
Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
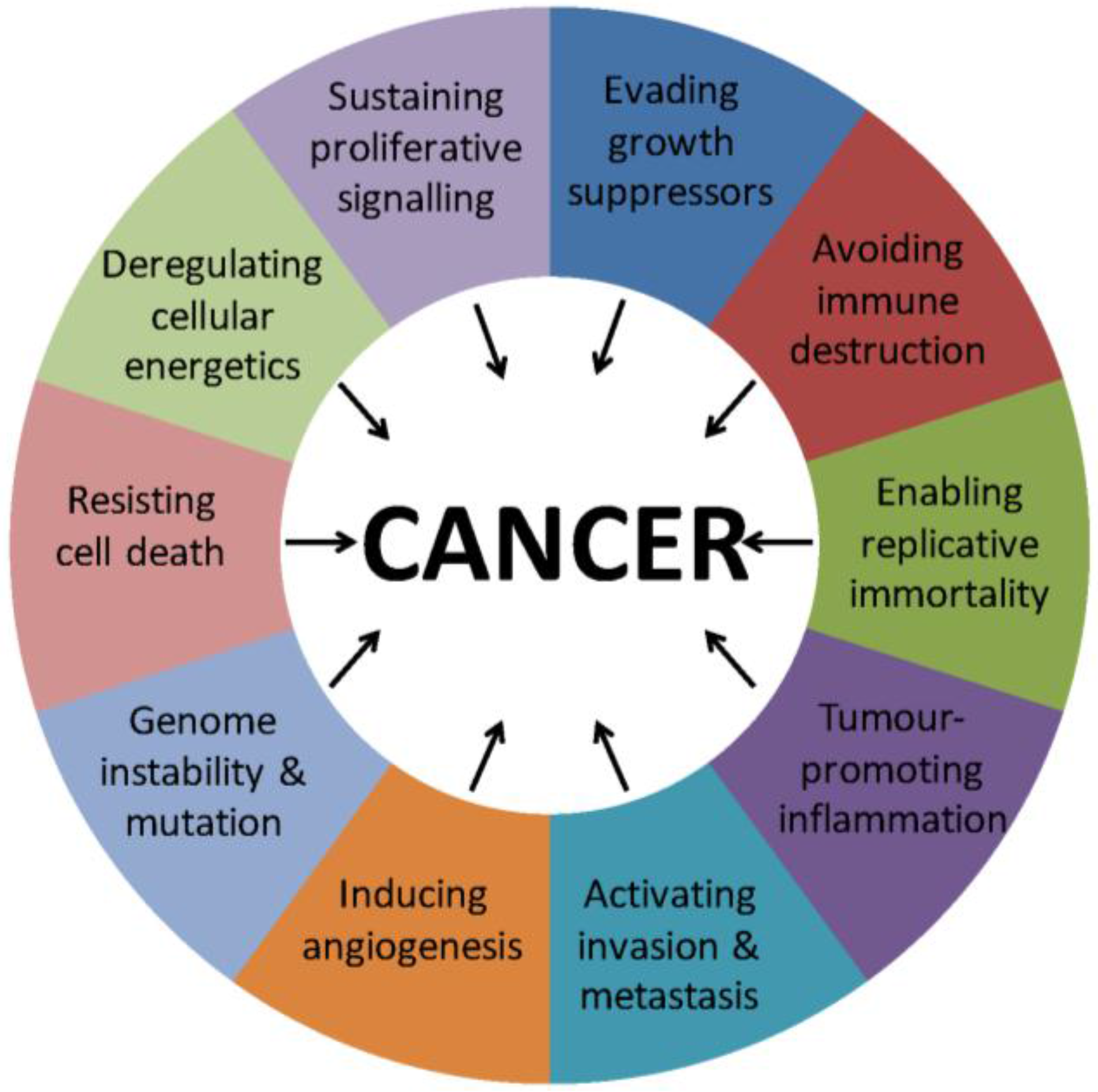
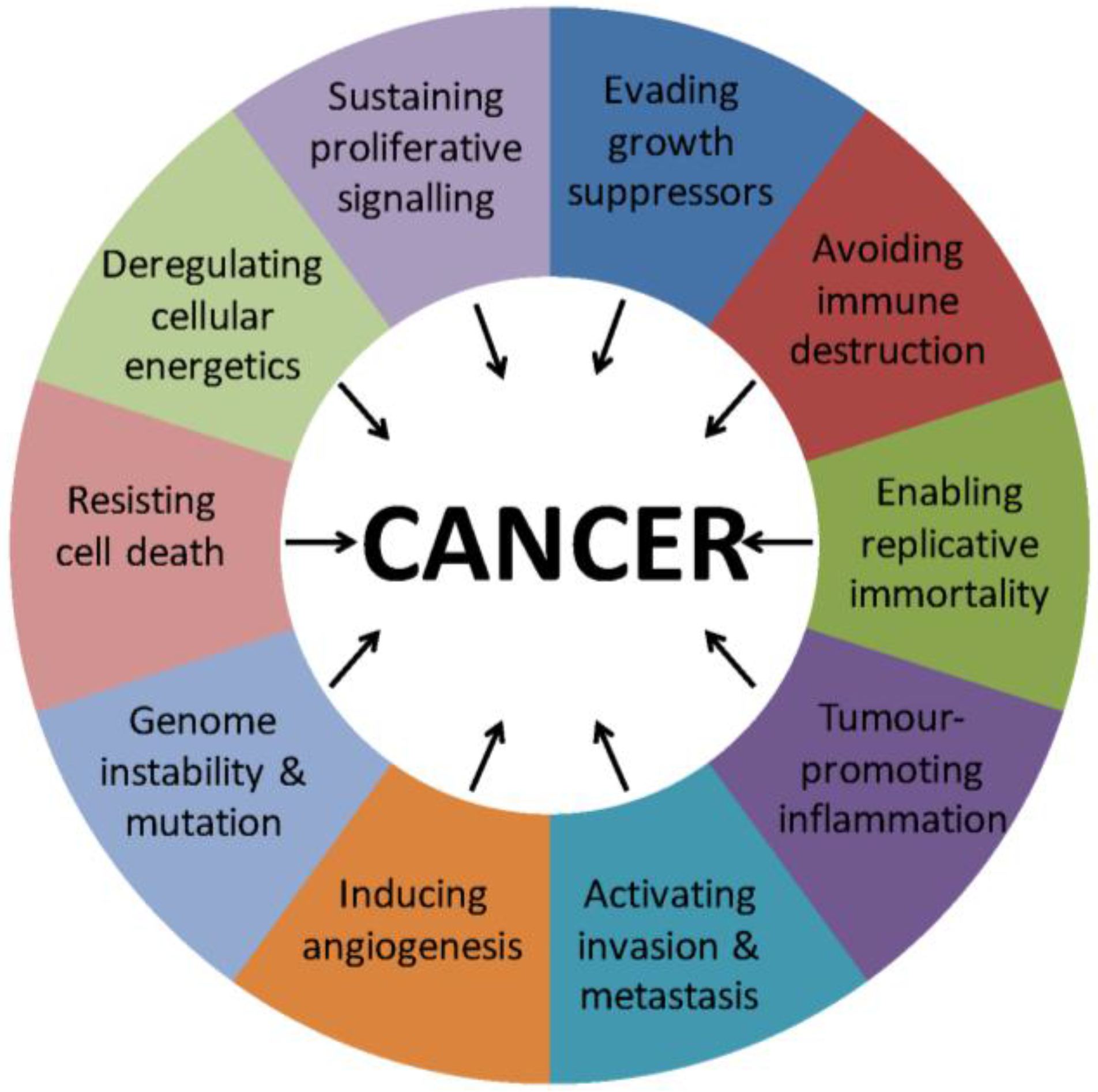
:1. Introduction
2. Nuclear Export Structures and Mechanisms
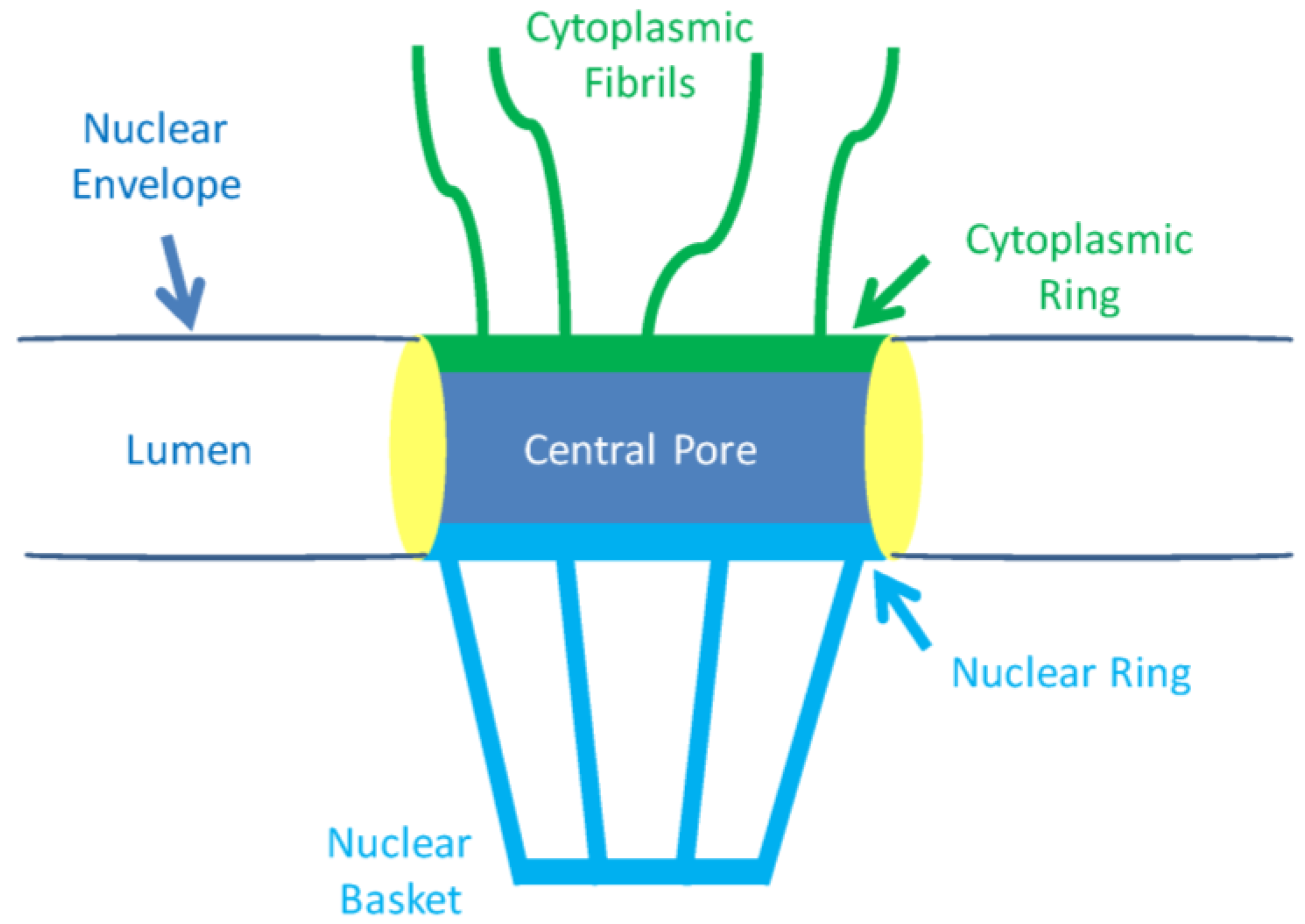
2.1. The Nuclear Pore and Nuclear Envelope
2.2. Nuclear Localisation and Export Signals
2.3. Karyopherins
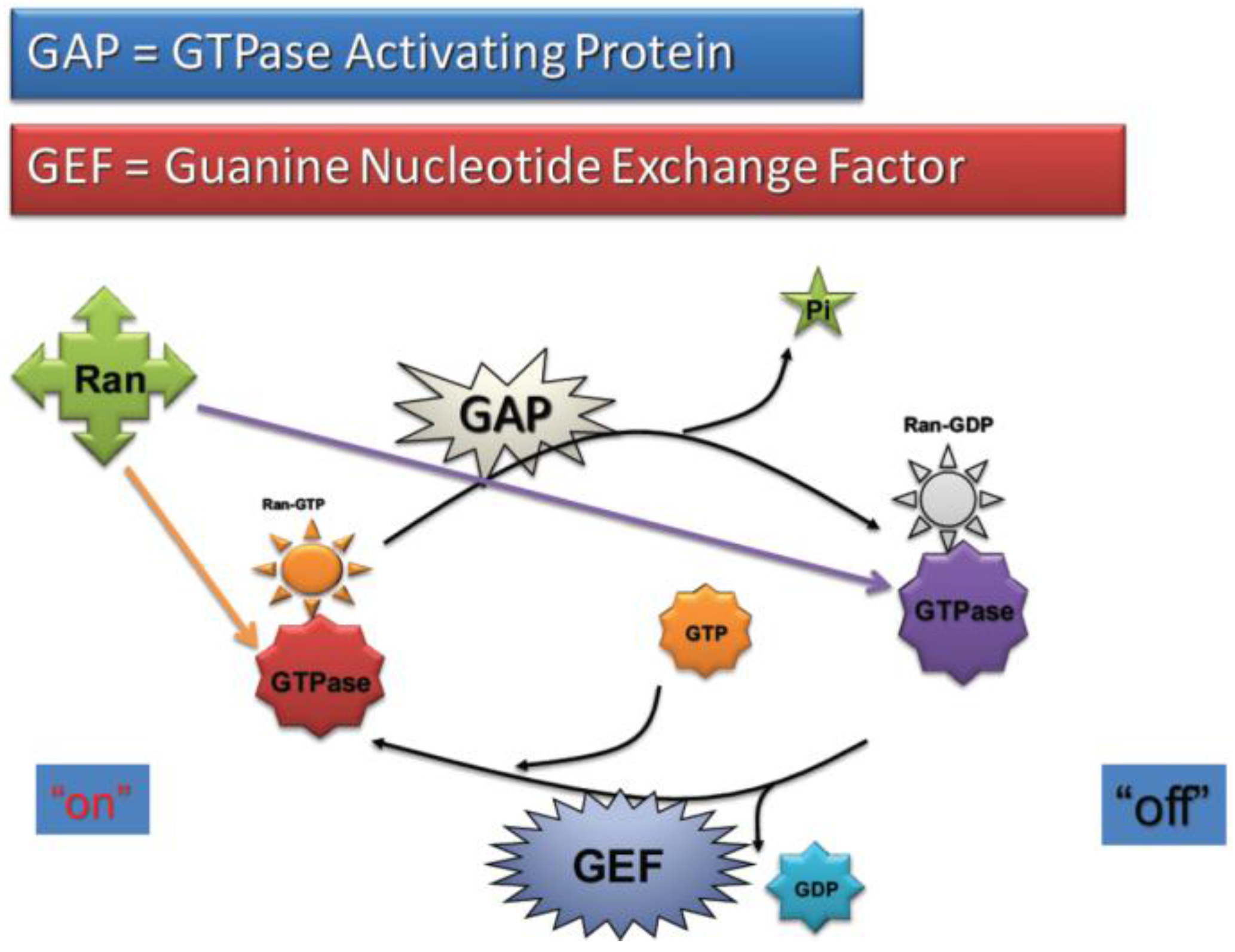
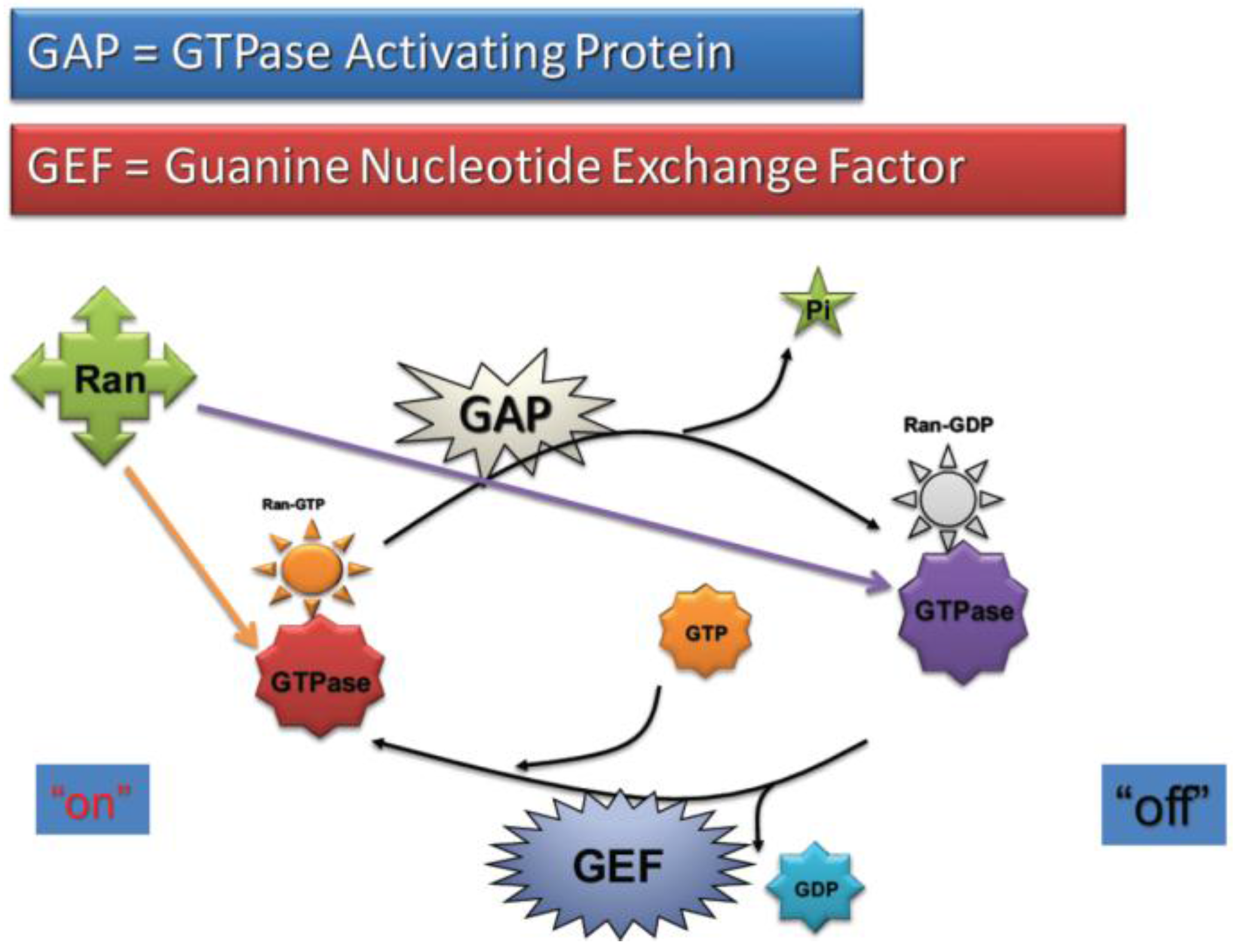
2.4. RanGTP
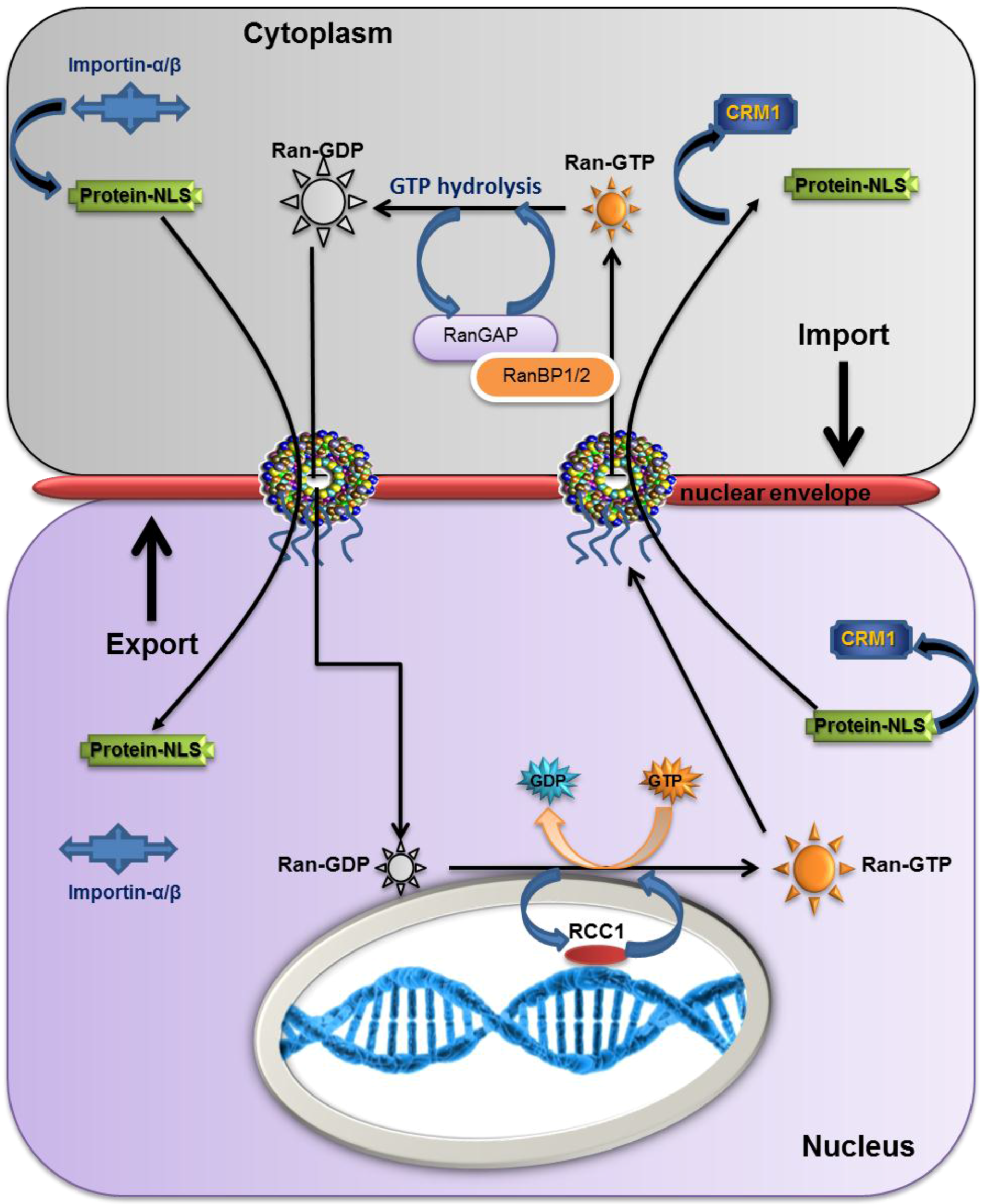
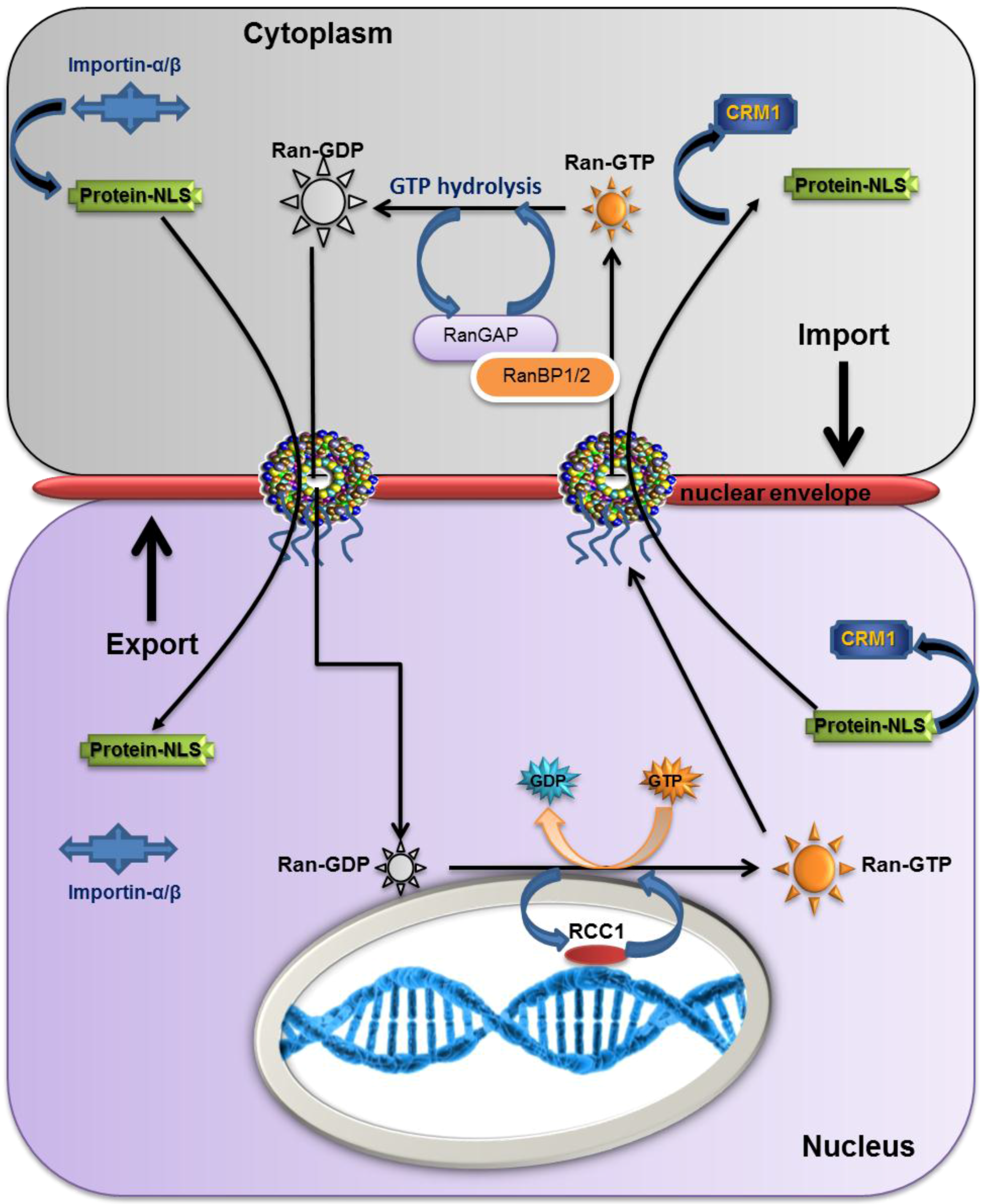
2.5. The Overall Export/Import Mechanism
3. The Role of Nuclear Export in Cancer and Drug Resistance
3.1. CRM1 and Cancer
3.2. CRM1 Inhibitors
4. Application of CRM1 Inhibitors
4.1. Topoisomerase
4.2. Galectin-3
4.3. Selective Inhibitors of Nuclear Export (SINE)
5. NES Inhibitors
6. Nup Dormancy and Resistance
7. Sgk1
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, F.A.; Kondrashov, A.S. Measurements of spontaneous rates of mutations in the recent past and the near future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Goymer, P. Natural selection: The evolution of cancer. Nature 2008, 454, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350. [Google Scholar] [PubMed]
- Ribatti, D. Novel angiogenesis inhibitors: Addressing the issue of redundancy in the angiogenic signaling pathway. Cancer Treat. Rev. 2011, 37, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.C.; Intapa, C.; Jabra-Rizk, M.A. “Persisters”: Survival at the cellular level. PLoS Pathog. 2011. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Hetzer, M.W. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008, 18, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Bono, F.; Jinek, M.; Conti, E. Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 2007, 76, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Terry, L.J.; Wente, S.R. Flexible gates: Dynamic topologies and functions for fg nucleoporins in nucleocytoplasmic transport. Eukaryot. Cell 2009, 8, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Raices, M.; D’Angelo, M.A. Nuclear pore complex composition: A new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell Biol. 2012, 13, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, C.; Li, H.; Lim, C.S. Model system to study classical nuclear export signals. AAPS PharmSci 2002. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.; Dilworth, S.M.; Laskey, R.A.; Dingwall, C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: Identification of a class of bipartite nuclear targeting sequence. Cell 1991, 64, 615–623. [Google Scholar] [CrossRef]
- Bogerd, H.P.; Fridell, R.A.; Benson, R.E.; Hua, J.; Cullen, B.R. Protein sequence requirements for function of the human T-cell leukemia virus type 1 rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol. Cell Biol. 1996, 16, 4207–4214. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Sullivan, D.M. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr. Med. Chem. 2008, 15, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Kutay, U.; Guttinger, S. Leucine-rich nuclear-export signals: Born to be weak. Trends Cell Biol. 2005, 15, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.; Zhang, C. Spatial and temporal coordination of mitosis by ran gtpase. Nat. Rev. Mol. Cell Biol. 2008, 9, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, F.R.; Ponstingl, H. Catalysis of guanine nucleotide exchange on ran by the mitotic regulator rcc1. Nature 1991, 354, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Renault, L.; Kuhlmann, J.; Henkel, A.; Wittinghofer, A. Structural basis for guanine nucleotide exchange on ran by the regulator of chromosome condensation (rcc1). Cell 2001, 105, 245–255. [Google Scholar] [CrossRef]
- Zhang, X.; Yamada, M.; Mabuchi, N.; Shida, H. Cellular requirements for CRM1 import and export. J. Biochem. 2003, 134, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Hurt, E. Gene regulation by nucleoporins and links to cancer. Mol. Cell 2010, 38, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Xylourgidis, N.; Sabri, N.; Uv, A.; Fornerod, M.; Samakovlis, C. The drosophila nucleoporin dnup88 localizes dnup214 and CRM1 on the nuclear envelope and attenuates nes-mediated nuclear export. J. Cell Biol. 2003, 163, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Matchett, K.B.; McFarlane, S.; Hamilton, S.E.; Eltuhamy, Y.S.; Davidson, M.A.; Murray, J.T.; Faheem, A.M.; El-Tanani, M. Ran gtpase in nuclear envelope formation and cancer metastasis. Adv. Exp. Med. Biol. 2014, 773, 323–351. [Google Scholar] [PubMed]
- Rodriguez, J.A.; Schuchner, S.; Au, W.W.; Fabbro, M.; Henderson, B.R. Nuclear-cytoplasmic shuttling of bard1 contributes to its proapoptotic activity and is regulated by dimerization with brca1. Oncogene 2004, 23, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Kau, T.R.; Way, J.C.; Silver, P.A. Nuclear transport and cancer: From mechanism to intervention. Nat. Rev. Cancer 2004, 4, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.; Wang, J.Y. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat. Med. 2001, 7, 228–234. [Google Scholar] [CrossRef] [PubMed]
- O’Brate, A.; Giannakakou, P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resist. Updat. 2003, 6, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Newlands, E.S.; Rustin, G.J.; Brampton, M.H. Phase i trial of elactocin. Br. J. Cancer 1996, 74, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Mirski, S.E.; Sparks, K.E.; Cole, S.P. Two cooh-terminal truncated cytoplasmic forms of topoisomerase II alpha in a vp-16-selected lung cancer cell line result from partial gene deletion and alternative splicing. Biochemistry 1997, 36, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Newlaczyl, A.U.; Yu, L.G. Galectin-3—A jack-of-all-trades in cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Song, Y.K.; Song, J.J.; Siervo-Sassi, R.R.; Kim, H.R.; Li, L.; Spitz, D.R.; Lokshin, A.; Kim, J.H. Reconstitution of galectin-3 alters glutathione content and potentiates trail-induced cytotoxicity by dephosphorylation of akt. Exp. Cell Res. 2003, 288, 21–34. [Google Scholar] [CrossRef]
- Hsu, D.K.; Yang, R.Y.; Pan, Z.; Yu, L.; Salomon, D.R.; Fung-Leung, W.P.; Liu, F.T. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am. J. Pathol. 2000, 156, 1073–1083. [Google Scholar] [CrossRef]
- Yamamoto-Sugitani, M.; Kuroda, J.; Ashihara, E.; Nagoshi, H.; Kobayashi, T.; Matsumoto, Y.; Sasaki, N.; Shimura, Y.; Kiyota, M.; Nakayama, R.; et al. Galectin-3 (gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2011, 108, 17468–17473. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Abdel-Azim, H.; Lim, M.; Arutyunyan, A.; von Itzstein, M.; Groffen, J.; Heisterkamp, N. Galectin-3 in pre-b acute lymphoblastic leukemia. Leukemia 2013, 27, 2385–2388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Hou, H.A.; Lee, M.C.; Liu, C.Y.; Jhuang, J.Y.; Lai, Y.J.; Lin, C.W.; Chen, H.Y.; Liu, F.T.; Chou, W.C.; et al. Higher bone marrow lgals3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood 2013, 121, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via wnt/beta-catenin signaling pathway. J. Hematol. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, M.; Tamayo, A.T.; Shacham, S.; Kauffman, M.; Lee, J.; Zhang, L.; Ou, Z.; Li, C.; Sun, L.; et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp. Hematol. 2013, 41, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Aboukameel, A.; Bao, B.; Sarkar, F.H.; Philip, P.A.; Kauffman, M.; Shacham, S.; Mohammad, R.M. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013, 144, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Carrasco, Y.P.; Hu, Y.; Guo, X.; Mirzaei, H.; Macmillan, J.; Chook, Y.M. Nuclear export inhibition through covalent conjugation and hydrolysis of leptomycin b by CRM1. Proc. Natl. Acad. Sci. USA 2013, 110, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Holloway, M.P.; Nguyen, K.; McCauley, D.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; Altura, R.A. Xpo1 (CRM1) inhibition represses stat3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Sanda, T.; Mansour, M.R.; Kentsis, A.; Montero, J.; Le, B.T.; Christie, A.L.; McCauley, D.; Rodig, S.J.; Kauffman, M.; et al. Kpt-330 inhibitor of CRM1 (xpo1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br. J. Haematol. 2013, 161, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Sun, Q.; Kentsis, A.; Farmer, A.; Zhang, Z.C.; Sanda, T.; Mansour, M.R.; Barcelo, C.; McCauley, D.; Kauffman, M.; et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2013, 27, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Yu, X.; Na, C.; Santhanam, R.; Shacham, S.; Kauffman, M.; Walker, A.; Klisovic, R.; Blum, W.; Caligiuri, M.; et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012, 120, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, J.; Sharma, A.; Kim, H.S.; Hammers, H.; Meeker, A.; De Marzo, A.; Carducci, M.; Kauffman, M.; Shacham, S.; Kachhap, S. Selective inhibitors of nuclear export (sine) as novel therapeutics for prostate cancer. Oncotarget 2014, 5, 6102–6112. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hattori, N.; Chien, W.; Sun, Q.; Sudo, M.; GL, E.L.; Ding, L.; Lim, S.L.; Shacham, S.; Kauffman, M.; et al. Kpt-330 has antitumour activity against non-small cell lung cancer. Br. J. Cancer 2014, 111, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bill, M.A.; Young, G.S.; La Perle, K.; Landesman, Y.; Shacham, S.; Kauffman, M.; Senapedis, W.; Kashyap, T.; Saint-Martin, J.R.; et al. Novel small molecule xpo1/CRM1 inhibitors induce nuclear accumulation of tp53, phosphorylated mapk and apoptosis in human melanoma cells. PLoS ONE 2014, 9, e102983. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gery, S.; Sun, H.; Shacham, S.; Kauffman, M.; Koeffler, H.P. Kpt-330 inhibitor of xpo1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (sine)––A novel class of anti-cancer agents. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Tortoreto, M.; Mancini, A.; Addis, A.; Di Cesare, E.; Lenzi, A.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. Xpo1/CRM1-selective inhibitors of nuclear export (sine) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (pca). J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gerecitano, J. Sine (selective inhibitor of nuclear export)-translational science in a new class of anti-cancer agents. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Knauer, S.K.; Kramer, O.H.; Knosel, T.; Engels, K.; Rodel, F.; Kovacs, A.F.; Dietmaier, W.; Klein-Hitpass, L.; Habtemichael, N.; Schweitzer, A.; et al. Nuclear export is essential for the tumor-promoting activity of survivin. FASEB J. 2007, 21, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Kalir, T.; Rahaman, J.; Dottino, P.; Kohtz, D.S. Alterations in nuclear pore architecture allow cancer cell entry into or exit from drug-resistant dormancy. Am. J. Pathol. 2012, 180, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.; Scumaci, D.; D’Antona, L.; Iuliano, R.; Menniti, M.; Di Sanzo, M.; Faniello, M.C.; Colao, E.; Malatesta, P.; Zingone, A.; et al. Sgk1 enhances ranbp1 transcript levels and decreases taxol sensitivity in rko colon carcinoma cells. Oncogene 2013, 32, 4572–4578. [Google Scholar] [CrossRef] [PubMed]
- Talarico, C.; D’Antona, L.; Scumaci, D.; Barone, A.; Gigliotti, F.; Fiumara, C.V.; Dattilo, V.; Gallo, E.; Visca, P.; Ortuso, F.; et al. Preclinical model in hcc: The sgk1 kinase inhibitor si113 blocks tumor progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget 2015, 6, 37511–37525. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Tanani, M.; Dakir, E.-H.; Raynor, B.; Morgan, R. Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy. Cancers 2016, 8, 35. https://doi.org/10.3390/cancers8030035
El-Tanani M, Dakir E-H, Raynor B, Morgan R. Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy. Cancers. 2016; 8(3):35. https://doi.org/10.3390/cancers8030035
Chicago/Turabian StyleEl-Tanani, Mohamed, El-Habib Dakir, Bethany Raynor, and Richard Morgan. 2016. "Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy" Cancers 8, no. 3: 35. https://doi.org/10.3390/cancers8030035