Interactions of Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (TRAIL) with the Immune System: Implications for Inflammation and Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discovery of TRAIL
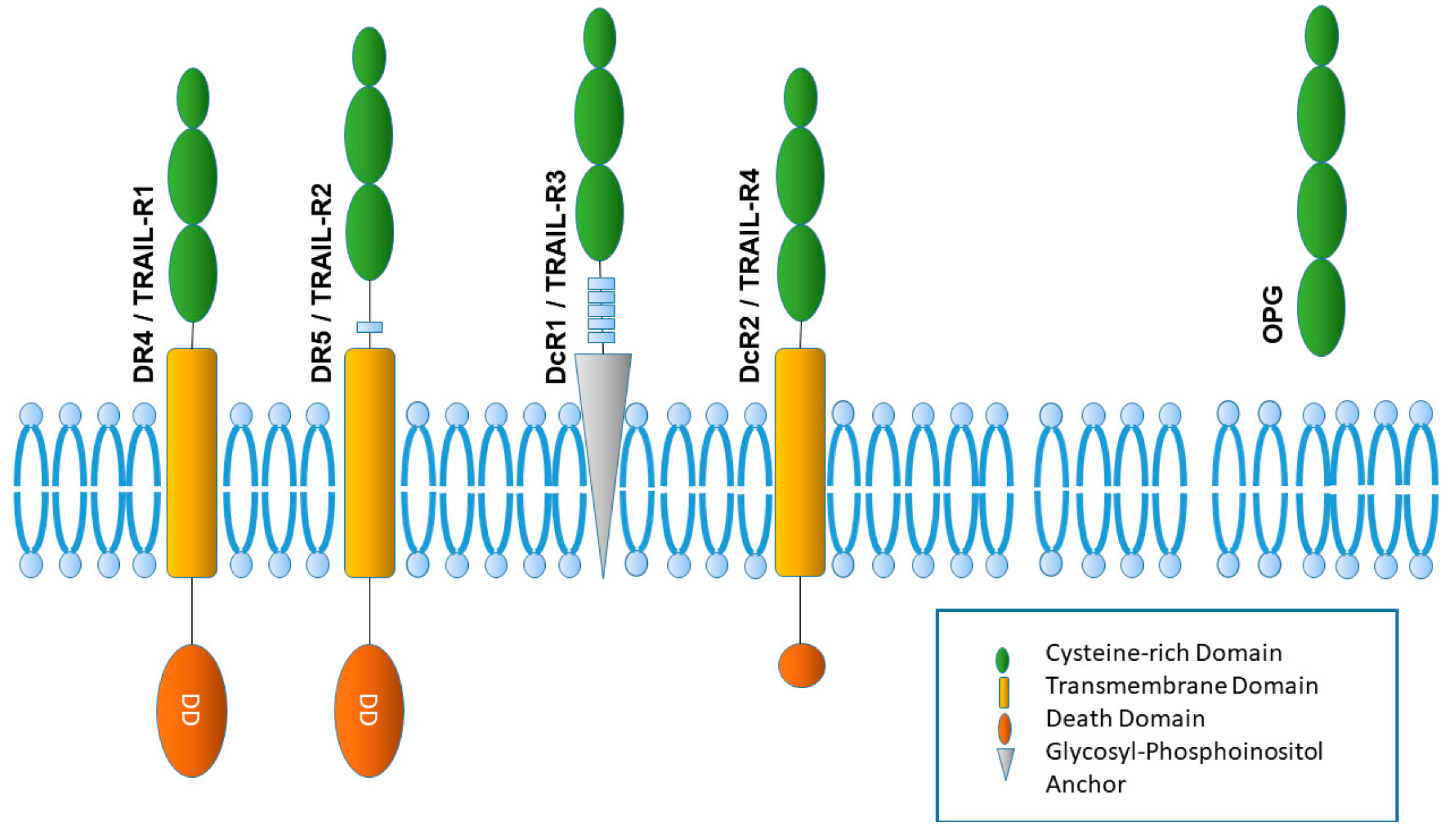
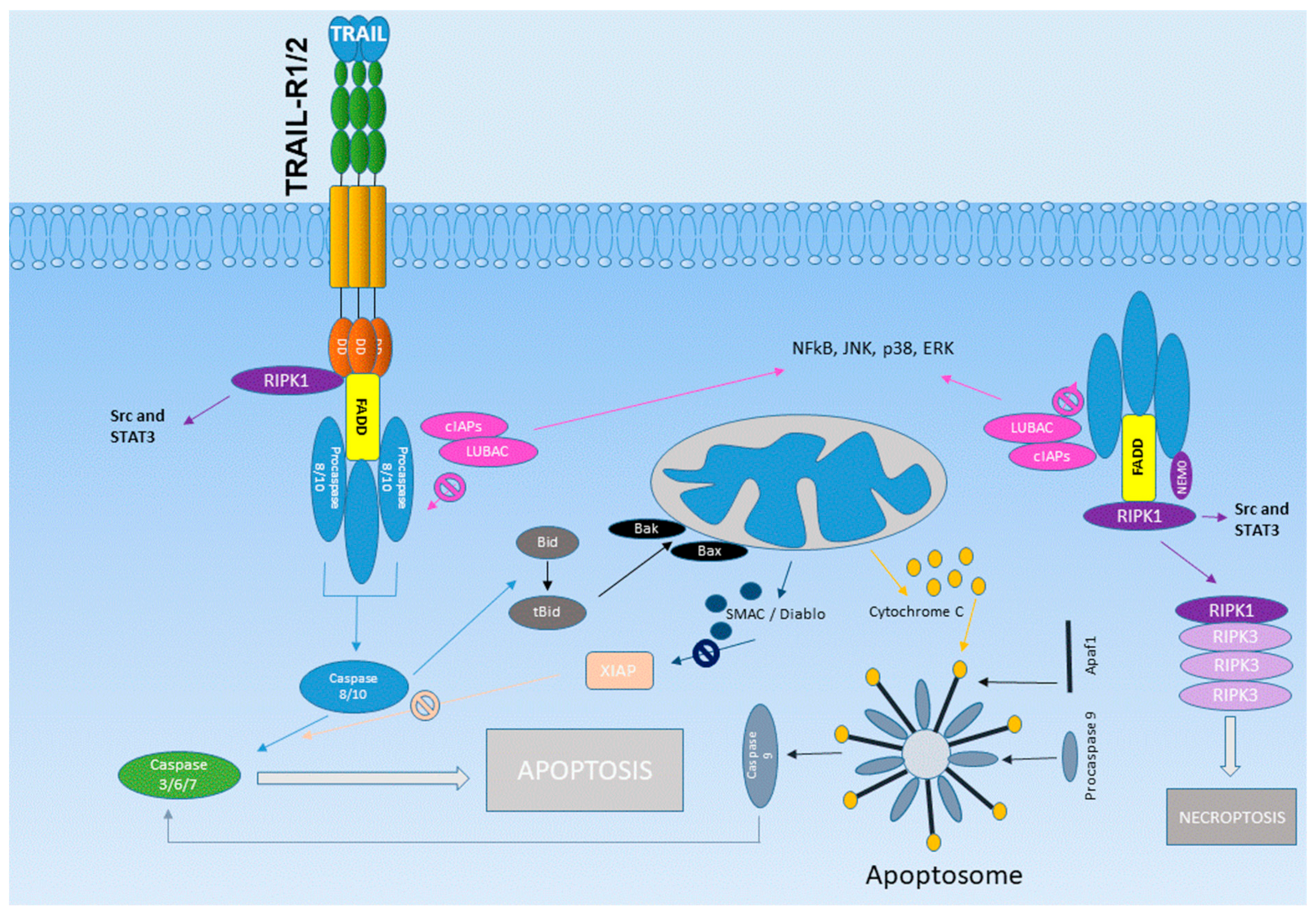
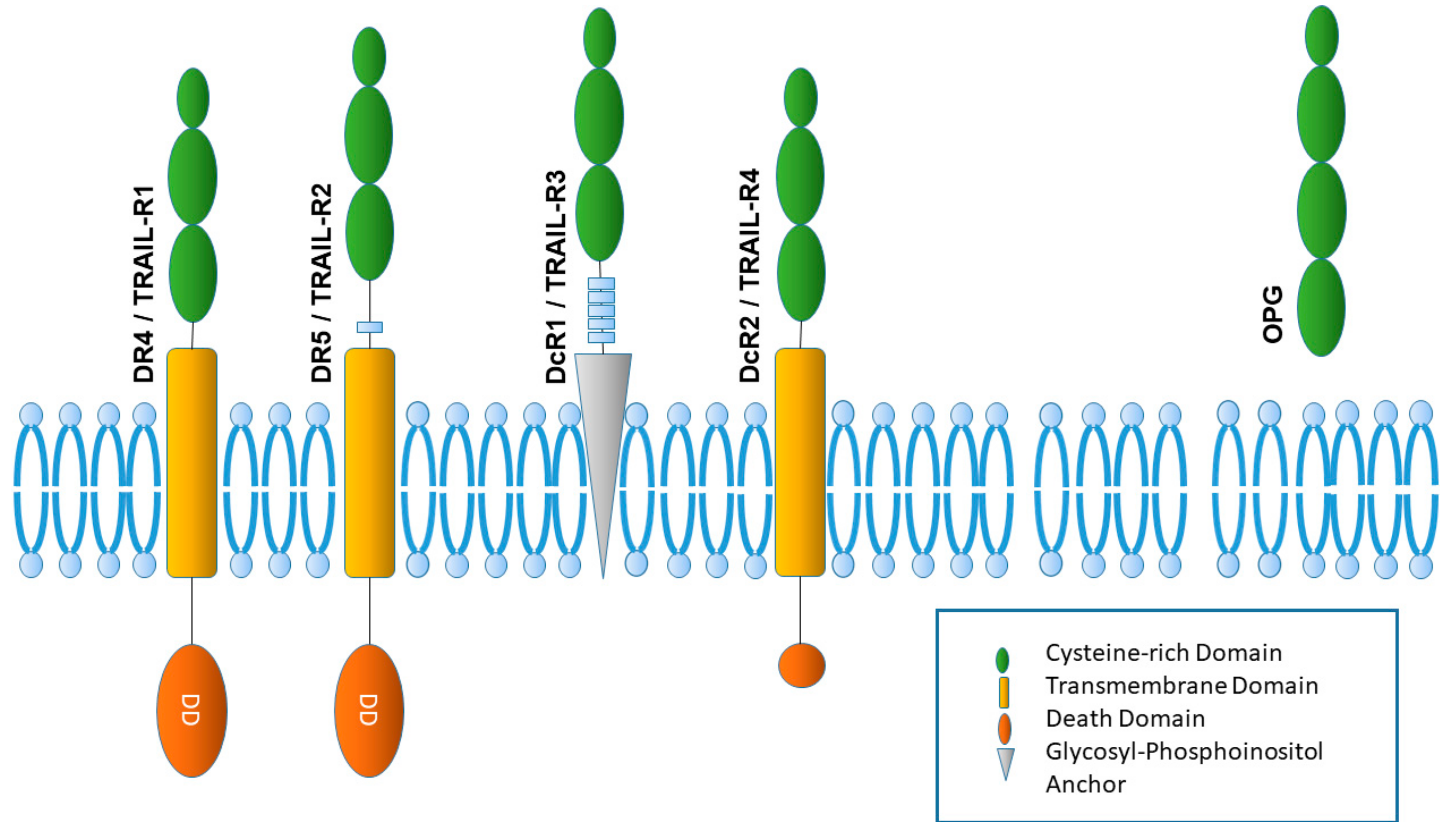
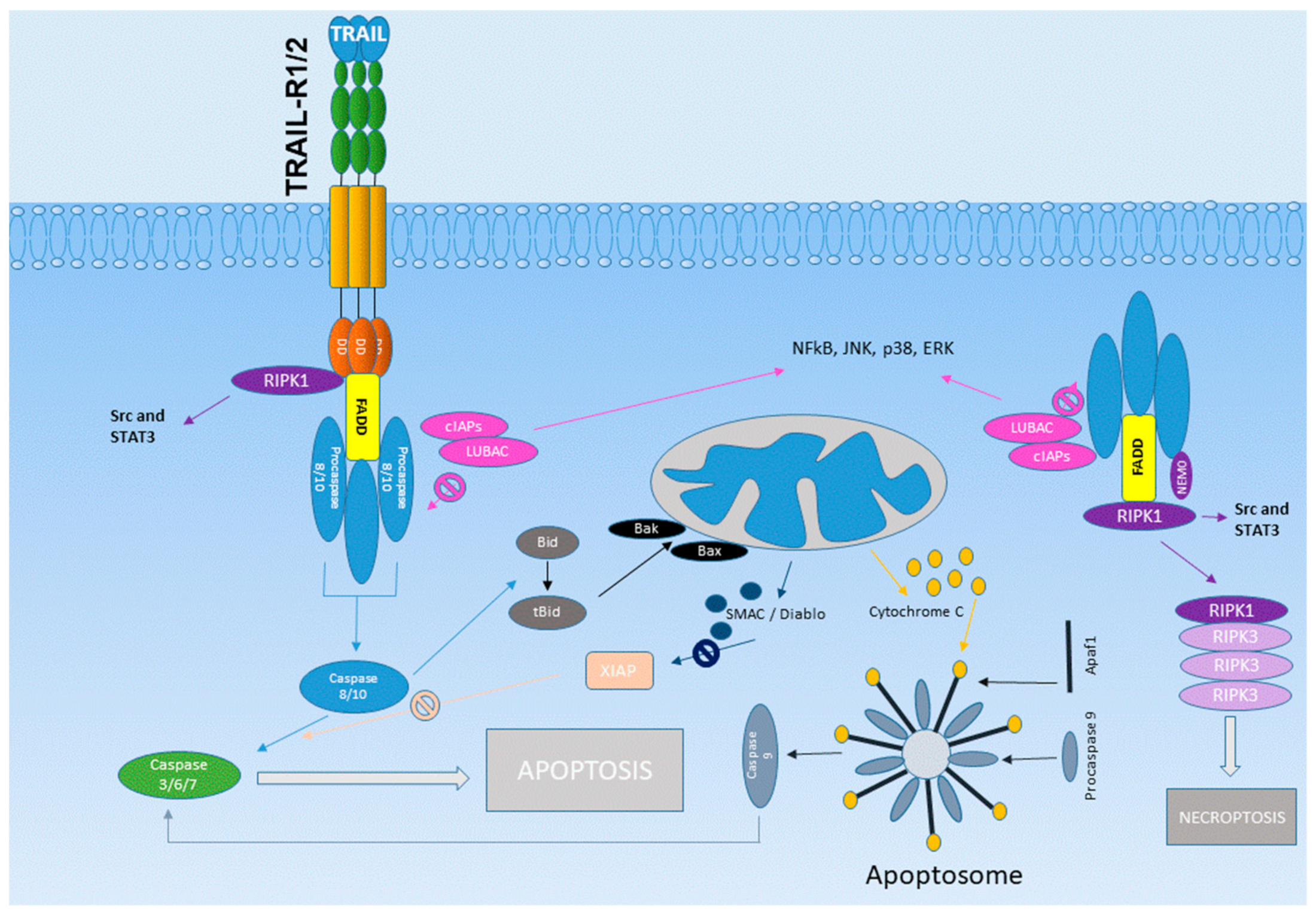
3. The TRAIL Receptor System
4. Clinical Development of TRAIL Agonists
5. Tumor-Promoting Side Effects of TRAIL
6. TRAIL: A Key Molecule for NK Cell-Mediated Immune Surveillance
7. TRAIL and Neutrophils: Lessons Learned from Models of Bacterial Infections
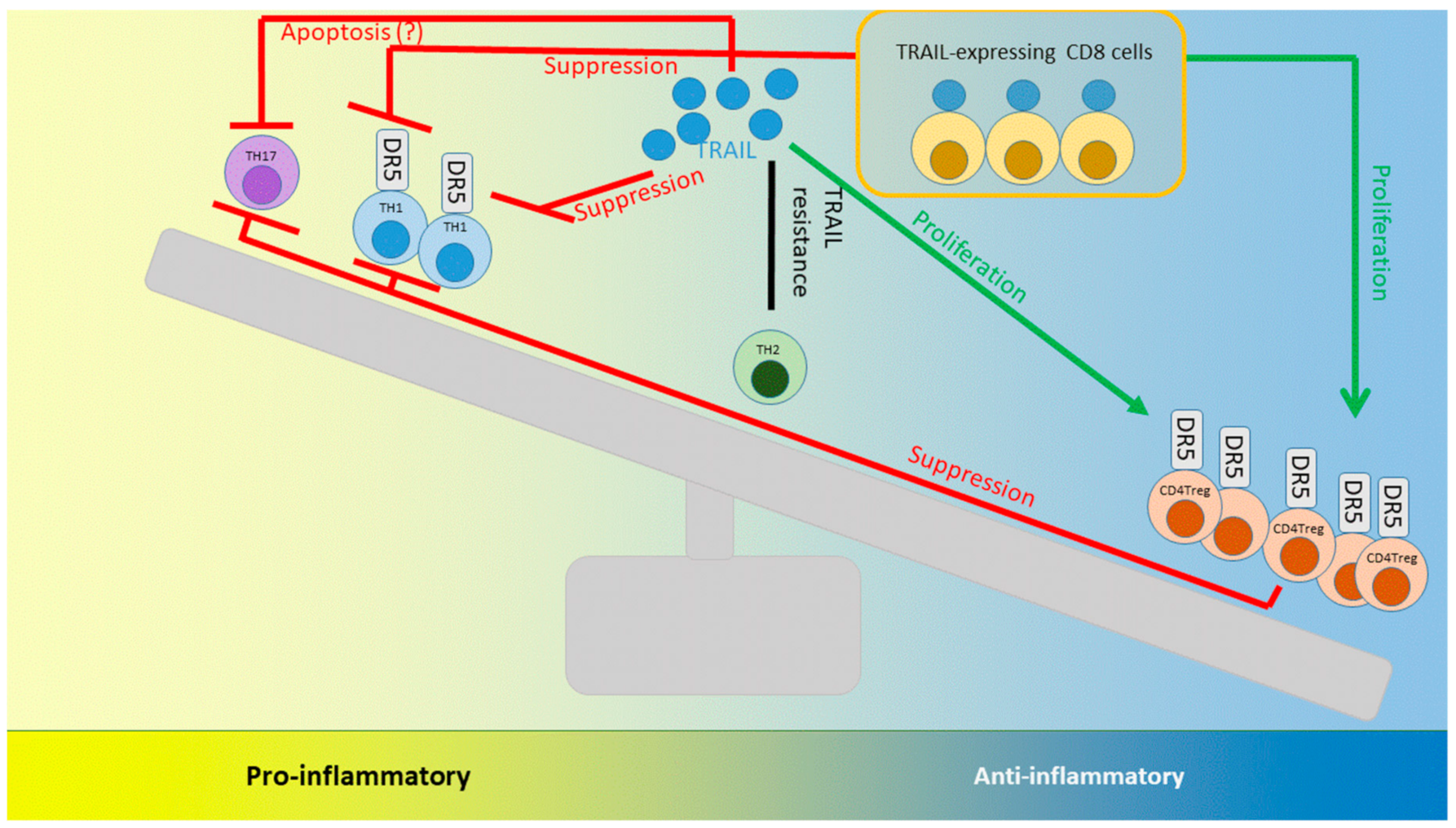
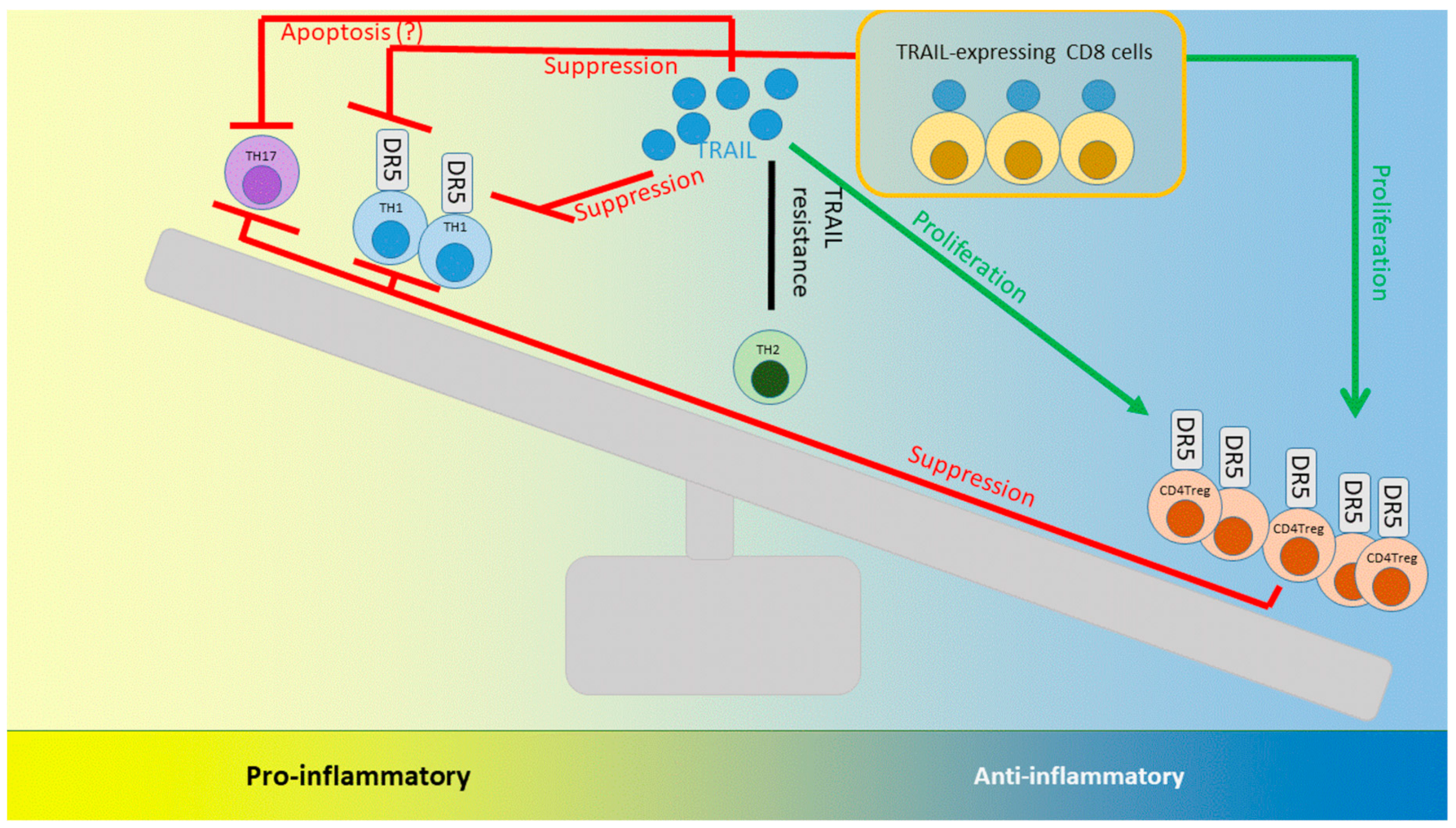
8. TRAIL and its Interactions with T cells: Lessons Learned from Autoimmunity and Infection
9. TRAIL’s Effects on Tumor Immunology
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Havell, E.A.; Fiers, W.; North, R.J. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J. Exp. Med. 1988, 167, 1067–1085. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Holoch, P.A.; Griffith, T.S. TNF-related apoptosis-inducing ligand (TRAIL): A new path to anti-cancer therapies. Eur. J. Pharmacol. 2009, 625, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretz, A.L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. TRAILblazing Strategies for Cancer Treatment. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Huang, A.; Skubatch, M.; Baldwin, D.; Yuan, J.; Gurney, A.; Goddard, A.D.; Godowski, P.; et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 1997, 7, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Ni, J.; Yu, G.; Wei, Y.F.; Dixit, V.M. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998, 424, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- James, B.R.; Griffith, T.S. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptotic pathways in cancer immunosurveillance: Molecular mechanisms and prospects for therapy. Res. Rep. Biochem. 2015, 5, 1–10. [Google Scholar]
- Shlyakhtina, Y.; Pavet, V.; Gronemeyer, H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017, 8, e3025. [Google Scholar] [CrossRef]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Stagg, J.; Yagita, H.; Okumura, K.; Smyth, M.J. Targeting death-inducing receptors in cancer therapy. Oncogene 2007, 26, 3745–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, S.; Philipp, S.; Davarnia, P.; Winoto-Morbach, S.; Roder, C.; Arenz, C.; Trauzold, A.; Kabelitz, D.; Schutze, S.; Kalthoff, H.; et al. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer 2014, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Partecke, L.I.; Roetz, F.; Fluhr, H.; Weiss, F.U.; Heidecke, C.D.; von Bernstorff, W. LPS promotes resistance to TRAIL-induced apoptosis in pancreatic cancer. Infect. Agents Cancer 2017, 12, 30. [Google Scholar] [CrossRef]
- Micheau, O. Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Gajan, A.; Chu, Q.; Xiong, H.; Wu, K.; Wu, G.S. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018, 37, 733–748. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Wajant, H. Molecular Mode of Action of TRAIL Receptor Agonists-Common Principles and Their Translational Exploitation. Cancers (Basel) 2019, 11, 954. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Fas and TRAIL ‘death receptors’ as initiators of inflammation: Implications for cancer. Semin Cell Dev. Biol. 2015, 39, 26–34. [Google Scholar] [CrossRef]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schutze, S.; Schafer, H.; Oestern, S.; Roder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Roder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogwater, F.J.; Nijkamp, M.W.; Smakman, N.; Steller, E.J.; Emmink, B.L.; Westendorp, B.F.; Raats, D.A.; Sprick, M.R.; Schaefer, U.; Van Houdt, W.J.; et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010, 138, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, V.; Kurz, A.; Bertsch, U.; Hubner, S.; Olempska-Muller, M.; Fritsch, J.; Hasler, R.; Pickl, A.; Fritsche, H.; Annewanter, F.; et al. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 2014, 146, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Zhang, B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Takeda, K.; Akiba, H.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Okumura, K.; Yagita, H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999, 163, 1906–1913. [Google Scholar]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef]
- Griffith, T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 1999, 189, 1343–1354. [Google Scholar] [CrossRef]
- Tu, M.M.; Mahmoud, A.B.; Makrigiannis, A.P. Licensed and Unlicensed NK Cells: Differential Roles in Cancer and Viral Control. Front. Immunol. 2016, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valipour, B.; Velaei, K.; Abedelahi, A.; Karimipour, M.; Darabi, M.; Charoudeh, H.N. NK cells: An attractive candidate for cancer therapy. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Dassler-Plenker, J.; Paschen, A.; Putschli, B.; Rattay, S.; Schmitz, S.; Goldeck, M.; Bartok, E.; Hartmann, G.; Coch, C. Direct RIG-I activation in human NK cells induces TRAIL-dependent cytotoxicity toward autologous melanoma cells. Int. J. Cancer 2019, 144, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Yamaguchi, N.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell. Immunol. 2001, 214, 194–200. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Jiao, Y.; Huntington, N.D.; Belz, G.T.; Seillet, C. Type 1 Innate Lymphoid Cell Biology: Lessons Learnt from Natural Killer Cells. Front. Immunol. 2016, 7, 426. [Google Scholar] [CrossRef]
- Mjosberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, G.; Ganter, S.; Barenwaldt, A.; Finke, D. NKp46 Calibrates Tumoricidal Potential of Type 1 Innate Lymphocytes by Regulating TRAIL Expression. J. Immunol. 2018, 200, 3762–3768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cziupka, K.; Busemann, A.; Partecke, L.I.; Potschke, C.; Rath, M.; Traeger, T.; Koerner, P.; von Bernstorff, W.; Kessler, W.; Diedrich, S.; et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) improves the innate immune response and enhances survival in murine polymicrobial sepsis. Crit. Care Med. 2010, 38, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Poetschke, C.; Partecke, L.I.; von Bernstorff, W.; Maier, S.; Broeker, B.M.; Heidecke, C.D. TRAIL induces neutrophil apoptosis and dampens sepsis-induced organ injury in murine colon ascendens stent peritonitis. PLoS ONE 2014, 9, e97451. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.E.; Marriott, H.M.; Lawrie, A.; Francis, S.E.; Sabroe, I.; Renshaw, S.A.; Dockrell, D.H.; Whyte, M.K. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 2011, 90, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Unsinger, J.; Kazama, H.; McDonough, J.S.; Griffith, T.S.; Hotchkiss, R.S.; Ferguson, T.A. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J. Immunol. 2010, 184, 6766–6772. [Google Scholar] [CrossRef] [PubMed]
- Weckmann, M.; Collison, A.; Simpson, J.L.; Kopp, M.V.; Wark, P.A.; Smyth, M.J.; Yagita, H.; Matthaei, K.I.; Hansbro, N.; Whitehead, B.; et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat. Med. 2007, 13, 1308–1315. [Google Scholar] [CrossRef]
- Ikeda, T.; Hirata, S.; Fukushima, S.; Matsunaga, Y.; Ito, T.; Uchino, M.; Nishimura, Y.; Senju, S. Dual effects of TRAIL in suppression of autoimmunity: The inhibition of Th1 cells and the promotion of regulatory T cells. J. Immunol. 2010, 185, 5259–5267. [Google Scholar] [CrossRef]
- Steinwede, K.; Henken, S.; Bohling, J.; Maus, R.; Ueberberg, B.; Brumshagen, C.; Brincks, E.L.; Griffith, T.S.; Welte, T.; Maus, U.A. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J. Exp. Med. 2012, 209, 1937–1952. [Google Scholar] [CrossRef] [Green Version]
- Renshaw, S.A.; Parmar, J.S.; Singleton, V.; Rowe, S.J.; Dockrell, D.H.; Dower, S.K.; Bingle, C.D.; Chilvers, E.R.; Whyte, M.K. Acceleration of human neutrophil apoptosis by TRAIL. J. Immunol. 2003, 170, 1027–1033. [Google Scholar] [CrossRef]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Hara, T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): A novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004, 64, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.M.; Qin, J.; Li, Y.C.; Wang, Y.; Li, D.; Shu, Y.; Luo, C.; Wang, S.S.; Chi, G.; Guo, F.; et al. IL-35 induces N2 phenotype of neutrophils to promote tumor growth. Oncotarget 2017, 8, 33501–33514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosevear, H.M.; Lightfoot, A.J.; O’Donnell, M.A.; Griffith, T.S. The role of neutrophils and TNF-related apoptosis-inducing ligand (TRAIL) in bacillus Calmette-Guerin (BCG) immunotherapy for urothelial carcinoma of the bladder. Cancer Metastasis Rev. 2009, 28, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.T.; Moore, J.M.; Luo, Y.; Chen, X.; Saltsgaver, N.A.; O’Donnell, M.A.; Griffith, T.S. Tumor necrosis factor-related apoptosis-inducing ligand: A novel mechanism for Bacillus Calmette-Guerin-induced antitumor activity. Cancer Res. 2004, 64, 3386–3390. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.P.; Moore, J.M.; Kemp, T.J.; Griffith, T.S. Identification of the mycobacterial subcomponents involved in the release of tumor necrosis factor-related apoptosis-inducing ligand from human neutrophils. Infect. Immun. 2007, 75, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Brincks, E.L.; Gurung, P.; Kucaba, T.A.; Ferguson, T.A. Systemic immunological tolerance to ocular antigens is mediated by TRAIL-expressing CD8+ T cells. J. Immunol. 2011, 186, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Tsai, H.F.; Wu, C.S.; Sung, C.C.; Hsu, P.N. TRAIL-Mediated Suppression of T Cell Receptor Signaling Inhibits T Cell Activation and Inflammation in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Chen, Y.; Goke, R.; Wilmen, A.; Seidel, C.; Goke, A.; Hilliard, B.; Chen, Y. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J. Exp. Med. 2000, 191, 1095–1104. [Google Scholar] [CrossRef]
- Ren, X.; Ye, F.; Jiang, Z.; Chu, Y.; Xiong, S.; Wang, Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ. 2007, 14, 2076–2084. [Google Scholar] [CrossRef]
- Zhang, X.R.; Zhang, L.Y.; Devadas, S.; Li, L.; Keegan, A.D.; Shi, Y.F. Reciprocal expression of TRAIL and CD95L in Th1 and Th2 cells: Role of apoptosis in T helper subset differentiation. Cell Death Differ. 2003, 10, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Oh, Y.; Park, O.; Foss, C.A.; Lim, S.M.; Jo, D.G.; Na, D.H.; Pomper, M.G.; Lee, K.C.; Lee, S. PEGylated TRAIL ameliorates experimental inflammatory arthritis by regulation of Th17 cells and regulatory T cells. J. Control. Release 2017, 267, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, C.; Weiswange, M.; Jeremias, I.; Bayer, C.; Grunert, M.; Debatin, K.M.; Strauss, G. TRAIL-receptor costimulation inhibits proximal TCR signaling and suppresses human T cell activation and proliferation. J. Immunol. 2014, 193, 4021–4031. [Google Scholar] [CrossRef] [PubMed]
- Hirata, S.; Senju, S.; Matsuyoshi, H.; Fukuma, D.; Uemura, Y.; Nishimura, Y. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programmed death-1 ligand. J. Immunol. 2005, 174, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Kucaba, T.A.; Schoenberger, S.P.; Ferguson, T.A.; Griffith, T.S. TRAIL-expressing CD8+ T cells mediate tolerance following soluble peptide-induced peripheral T cell deletion. J. Leukoc. Biol. 2010, 88, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Kazama, H.; VanOosten, R.L.; Earle, J.K., Jr.; Herndon, J.M.; Green, D.R.; Ferguson, T.A. Apoptotic cells induce tolerance by generating helpless CD8+ T cells that produce TRAIL. J. Immunol. 2007, 178, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, R.K.; Skaggs, B.J.; La Cava, A.; Hahn, B.H.; Singh, R.P. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun. Rev. 2010, 9, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, R.J.; Lineburg, K.E.; Kuns, R.D.; Wilson, Y.A.; Raffelt, N.C.; Olver, S.D.; Varelias, A.; Alexander, K.A.; Teal, B.E.; Sparwasser, T.; et al. Identification and expansion of highly suppressive CD8(+)FoxP3(+) regulatory T cells after experimental allogeneic bone marrow transplantation. Blood 2012, 119, 5898–5908. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Stollhof, L.; Poetschke, C.; von Bernstorff, W.; Partecke, L.I.; Diedrich, S.; Maier, S.; Broker, B.M.; Heidecke, C.D. TNF-related apoptosis-inducing ligand deficiency enhances survival in murine colon ascendens stent peritonitis. J. Inflamm. Res. 2016, 9, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Rai, D.; Condotta, S.A.; Babcock, J.C.; Badovinac, V.P.; Griffith, T.S. Immune unresponsiveness to secondary heterologous bacterial infection after sepsis induction is TRAIL dependent. J. Immunol. 2011, 187, 2148–2154. [Google Scholar] [CrossRef]
- Brincks, E.L.; Katewa, A.; Kucaba, T.A.; Griffith, T.S.; Legge, K.L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 2008, 181, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Peteranderl, C.; Morales-Nebreda, L.; Selvakumar, B.; Lecuona, E.; Vadasz, I.; Morty, R.E.; Schmoldt, C.; Bespalowa, J.; Wolff, T.; Pleschka, S.; et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Investig. 2016, 126, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I.S.; Wikstrom, M.E.; Brizard, G.; Coudert, J.D.; Estcourt, M.J.; Manzur, M.; O’Reilly, L.A.; Smyth, M.J.; Trapani, J.A.; Hill, G.R.; et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 2014, 41, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Loewendorf, A.; Wang, Q.; McDonald, B.; Redwood, A.; Benedict, C.A. Inhibition of the TRAIL death receptor by CMV reveals its importance in NK cell-mediated antiviral defense. PLoS Pathog. 2014, 10, e1004268. [Google Scholar] [CrossRef] [PubMed]
- Picarda, G.; Ghosh, R.; McDonald, B.; Verma, S.; Thiault, N.; El Morabiti, R.; Griffith, T.S.; Benedict, C.A. Cytomegalovirus evades TRAIL-mediated innate lymphoid cell 1 defenses. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zerafa, N.; Westwood, J.A.; Cretney, E.; Mitchell, S.; Waring, P.; Iezzi, M.; Smyth, M.J. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J. Immunol. 2005, 175, 5586–5590. [Google Scholar] [CrossRef]
- Sedger, L.M.; Glaccum, M.B.; Schuh, J.C.; Kanaly, S.T.; Williamson, E.; Kayagaki, N.; Yun, T.; Smolak, P.; Le, T.; Goodwin, R.; et al. Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur. J. Immunol. 2002, 32, 2246–2254. [Google Scholar] [CrossRef]
- Finnberg, N.; Klein-Szanto, A.J.; El-Deiry, W.S. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J. Clin. Investig. 2008, 118, 111–123. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailey, S.L.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schutz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Investig. 2008, 118, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.H.; Trauzold, A.; Roder, C.; Pan, G.; Zheng, C.; Kalthoff, H. The potential molecular mechanism of overexpression of uPA, IL-8, MMP-7 and MMP-9 induced by TRAIL in pancreatic cancer cell. Hepatobiliary Pancreat. Dis Int. 2008, 7, 201–209. [Google Scholar] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, K.; Normann, L.; Sendler, M.; Kading, A.; Heidecke, C.D.; Partecke, L.I.; von Bernstorff, W. TRAIL Promotes Tumor Growth in a Syngeneic Murine Orthotopic Pancreatic Cancer Model and Affects the Host Immune Response. Pancreas 2016, 45, 401–408. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyer, K.; Baukloh, A.-K.; Stoyanova, A.; Kamphues, C.; Sattler, A.; Kotsch, K. Interactions of Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (TRAIL) with the Immune System: Implications for Inflammation and Cancer. Cancers 2019, 11, 1161. https://doi.org/10.3390/cancers11081161
Beyer K, Baukloh A-K, Stoyanova A, Kamphues C, Sattler A, Kotsch K. Interactions of Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (TRAIL) with the Immune System: Implications for Inflammation and Cancer. Cancers. 2019; 11(8):1161. https://doi.org/10.3390/cancers11081161
Chicago/Turabian StyleBeyer, Katharina, Ann-Kathrin Baukloh, Ani Stoyanova, Carsten Kamphues, Arne Sattler, and Katja Kotsch. 2019. "Interactions of Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (TRAIL) with the Immune System: Implications for Inflammation and Cancer" Cancers 11, no. 8: 1161. https://doi.org/10.3390/cancers11081161