and highlighted previously unidentified mechanisms by which oxidative stress contributes to these diseases.
14. Conclusions
Neurodegenerative diseases are life-threatening disorders, rapidly spreading in the aged population, with the number of cases rapidly increasing worldwide. These diseases impose a substantial health burden both on the patients, their families and society. The mechanisms implicated in the pathogenesis and development of neurodegenerative diseases have not yet been completely understood. However, some biological mechanisms have been proposed for the development and pathogenesis of these diseases, of which functional disorders of mitochondria and oxidative stress are key mechanisms considered to be involved in the progression of these diseases. In brain tissues, reactive oxygen species are generated from mitochondrial disorders, dopamine metabolism and inflammatory neurons. Therefore, protective biological mechanisms, which contribute to the modulation of these biological processes, are an important area for future studies. Based on the results of previous studies, various therapeutic approaches such as restoring functions of mitochondria and reducing oxidative damage have been developed. However, despite encouraging outcomes in model organisms, many clinical trials have failed to describe an influence on the development of disease. Failures from these approaches analyzed so far should guide future, newer strategies.
Author Contributions
S.K., F.W. and H.C. reviewed the literature, designed the figures and co-wrote the manuscript.
Funding
We are grateful for funding support from the National Key Research and Development Program of China (No. 2016YFC1302204 and 2017YFC1308600 to H. Cui), the National Natural Science Foundation of China (No. 81672502 to H. Cui).
Acknowledgments
The authors wish to acknowledge Muhammad Nadeem Abbas of the State Key Laboratory of Silkworm Biology, Southwest University for helping to revise the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging and development. Cell 2000, 125, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Wang, L.; Wei, G.Q.; Qian, C.; Dai, L.S.; Sun, Y.; Abbas, M.N.; Zhu, B.J.; Liu, C.L. Characterization of the complete mitochondrial genome of Leucoma salicis (Lepidoptera: Lymantriidae) and comparison with other lepidopteran insects. Sci. Rep. 2016, 6, 39153. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, structure and function. J. Physiol. 2011, 589, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Aspects Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarewicz, M.; Borek, A.; Cieluch, E.; Swierczek, M.; Osyczka, A. Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc 1. Implications for the mechanism of superoxide production. Biochim. Biophys. Acta 2010, 1797, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress and aging. Free Rad. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Migliorea, L.; Coppedè, F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Toxicol. Environ. Mutagen. 2009, 674, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; McMurray, C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta 1998, 1366, 211–223. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.S.; Zhou, X.D.; Kausar, S.; Abbas, M.N.; Wu, L.; Zhou, H.L. Mitochondrial genome of Diaphania indica (saunders) (Lepidoptera: Pyraloidea) and implications for its phylogeny. Intl. J. Biol. Macromol. 2018, 108, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Collins, S.R.; Cassidy-Stone, A.; Hummel, E.; DeVay, R.M.; Lackner, L.L.; Westermann, B.; Schuldiner, M.; Weissman, J.S.; Nunnari, J. A mitochondrialfocused genetic interaction map reveals a scaffoldlike complex required for inner membrane organization in mitochondria. J. Cell Biol. 2011, 195, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Von der Malsburg, K.; Müller, J.M.; Bohnert, M.; Oeljeklaus, S.; Kwiatkowska, P.; Becker, T.; Loniewska-Lwowska, A.; Wiese, S.; Rao, S.; Milenkovic, D.; et al. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 2011, 21, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.A.; Ellisman, M.H.; Fox, D.A. Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: Bioenergetic and functional implications. Mol. Vision 2003, 9, 60–73. [Google Scholar]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. Sci. 2016, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; Lin, S.S.; Chen, J.M.; Tsai, H.Z.; Hsieh, T.S.; Fu, C.Y. Electron tomographic analysis reveals ultrastructural features of mitochondrial cristae architecture which reflect energetic state and aging. Sci. Rep. 2017, 7, 45474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.S.; Kausar, S.; Abbas, M.N.; Wang, T.T. Complete sequence and characterization of the Ectropis oblique mitochondrial genome and its phylogenetic implications. Int. J. Biol. Macromol. 2018, 107, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Sun, Y.X.; Yang, L.L.; Wu, L.; Abbas, M.N.; Chen, C.; Gao, J.; Li, X.K.; Liu, C.L.; Dai, L.S. Characterization of the complete mitochondrial genome of Biston marginata (Lepidoptera: Geometridae) and phylogenetic analysis among lepidopteran insects. Int. J. Biol. Macromol. 2018, 113, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiol. Pathophysiol. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef] [PubMed]
- Warnaua, J.; Sharmac, V.; Gamiz-Hernandez, A.P.; Luca, A.D.; Haapanen, O.; Vattulainen, I.; Wikström, M.; Hummer, G.; Kaila, V.R.I. Redox-coupled quinone dynamics in the respiratory complex I. Proc. Natl. Acad. Sci. USA 2018, 115, E8413–E8420. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, K.; Koziel, A.; Jarmuszkiewicz, W. The interplay between mitochondrial reactive oxygen species formation and the coenzyme Q reduction level. Redox Biol. 2018, 18, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. Role of mitochondria in oxidative stress and aging. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef]
- Galkin, A.; Brandt, U. Superoxide radical formation by pure Complex I (NADH:Ubiquinone Oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [PubMed]
- Degli, E.M.; Ngo, A.; Ghelli, A.; Benelli, B.; Carelli, V.; McLennan, H.; Lirnnane, A.W. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch. Biochem. Biophys. 1996, 330, 395–400. [Google Scholar]
- Takeshige, K.; Minakami, S. NADH and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J. 1979, 180, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef] [PubMed]
- 35 Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at Complex I. The J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.; Barja, G. Localization of the site of oxygen radical generation inside the Complex I of heart and nonsynaptic brain mammalian mitochondria. J. Bioenerg. Biomembr. 2000, 32, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2011, 286, 27103–27110. [Google Scholar] [CrossRef] [PubMed]
- McLennan, H.R.; Degli, E.M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr. 2000, 32, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH: Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. PNAS 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.; Wang, E. Modelling the effects of age-related mtDNA mutation accumulation: Complex I defciency, superoxide and cell death. Biochim. Biophys. Acta 1995, 1271, 171–176. [Google Scholar] [CrossRef]
- Votyakova, T.V.; Reynolds, I.J. ΔΨ-dependent and independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Hernandez-Esquivel, L.; Rivero-Segura, N.A.; Marin-Hernandez, A.; Neuzil, J.; Ralph, S.J.; Rodriguez-Enriquez, S. Reactive oxygen species are generated by the respiratory complex II – evidence for lack of contribution of the reverse electron flow in Complex I. FEBS J. 2013, 280, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Moreno-Sanchez, R.; Neuzil, J.; Rodriguez-Enriquez, S. Inhibitors of succinate: Quinone reductase/complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm. Res. 2011, 28, 2695–2730. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Miyazawa, M.; Onodera, A.; Yasuda, K.; Kawabe, N.; Kirinashizawa, M.; Yoshimura, S.; Maruyama, N.; Hartman, P.S.; Ishii, N. Mitochondrial reactive oxygen species generation by the SDHC V69E mutation causes low birth weight and neonatal growth retardation. Mitochondrion 2011, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.M.; Aykin-Burns, N.; Dayal, D.; Coleman, M.C.; Domann, F.E.; Spitz, D.R. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2 − degrees and H2O2. Free Rad. Biol. Med. 2012, 52, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Fujii, M.; Hartman, P.S.; Tsuda, M.; Yasuda, K.; Senoo-Matsuda, N.; Yanase, S.; Ayusawa, D.; Suzuki, K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 1998, 394, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Senoo-Matsuda, N.; Yasuda, K.; Tsuda, M.; Ohkubo, T.; Yoshimura, S.; Nakazawa, H.; Hartman, P.S.; Ishii, N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion over production and abnormal energy metabolism in Caenorhabditis elegans. J. Biol. Chem. 2001, 276, 41553–41558. [Google Scholar] [CrossRef] [PubMed]
- Borek, A.; Sarewicz, M.; Osyczka, A. Movement of the iron–sulfur head domain of cytochrome bc1 transiently opens the catalytic Qo site for reaction with oxygen. Biochemistry 2008, 47, 12365–12370. [Google Scholar] [CrossRef] [PubMed]
- Gurung, B.; Yu, L.; Yu, C.A. Stigmatellin induces reduction of iron–sulfur protein in the oxidized cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 28087–28094. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Azzi, A.; Flohe, L. Mitochondrial H2O2 formation: Relationship with energy conservation. FEBS Lett. 1973, 33, 84–87. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem. Biophys. Res. Commun. 2001, 281, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc 1 complex. Biochemistry 2002, 41, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- Osyczka, A.; Moser, C.C.; Daldal, F.; Dutton, P.L. Reversible redox energy coupling in electron transfer chains. Nature 2004, 427, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. AJP Cell. Physiol. 2008, 294, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Calmettes, G.; John, S.A.; Weiss, J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of Complex III. J. Biol. Chem. 2017, 292, 9882–9895. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Roberts, A.G.; Bowman, M.K.; Kramer, D.M. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 2003, 42, 6493–6499. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The sources of reactive oxygen species and its possible role in the pathogenesis of Parkinson’s disease. Parkinson’s Dis. 2018, 9163040, 9. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect. 1994, 102, 5–12. [Google Scholar] [PubMed]
- Jodeiri, F.M.; Ghaedi, K. Huntington’s disease and mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, W.J.; Switonski, P.M.; Krzyzosiak, W.J.; Figlerowicz, M.; Figiel, M. Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis. Model. Mech. 2015, 8, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 8590578, 15. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorder. Cell 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1 α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- De Maagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Massano, J.; Bhatia, K.P. Clinical approach to Parkinson’s disease: Features, diagnosis, and principles of management. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Carreras, M.; Franco, M.C.; Peralta, J.G.; Poderoso, J.J. Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol. Aspects Med. 2004, 25, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Clifford, W.; Shults, M.D. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Davis, R.E.; Tuttle, J.B.; Trimmer, P.A.; Jason, P.; Sheehan, B.S. Origin and functional consequences of the Complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Castillo, M.D.C.L.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–70015. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Brannock, C. Invited review free radicals and the pathobiology of brain dopamine systems. Neurochem. Int. 1998, 32, 117–131. [Google Scholar] [CrossRef]
- Gao, L.; Laude, K.; Cai, H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet. Clin. North Am. Small Anim. Pr. 2008, 38, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Shima, T.; Stroppolo, A.; Goj, C.; Battiston, G.A.; Gerbasi, R.; Sarna, T.; Swartz, H.M. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience 1996, 73, 407–415. [Google Scholar] [CrossRef]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 4, a009332. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Molecular mechanism of the relation of monoamine oxidase inhibitors to Parkinson’s disease: Possible implications of glial cells. J. Neural. Transm. 2006, 71, 53–65. [Google Scholar]
- Meiser, J.; Weind, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, H.; Yasuda, T. Iron accumulation in Parkinson’s disease. J. Neural Transm. 2012, 119, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Núñez, M.T. Parkinson’s disease: The mitochondria-iron link. Parkinson’s Dis. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Atasoy, H.T.; Nuyan, O.; Tunc, T.; Yorubulut, M.; Unal, A.E.; Inan, L.E. T2-weighted MRI in Parkinson’s disease; substantia nigra pars compacta hypointensity correlates with the clinical scores. Neurol. India 2004, 52, 332–337. [Google Scholar] [PubMed]
- Shichir, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2003, 86, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzerd, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Floor, E.; Wetzel, M.G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998, 70, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Danielson, S.R.; Andersen, J.K. Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic. Biol. Med. 2008, 44, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Soundararajan, C.C.; Vivekanandhan, S.; Behari, M. Erythrocyte antioxidant enzymes in Parkinson’s disease. Indian J. Med. Res. 2005, 121, 111–115. [Google Scholar] [PubMed]
- Beal, M.F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Rajasankar, S. Parkinson’s disease: Oxidative stress and therapeutic approaches. Neurol. Sci. 2010, 31, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Linking selective vulnerability to cell death mechanisms in Parkinson’s disease. Am. J. Pathol. 2007, 170, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Liu, J.L.; Wu, Y.R.; Chen, Y.C.; Cheng, H.S.; Cheng, M.L.; Chiu, D.T.Y. Increased oxidative damage in peripheral blood correlates with severity of Parkinson’s disease. Neurobiol. Dis. 2009, 33, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Anti-Oxidants in Parkinson’s disease therapy: A critical point of view. Curr. Neuropharmacol. 2016, 14, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Tasset, I.; Sanchez, F.; Tunez, I. The molecular bases of Huntington’s disease: The role played by oxidative stress. Rev. Neurol. 2009, 49, 424–429. [Google Scholar] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Brocardo, P.S.; Christie, B.R. The role of oxidative stress in Huntington’s disease: Are antioxidants good therapeutic candidates? Curr. Drug Targets 2014, 15, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzlerc, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M.; et al. The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Human Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Deckel, A.W.; Tang, V.; Nuttal, D.; Gary, K.; Elder, R. Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington’s disease transgenic mice. Brain Res. 2002, 939, 7686. [Google Scholar] [CrossRef]
- Perez-Severiano, F.; Escalante, B.; Vergara, P.; Rios, C.; Segovia, J. Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res. 2002, 951, 36–42. [Google Scholar] [CrossRef]
- Perez-Severiano, F.; Santamaria, A.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Rios, C.; Segovia, J. Increased formation of reactive oxygen species, but no changes in glutathione peroxidase activity, in striata of mice transgenic for the Huntington’s disease mutation. Neurochem. Res. 2004, 29, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Pitts, A.; Dailey, K.; Newington, J.T.; Chien, A.; Arseneault, R.; Cann, T.; Thompson, L.M.; Cumming, R.C. Dithiol-based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J. Biol. Chem. 2012, 287, 22717–22729. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dara, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Wu, Y.R.; Chengetal, M.L. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophysi. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kumar, P.; Malik, J. Plants and phytochemicals for Huntington’s disease. Pharmacog. Rev. 2013, 7, 81–91. [Google Scholar]
- Klepac, N.; Relja, M.; Klepac, R.; Hecimovic, S.; Babic, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Goula, A.V.; Berquist, B.R.; Wilson, D.M., III; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009, 5, e1000749. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Rosenstock, T.R.; Cunha-Oliveira, T.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radcal. Biol. Med. 2012, 53, 1857–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.; Barrero, F.J.; Morales, B.; Luna, J.D.; Ramirez, M.; Vives, F. Oxidative stress and plasma amino peptidase activity in Huntington’s disease. J. Neural Transm. 2010, 117, 325–332. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The major sites for the production of reactive oxygen species in a mitochondrion.
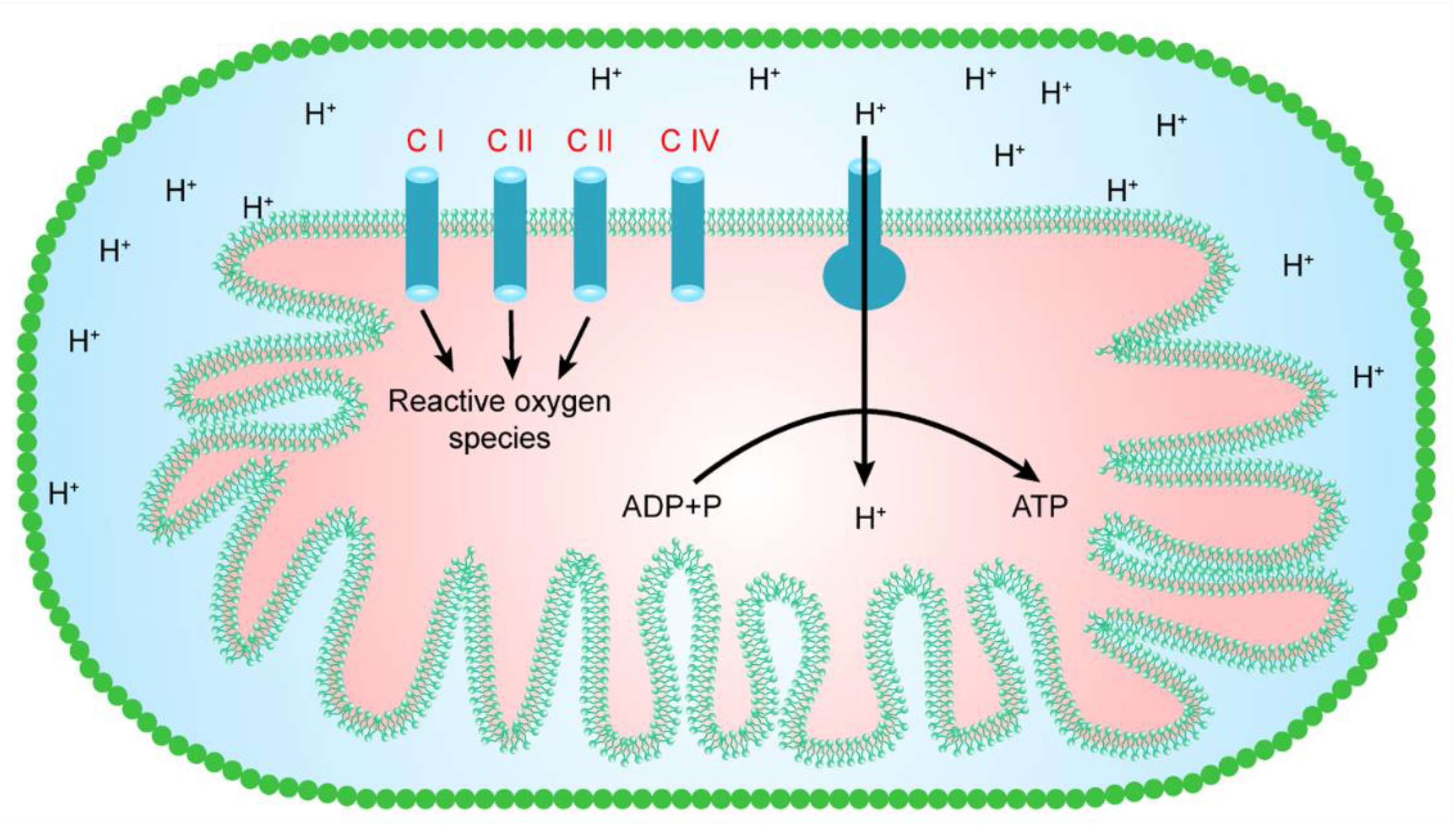
Figure 2.
The causes of oxidative stress in neurodegenerative diseases.
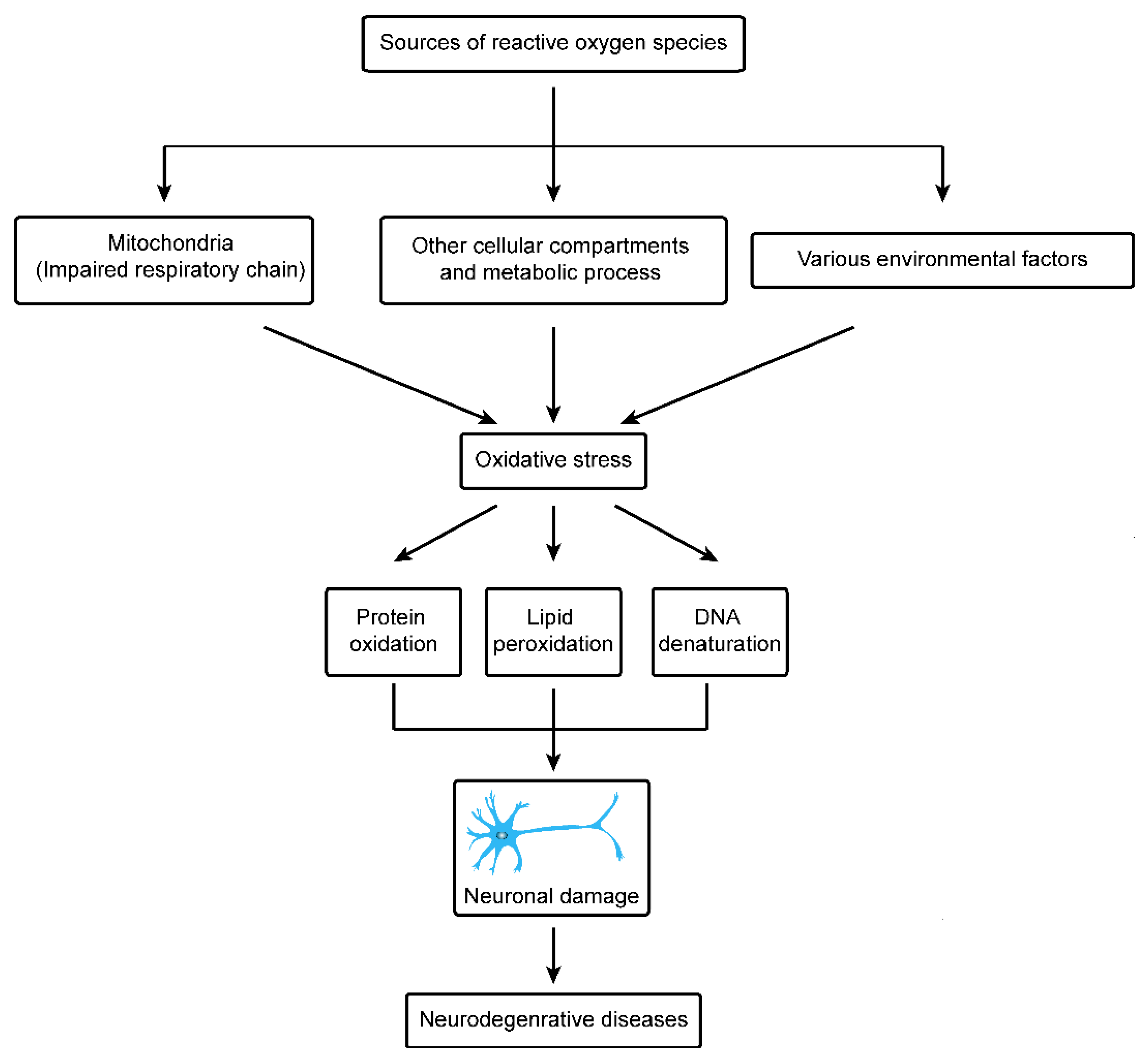
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. https://doi.org/10.3390/cells7120274
AMA Style
Kausar S, Wang F, Cui H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells. 2018; 7(12):274. https://doi.org/10.3390/cells7120274
Chicago/Turabian StyleKausar, Saima, Feng Wang, and Hongjuan Cui. 2018. "The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases" Cells 7, no. 12: 274. https://doi.org/10.3390/cells7120274
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.