Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
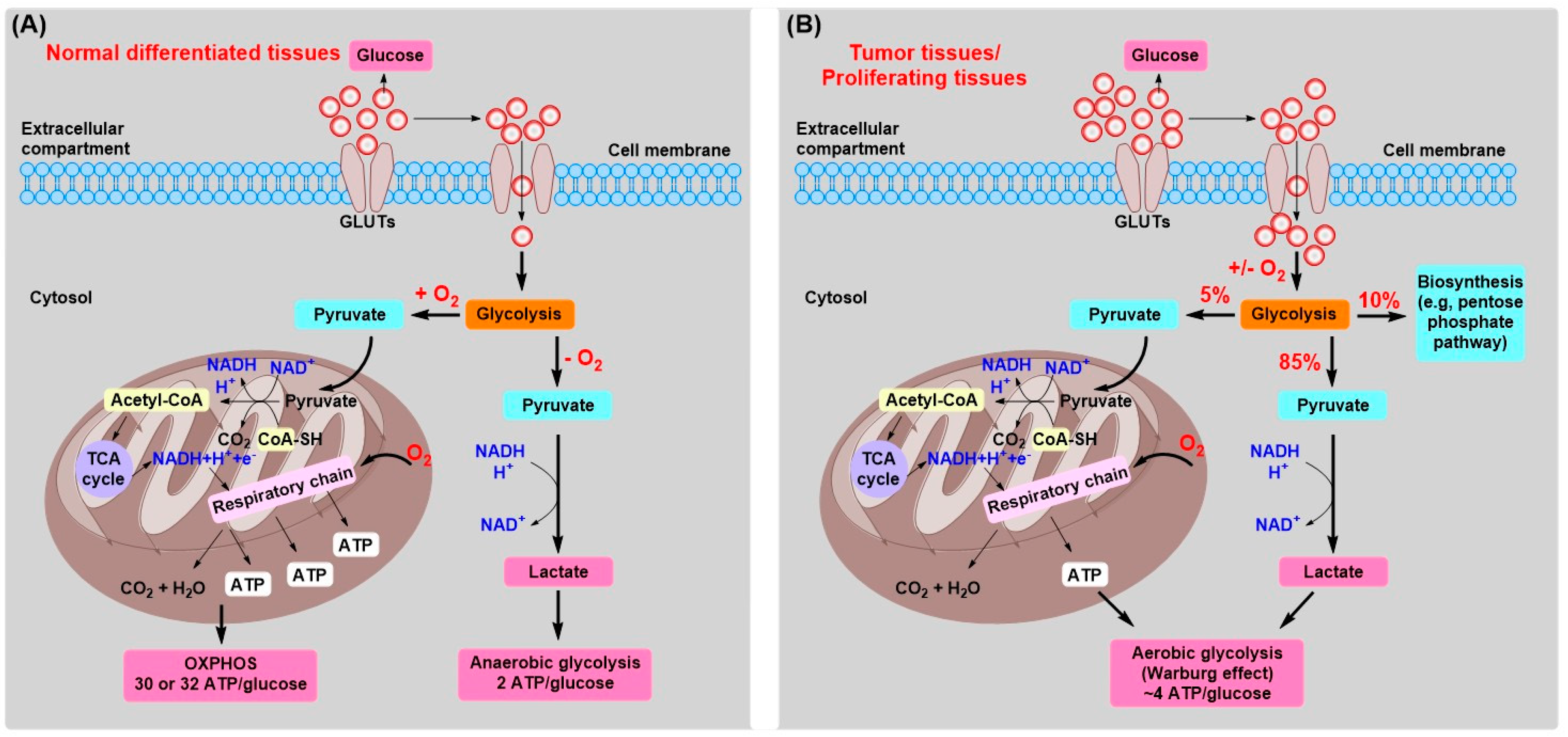
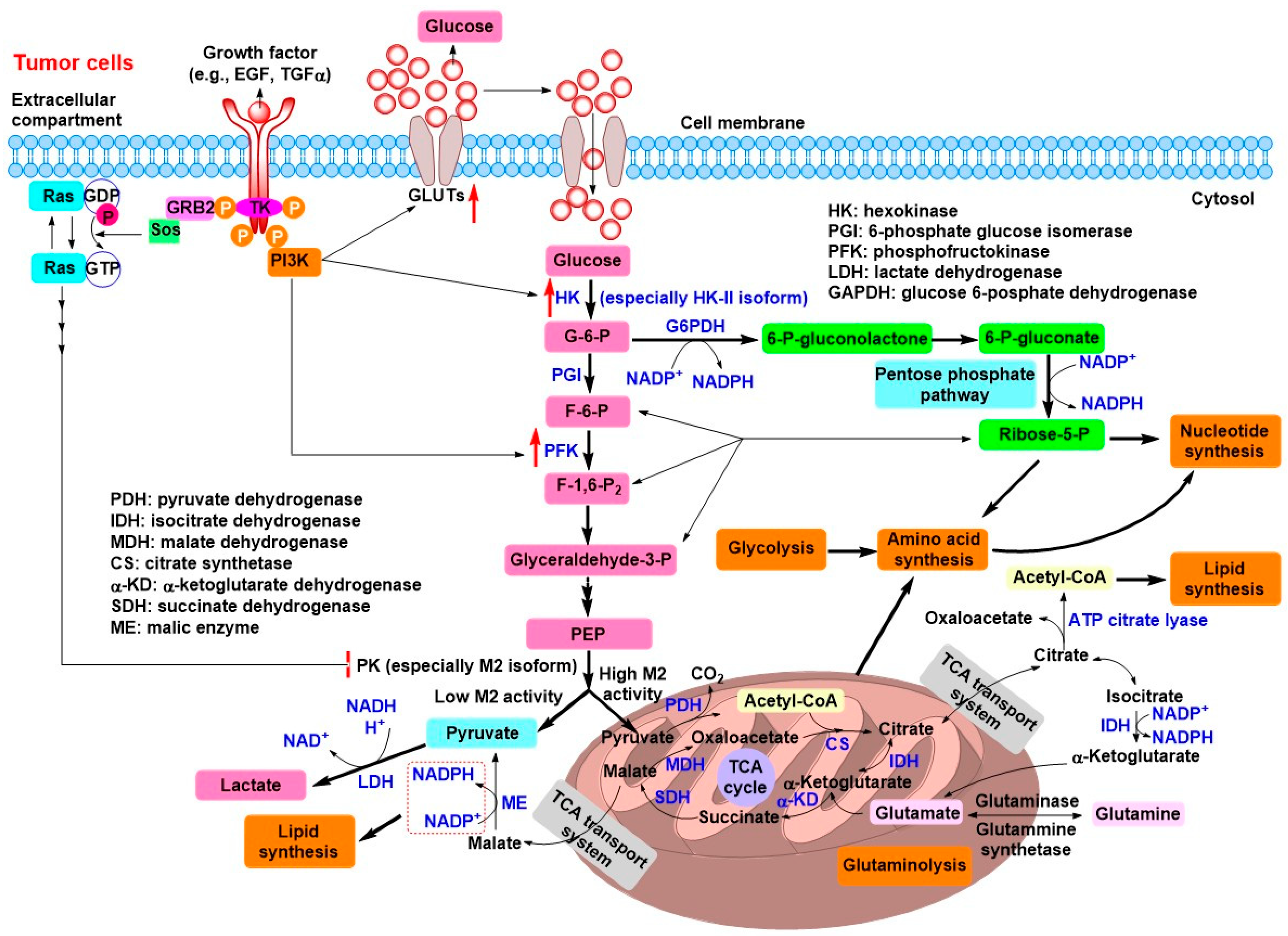
2. Tumor Energy Metabolism: Target for Tumor Chemotherapy
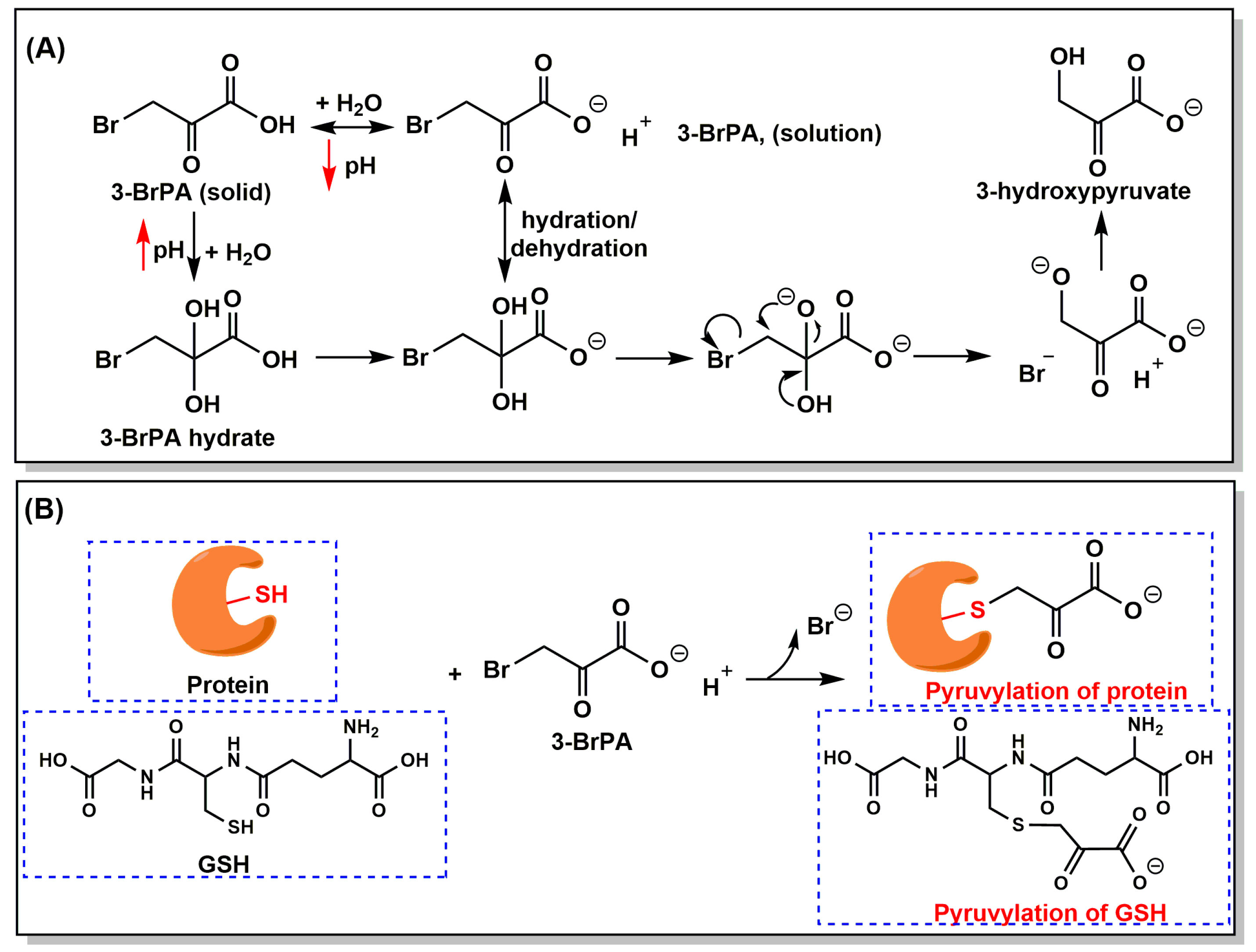
3. Properties, Mechanism of Action, and Cellular Targets of 3-BrPA
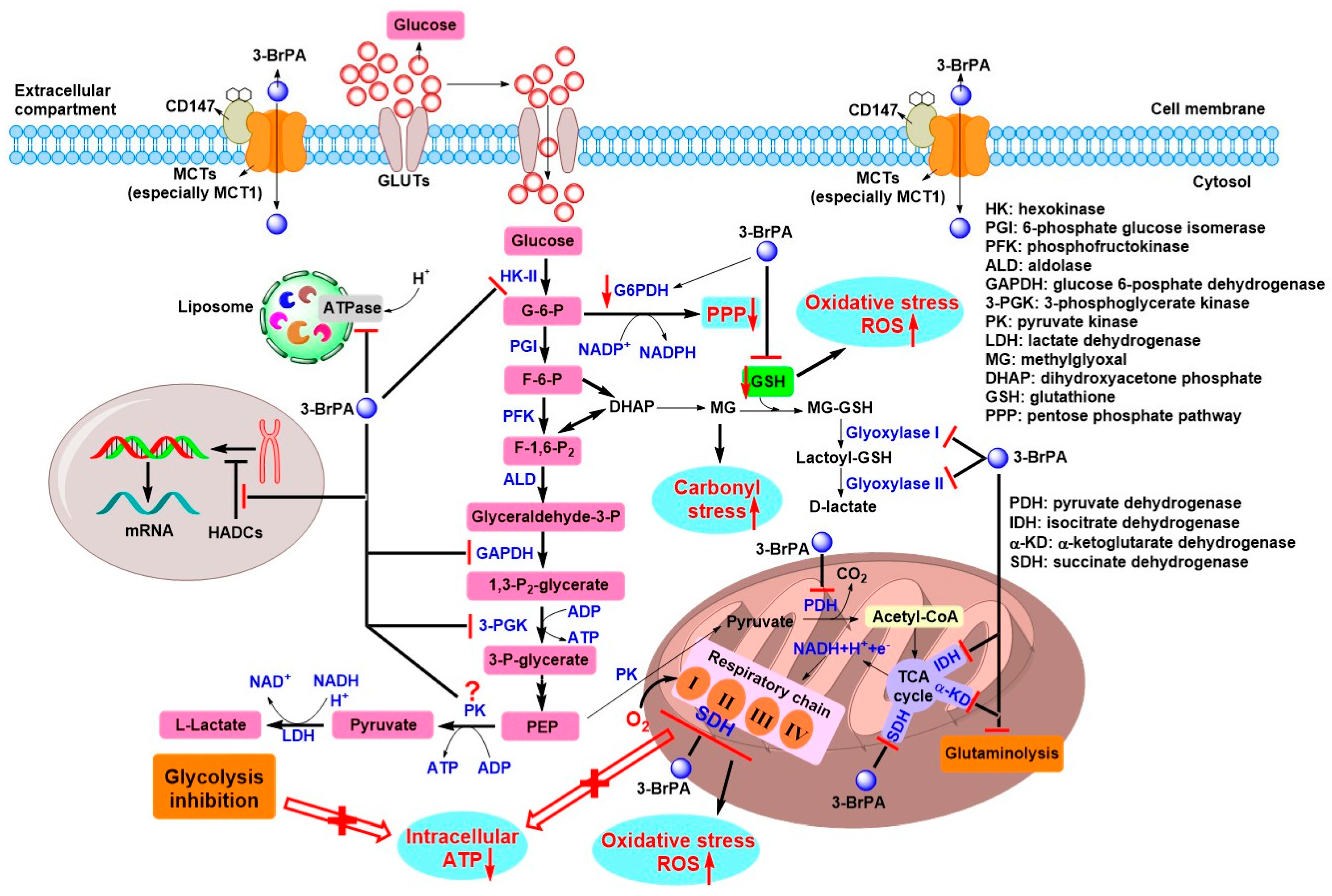
4. Antitumor Effects of 3-BrPA and the Underlying Mechanism
4.1. Initial Antitumor Studies and Cell Death Induced by 3-BrPA
4.2. Role of GSH and ROS on the Antitumor Effects of 3-BrPA
4.3. Specific Tumor Selectivity of 3-BrPA
4.4. Chemosensitivity of 3-BrPA with Other Antitumor Drugs In Vitro and In Vivo
4.5. Clinical Studies of 3-BrPA for Tumor Treatment
5. Novel Chemotherapeutic Strategies of 3-BrPA
6. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Expo. Tumor Res. 1978, 22, 190–274. [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Cuezva, J.M. Oxidative Phosphorylation and Cancer: The Ongoing Warburg Hypothesis; Humana Press Inc: Totowa, NJ, USA, 2009; pp. 1–18. [Google Scholar]
- Grander, D. How do mutated oncogenes and tumor suppressor genes cause cancer? Med. Oncol. 1998, 15, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [PubMed]
- Cardaci, S.; Desideri, E.; Ciriolo, M.R. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J. Bioenerg. Biomembr. 2012, 44, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lis, P.; Dylag, M.; Niedzwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ulaszewski, S. The HK2 dependent “Warburg effect” and mitochondrial oxidative phosphorylation in cancer: Targets for effective therapy with 3-bromopyruvate. Molecules 2016, 21, 1730. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2008, 17, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [PubMed]
- Kim, W.; Yoon, J.H.; Jeong, J.M.; Cheon, G.J.; Lee, T.S.; Yang, J.I.; Park, S.C.; Lee, H.S. Apoptosis-inducing antitumor efficacy of hexokinase II inhibitor in hepatocellular carcinoma. Mol. Cancer Ther. 2007, 6, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R.; et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Zhou, J.; Zhou, Q.; Pan, F.; Zhong, D.; Liang, H. Silencing hexokinase II gene sensitizes human colon cancer cells to 5-fluorouracil. Hepato-Gastroenterol. 2009, 56, 355–360. [Google Scholar]
- Nakashima, R.A.; Mangan, P.S.; Colombini, M.; Pedersen, P.L. Hexokinase receptor complex in hepatoma mitochondria—Evidence from N,N′-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry 1986, 25, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy-metabolism of tumor-cells—Requirement for a form of hexokinase with a propensity for mitochondrial binding. J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar] [PubMed]
- Nakashima, R.A.; Paggi, M.G.; Scott, L.J.; Pedersen, P.L. Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res. 1988, 48, 913–919. [Google Scholar] [PubMed]
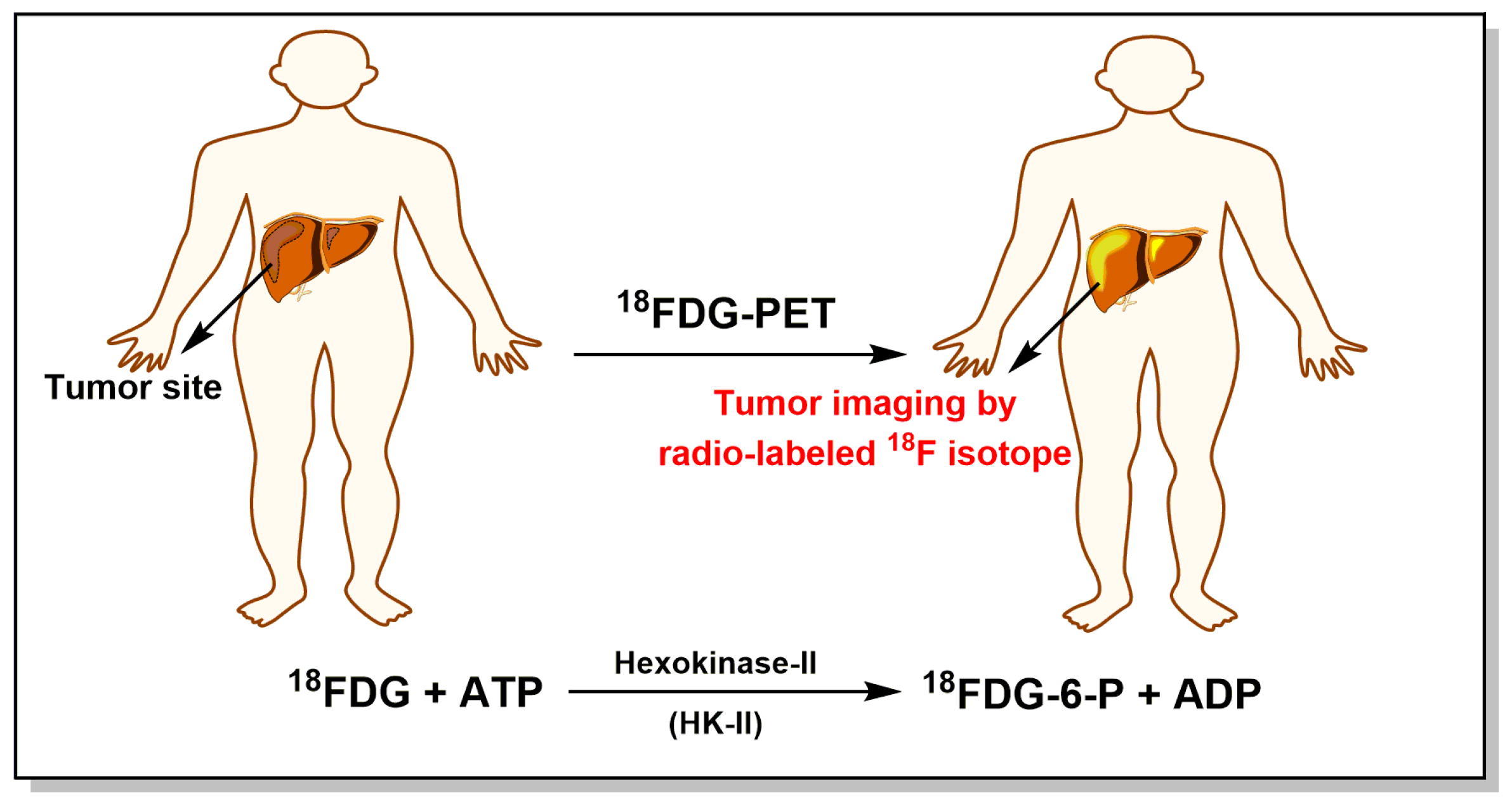
- Ding, J.J.; Chen, Y.L.; Zhou, S.H.; Zhao, K. Positron emission tomography/computed tomography in the diagnosis, staging, and prognostic evaluation of natural killer/T-cell lymphoma. J. Int. Med. Res. 2018, 46, 4920–4929. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Suzuki, S.; Matsutani, N.; Kawamura, M. 18F-fluorodeoxyglucose positron emission tomography/computed tomography in the evaluation of clinically node-negative non-small cell lung cancer. Thorac. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Cascio, M.B.; Sawa, A. GAPDH as a sensor of NO stress. BBA-Mol. Basis Dis. 2006, 1762, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Liu, S.; Sun, M.Z. Novel insight into the role of GAPDH playing in tumor. Clin. Transl. Oncol. 2013, 15, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yuan, S.; Hu, Y.; Zhang, H.; Wu, W.; Zeng, Z.; Yang, J.; Yun, J.; Xu, R.; Huang, P. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-bromopyruvate propyl ester. J. Bioenerg. Biomembr. 2012, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.; Pellin-Carcelen, A.; Agusti, J.; Murgui, A.; Jorda, E.; Pellin, A.; Monteagudo, C. Deregulation of glyceraldehyde-3-phosphate dehydrogenase expression during tumor progression of human cutaneous melanoma. Anticancer Res. 2015, 35, 439–444. [Google Scholar]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The expression of glyceraldehyde-3-phosphate dehydrogenase associated cell cycle (GACC) genes correlates with cancer stage and poor survival in patients with solid tumors. PLoS ONE 2013, 8, e61262. [Google Scholar] [CrossRef]
- Gupta, V.; Bamezai, R.N.K. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef]
- Presek, P.; Reinacher, M.; Eigenbrodt, E. Pyruvate-kinase type M2 is phosphorylated at tyrosine residues in cells transformed by Rous-sarcoma virus. FEBS Lett. 1988, 242, 194–198. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Durr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Ma, F.; Wang, Y.; Hao, L.; Zeng, H.; Jia, C.; Wang, Y.; Liu, P.; Ong, I.M.; Li, B.; et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017, 19, 1358–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Azevedo-Silva, J.; Queiros, O.; Baltazar, F.; Uaszewski, S.; Goffeau, A.; Ko, Y.H.; Pedersen, P.L.; Preto, A.; Casal, M. The anticancer agent 3-bromopyruvate: A simple but powerful molecule taken from the lab to the bedside. J. Bioenerg. Biomembr. 2016, 48, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Gwak, G.Y.; Yoon, J.H.; Kim, K.M.; Lee, H.S.; Chung, J.W.; Gores, G.J. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 2005, 42, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Shoshan, M.C. 3-Bromopyruvate: Targets and outcomes. J. Bioenerg. Biomembr. 2012, 44, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticanc. 2004, 4, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.P.; Rabin, B.R. Effects of bromopyruvate on the control and catalytic properties of glutamate dehydrogenase. Eur. J. Biochem. 1969, 11, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Sieber, M.; Schellenberger, A. The carbonyl reactivity of 3-bromopyruvate and related compounds. Bioorgan. Chem. 1982, 11, 478–484. [Google Scholar] [CrossRef]
- Glick, M.; Biddle, P.; Jantzi, J.; Weaver, S.; Schirch, D. The antitumor agent 3-bromopyruvate has a short half-life at physiological conditions. Biochem. Biophys. Res. Commun. 2014, 452, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Meloche, H.P.; Monti, C.T.; Hogueangeletti, R.A. Identification of bromopyruvate-sensitive glutamate within active-site of 2-keto-3-deoxygluconate-6-P aldolase. Biochem. Biophys. Res. Commun. 1978, 84, 589–594. [Google Scholar] [CrossRef]
- Bailey, C.T.; Patch, M.G.; Carrano, C.J. Affinity labels for the anion-binding site in ovotransferrin. Biochemistry 1988, 27, 6276–6282. [Google Scholar] [CrossRef] [PubMed]
- Fonda, M.L. Bromopyruvate inactivation of glutamate apodecarboxylase—Kinetics and specificity. J. Biol. Chem. 1976, 251, 229–235. [Google Scholar] [PubMed]
- Ko, Y.H.; McFadden, B.A. Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate. Arch. Biochem. Biophys. 1990, 278, 373–380. [Google Scholar] [CrossRef]
- Kratky, M.; Vinsova, J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, K.; Dylag, M.; Augustyniak, D.; Majkowska-Skrobek, G.; Cal-Bakowska, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ulaszewski, S. Glutathione may have implications in the design of 3-bromopyruvate treatment protocols for both fungal and algal infections as well as multiple myeloma. Oncotarget 2016, 7, 65614–65626. [Google Scholar] [CrossRef] [PubMed]
- Dylag, M.; Lis, P.; Niedwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ulaszewski, S. 3-Bromopyruvate: A novel antifungal agent against the human pathogen Cryptococcus neoformans. Biochem. Biophys. Res. Commun. 2013, 434, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Jagielski, T.; Niedzwiecka, K.; Roeske, K.; Dylag, M. 3-Bromopyruvate as an alternative option for the treatment of protothecosis. Front. Pharmacol. 2018, 9, 375. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.P.; Reynafarje, B.; Pedersen, P.L. Glucose catabolism in African trypanosomes—Evidence that the terminal step is catalyzed by a pyruvate transporter capable of facilitating uptake of toxic analogs. J. Biol. Chem. 1993, 268, 3654–3661. [Google Scholar] [PubMed]
- de Lima, L.P.O.; Seabra, S.H.; Carneiro, H.; Barbosa, H.S. Effect of 3-bromopyruvate and atovaquone on infection during in vitro interaction of Toxoplasma gondii and LLC-MK2 Cells. Antimicrob. Agents Chemther. 2015, 59, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Lu, W.; Huang, P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. BBA-Bioenergetics 2009, 1787, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Pandey, S.K.; Kumar, A.; Kujur, P.K.; Singh, R.P.; Singh, S.M. Antitumor and chemosensitizing action of 3-bromopyruvate: Implication of deregulated metabolism. Chem.-Biol. Interact. 2017, 270, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kujur, P.K.; Pandey, S.K.; Goel, Y.; Maurya, B.N.; Verma, A.; Kumar, A.; Singh, R.P.; Singh, S.M. Antitumor action of 3-bromopyruvate implicates reorganized tumor growth regulatory components of tumor milieu, cell cycle arrest and induction of mitochondria-dependent tumor cell death. Toxicol. Appl. Pharm. 2018, 339, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Attia, Y.M.; El-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838. [Google Scholar] [CrossRef] [PubMed]
- Pereira da Silva, A.P.; El-Bacha, T.; Kyaw, N.; dos Santos, R.S.; da-Silva, W.S.; Almeida, F.C.L.; Da Poian, A.T.; Galina, A. Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem. J. 2009, 417, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F.H.; Kunjithapatham, R.; Buijs, M.; Vossen, J.A.; Tchernyshyov, I.; Cole, R.N.; Syed, L.H.; Rao, P.P.; Ota, S.; et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009, 29, 4909–4918. [Google Scholar] [PubMed]
- Ehrke, E.; Arend, C.; Dringen, R. 3-Bromopyruvate inhibits glycolysis, depletes cellular glutathione, and compromises the viability of cultured primary rat astrocytes. J. Neurosci. Res. 2015, 93, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Vacca, R.A.; de Bari, L. 3-Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J. Bioenerg. Biomembr. 2015, 47, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Yu, J.; Nigjeh, E.N.; Wang, W.; Myint, P.T.; Zandi, E.; Hofman, F.M.; Schonthal, A.H. A perillyl alcohol-conjugated analog of 3-bromopyruvate without cellular uptake dependency on monocarboxylate transporter 1 and with activity in 3-BP-resistant tumor cells. Cancer Lett. 2017, 400, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Dell’Antone, P. Targets of 3-bromopyruvate, a new, energy depleting, anticancer agent. Med. Chem. 2009, 5, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Torbenson, M.S.; Rao, P.P.; Carson, K.A.; Buijs, M.; Vali, M.; Geschwind, J.F.H. Human hepatocellular carcinoma in a mouse model: Assessment of tumor response to percutaneous ablation by using glyceraldehyde-3-phosphate dehydrogenase antagonists. Radiology 2012, 262, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Acan, N.L.; Ozer, N. Modification of human erythrocyte pyruvate kinase by an active site-directed reagent: Bromopyruvate. J. Enzym. Inhib. 2001, 16, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.L.; Suelter, C.H. Modification of yeast pyruvate-kinase by an active site-directed reagent, bromopyruvate. J. Biol. Chem. 1979, 254, 1811–1815. [Google Scholar] [PubMed]
- Vander Heiden, M.G.; Christofk, H.R.; Schuman, E.; Subtelny, A.O.; Sharfi, H.; Harlow, E.E.; Xian, J.; Cantley, L.C. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem. Pharmacol. 2010, 79, 1118–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardim-Messeder, D.; Moreira-Pacheco, F. 3-Bromopyruvic acid inhibits tricarboxylic acid cycle and glutaminolysis in HepG2 Cells. Anticancer Res. 2016, 36, 2233–2241. [Google Scholar] [PubMed]
- Lowe, P.N.; Perham, R.N. Bromopyruvate as an active-site-directed inhibitor of the pyruvate-dehydrogenase multienzyme complex from Escherichia coli. Biochemistry 1984, 23, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Ali, M.S.; Patel, M.S. Involvement of alpha-cysteine-62 and beta-tryptophan-135 in human pyruvate dehydrogenase catalysis. Arch. Biochem. Biophys. 1999, 369, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, B.M.; Felberg, N.T.; Hollocher, T.C. The inactivation of succinate dehydrogenase by bromopyruvate. Biochim. Biophys. Acta 1971, 227, 219–231. [Google Scholar] [CrossRef]
- Kwiatkowska, E.; Wojtala, M.; Gajewska, A.; Soszynski, M.; Bartosz, G.; Sadowska-Bartosz, I. Effect of 3-bromopyruvate acid on the redox equilibrium in non-invasive MCF-7 and invasive MDA-MB-231 breast cancer cells. J. Bioenerg. Biomembr. 2016, 48, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Z.; Xin, H.; Nickoloff, B.J. 3-Bromopyruvate induces necrotic cell death in sensitive melanoma cell lines. Biochem. Biophys. Res. Commun. 2010, 396, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Szewczyk, R.; Jaremko, L.; Jaremko, M.; Bartosz, G. Anticancer agent 3-bromopyruvic acid forms a conjugate with glutathione. Pharmacol. Rep. 2016, 68, 502–505. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M.; Baghdadi, H.; Zolaly, M.; Almaramhy, H.H.; Ayat, M.; Donki, J.G. The promising anticancer drug 3-bromopyruvate is metabolized through glutathione conjugation which affects chemoresistance and clinical practice: An evidence-based view. Med. Hypotheses 2017, 100, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Jo, A.; Lee, S.; Bin Kim, J.; Chang, Y.; Nam, J.Y.; Cho, H.; Cho, Y.Y.; Cho, E.J.; Lee, J.H.; et al. 3-Bromopyruvate and buthionine sulfoximine effectively kill anoikis-resistant hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0174271. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Karunakaran, S.K.; Itagaki, S.; Gopal, E.; Elangovan, S.; Prasad, P.D.; Ganapathy, V. Transport by SLC5A8 with subsequent inhibition of histone deacetylase 1 (HDAC1) and HDAC3 underlies the antitumor activity of 3-bromopyruvate. Cancer 2009, 115, 4655–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Antone, P. Inactivation of H+-vacuolar ATPase by the energy blocker 3-bromopyruvate, a new antitumour agent. Life Sci. 2006, 79, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, J.F. H.; Ko, Y.H.; Torbenson, M.S.; Magee, C.; Pedersen, P.L. Novel therapy for liver cancer: Direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res. 2002, 62, 3909–3913. [Google Scholar] [PubMed]
- Ko, Y.H.; Smith, B.L.; Wang, Y.C.; Pomper, M.G.; Rini, D.A.; Torbenson, M.S.; Hullihen, J.; Pedersen, P.L. Advanced cancers: Eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem. Biophys. Res. Commun. 2004, 324, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Zhang, X.D.; Lemoisson, E.; Louis, M.H.; Allouche, S.; Lincet, H.; Poulain, L. Experimental results using 3-bromopyruvate in mesothelioma: In vitro and in vivo studies. J. Bioenerg. Biomembr. 2012, 44, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Calvino, E.; Estan, M.C.; Sanchez-Martin, C.; Brea, R.; de Blas, E.; Boyano-Adanez, M.D.; Rial, E.; Aller, P. Regulation of death induction and chemosensitizing action of 3-bromopyruvate in myeloid leukemia cells: Energy depletion, oxidative stress, and protein kinase activity modulation. J. Pharmacol. Exp. Ther. 2014, 348, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pan, J.; North, P.E.; Yang, S.; Lubet, R.A.; Wang, Y.; You, M. Aerosolized 3-bromopyruvate inhibits lung tumorigenesis without causing liver toxicity. Cancer Prev. Res. 2012, 5, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.J.; Li, S.S.; Zhang, D.P.; Liu, T.J.; Yu, M.; Wang, F. Separate and concurrent use of 2-deoxy-d-glucose and 3-bromopyruvate in pancreatic cancer cells. Oncol. Rep. 2013, 29, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Ihrlund, L.S.; Hernlund, E.; Khan, O.; Shoshan, M.C. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2008, 2, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiasserini, D.; Davidescu, M.; Orvietani, P.L.; Susta, F.; Macchioni, L.; Petricciuolo, M.; Castigli, E.; Roberti, R.; Binaglia, L.; Corazzi, L. 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteom. 2017, 152, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Messner, K.R.; Imlay, J.A. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 2002, 277, 42563–42571. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family-Role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Myeroff, L.; Smiraglia, D.; Romero, M.F.; Pretlow, T.P.; Kasturi, L.; Lutterbaugh, J.; Rerko, R.M.; Casey, G.; Issa, J.P.; et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc. Natl. Acad. Sci. USA 2003, 100, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Gopal, E.; Martin, P.M.; Itagaki, S.; Miyauchi, S.; Prasad, P.D. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008, 10, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Silva, J.; Queiros, O.; Ribeiro, A.; Baltazar, F.; Young, K.H.; Pedersen, P.L.; Preto, A.; Casal, M. The cytotoxicity of 3-bromopyruvate in breast cancer cells depends on extracellular pH. Biochem. J. 2015, 467, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G.; Peng, Y.Z. The specific role of O6-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G. The induction and repair of DNA interstrand crosslinks and implications in cancer chemotherapy. Anti-Cancer Agents Med. Chem. 2016, 16, 221–246. [Google Scholar]
- Sun, G.H.; Fan, T.J.; Zhao, L.J.; Zhou, Y.; Zhong, R.G. The potential of combi-molecules with DNA-damaging function as anticancer agents. Future Med. Chem. 2017, 9, 403–435. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhao, L.J.; Fan, T.J.; Li, S.S.; Zhong, R.G. Investigations on the effect of O6-benzylguanine on the formation of dG-dC interstrand cross-links induced by chloroethylnitrosoureas in human glioma cells using stable isotope dilution high-performance liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2014, 27, 1253–1262. [Google Scholar] [PubMed]
- Kapp, N.; Stander, X.X.; Stander, B.A. Synergistic in vitro effects of combining an antiglycolytic, 3-bromopyruvate, and a bromodomain-4 inhibitor on U937 myeloid leukemia cells. Anti-Cancer Drug. 2018, 29, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.H.; Bloomston, M.; Zhang, T.; Frankel, W.L.; Jia, G.; Wang, B.; Hall, N.C.; Koch, R.M.; Cheng, H.; Knopp, M.V.; et al. Synergistic antipancreatic tumor effect by simultaneously targeting hypoxic cancer cells with HSP90 inhibitor and glycolysis inhibitor. Clin. Cancer Res. 2008, 14, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Tozzi, F.; Chen, J.Y.; Fan, F.; Xia, L.; Wang, J.R.; Gao, G.; Zhang, A.J.; Xia, X.F.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Lopes, G.D.; Dicksey, J.S.; Peters, W.P.; Palalay, M.; Chang, A.Y. Final results of a prematurely discontinued Phase 1/2 study of eniluracil with escalating doses of 5-fluorouracil administered orally in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2011, 68, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.L.; Ma, L.Y.; Liu, F.; Zhang, Z.R.; Zhao, S.R.; Huo, Q.; Zhang, P.; Zheng, H.L.; Liu, H. Synergistic antitumor effect of 3-bromopyruvate and 5-fluorouracil against human colorectal cancer through cell cycle arrest and induction of apoptosis. Anti-Cancer Drug. 2017, 28, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Wei, L.; Zhang, X.J.; Liu, X.F.; Chen, Y.S.; Zhang, S.; Zhou, L.Z.; Li, Q.X.; Pan, Q.; Zhao, S.R.; et al. 3-Bromopyruvate sensitizes human breast cancer cells to TRAIL-induced apoptosis via the phosphorylated AMPK-mediated upregulation of DR5. Oncol. Rep. 2018, 40, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS ONE 2011, 6, e27222. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.F.; Qiu, Y.Y.; Yu, S.T.; Clark, S.; Chu, F.; Madonna, M.B. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J. Pediatr. Surg. 2014, 49, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Y.M.; Hong, H.Y.; Zhao, S.R.; Zou, X.; Ma, R.Q.; Jiang, C.C.; Wang, Z.W.; Li, H.B.; Liu, H. 3-bromopyruvate enhanced daunorubicin-induced cytotoxicity involved in monocarboxylate transporter 1 in breast cancer cells. Am. J. Cancer Res. 2015, 5, 2673–2685. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xu, J.; Yuan, W.Q.; Wu, B.J.; Wang, H.; Liu, G.Q.; Wang, X.X.; Du, J.; Cai, S.H. The reversal effects of 3-bromopyruvate on multidrug resistance in vitro and in vivo derived from human breast MCF-7/ADR cells. PLoS ONE 2014, 9, e112132. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Grebowski, J.; Kepka, E.; Studzian, M.; Bartosz, G.; Pulaski, L. ABCB1-overexpressing MDCK-II cells are hypersensitive to 3-bromopyruvic acid. Life Sci. 2016, 162, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, Y.; Kobayashi, M.; Ideno, M.; Narumi, K.; Furugen, A.; Iseki, K. Valproate sensitizes human glioblastoma cells to 3-bromopyruvate-induced cytotoxicity. Int. J. Pharm. 2018, 551, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Vali, M.; Liapi, E.; Kowalski, J.; Hong, K.; Khwaja, A.; Torbenson, M.S.; Georgiades, C.; Geschwind, J.F.H. Intraarterial therapy with a new potent inhibitor of tumor metabolism (3-bromopyruvate): Identification of therapeutic dose and method of injection in an animal model of liver cancer. J. Vasc. Interv. Radiol. 2007, 18, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Buijs, M.; Wijlemans, J.W.; Kwak, B.K.; Ota, S.; Geschwind, J.F.H. Antiglycolytic therapy combined with an image-guided minimally invasive delivery strategy for the treatment of breast cancer. J. Vasc. Interv. Radiol. 2013, 24, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.M.; Chung, J.W.; Jae, H.J.; Eh, H.; Son, K.R.; Lee, K.C.; Park, J.H. Local toxicity of hepatic arterial infusion of hexokinase II inhibitor, 3-bromopyruvate: In vivo investigation in normal rabbit model. Acad. Radiol. 2007, 14, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kunjithapatham, R.; Geschwind, J.F.H.; Rao, P.P.; Boronina, T.N.; Cole, R.N. Systemic administration of 3-bromopyruvate reveals its interaction with serum proteins in a rat model. BMC Res. Notes 2013, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M.; Mohamed, W.G.; Seddik, M.A.H.; Ahmed, A.S.A.; Mahmoudi, A.G.; Amer, W.H.; Nabo, M.M.H.; Hamed, A.R.; Ahmed, N.S.; Abd-Allah, A.A.R. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: A concise literature review and case study. Chin. J. Cancer 2014, 33, 356–364. [Google Scholar] [PubMed]
- Banfalvi, T.; Edesne, M.B.; Gergye, M.; Udvarhelyi, N.; Orosz, Z.; Gilde, K.; Kremmer, T.; Otto, S.; Timar, J. Laboratory markers of melanoma progression. Magy. Onkol. 2003, 47, 89–104. [Google Scholar] [PubMed]
- Agarwala, S.S.; Keilholz, U.; Gilles, E.; Bedikian, A.Y.; Wu, J.; Kay, R.; Stein, C.A.; Itri, L.M.; Suciu, S.; Eggermont, A.M.M. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur. J. Cancer 2009, 45, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Pandey, S.K.; Goel, Y.; Kujur, P.K.; Maurya, B.N.; Verma, A.; Kumar, A.; Singh, R.P.; Singh, S.M. Protective and recuperative effects of 3-bromopyruvate on immunological, hepatic and renal homeostasis in a murine host bearing ascitic lymphoma: Implication of niche dependent differential roles of macrophages. Biomed. Pharmacother. 2018, 99, 970–985. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.M. Enhancing anticancer effects, decreasing risks and solving practical problems facing 3-bromopyruvate in clinical oncology: 10 years of research experience. Int. J. Nanomed. 2018, 13, 4699–4709. [Google Scholar] [CrossRef] [PubMed]
- Wicks, R.T.; Azadi, J.; Mangraviti, A.; Zhang, I.; Hwang, L.; Joshi, A.; Bow, H.; Hutt-Cabezas, M.; Martin, K.L.; Rudek, M.A.; et al. Local delivery of cancer-cell glycolytic inhibitors in high-grade glioma. Neuro-Oncology 2015, 17, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Feldwisch-Drentrup, H. Candidate Cancer Drug Suspected after Death of Three Patients at an Alternative Medicine Clinic. Available online: http://dx.doi.org/10.1126/science.aah7192 (accessed on 12 August 2016).
- Chapiro, J.; Sur, S.; Savic, L.J.; Ganapathy-Kanniappan, S.; Reyes, J.; Duran, R.; Thiruganasambandam, S.C.; Moats, C.R.; Lin, M.; Luo, W.; et al. Systemic delivery of microencapsulated 3-bromopyruvate for the therapy of pancreatic Cancer. Clin. Cancer Res. 2014, 20, 6406–6417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pan, J.; Lubet, R.A.; Komas, S.M.; Kalyanaraman, B.; Wang, Y.; You, M. Enhanced antitumor activity of 3-bromopyruvate in combination with rapamycin in vivo and in vitro. Cancer Prev. Res. 2015, 8, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.G.; Zage, P.E.; Akers, L.J.; Ghisoli, M.L.; Chen, Z.; Fang, W.; Kannan, S.; Graham, T.; Zeng, L.; Franklin, A.R.; et al. The combination of the novel glycolysis inhibitor 3-BrOP and rapamycin is effective against neuroblastoma. Investig. New Drugs 2012, 30, 191–199. [Google Scholar] [CrossRef] [PubMed]
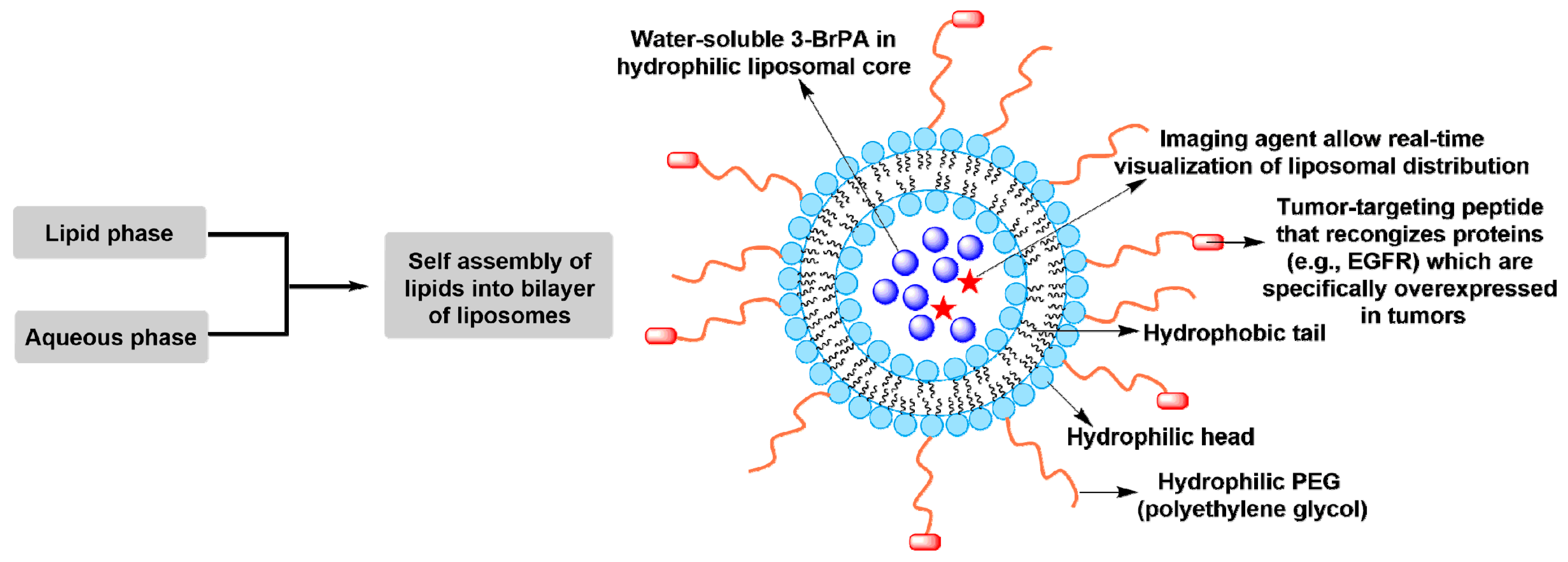
- Gandham, S.K.; Talekar, M.; Singh, A.; Amiji, M.M. Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int. J. Nanomed. 2015, 10, 4405–4423. [Google Scholar]
- Zhang, Y.; Wei, J.; Xu, J.; Leong, W.S.; Liu, G.; Ji, T.; Cheng, Z.; Wang, J.; Lang, J.; Zhao, Y.; et al. Suppression of tumor energy supply by liposomal nanoparticle-mediated inhibition of aerobic glycolysis. ACS Appl. Mater. Int. 2018, 10, 2347–2353. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. https://doi.org/10.3390/cancers11030317
Fan T, Sun G, Sun X, Zhao L, Zhong R, Peng Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers. 2019; 11(3):317. https://doi.org/10.3390/cancers11030317
Chicago/Turabian StyleFan, Tengjiao, Guohui Sun, Xiaodong Sun, Lijiao Zhao, Rugang Zhong, and Yongzhen Peng. 2019. "Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment" Cancers 11, no. 3: 317. https://doi.org/10.3390/cancers11030317