Anatomic Origin of Osteochondrogenic Progenitors Impacts Sensitivity to EWS-FLI1-Induced Transformation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
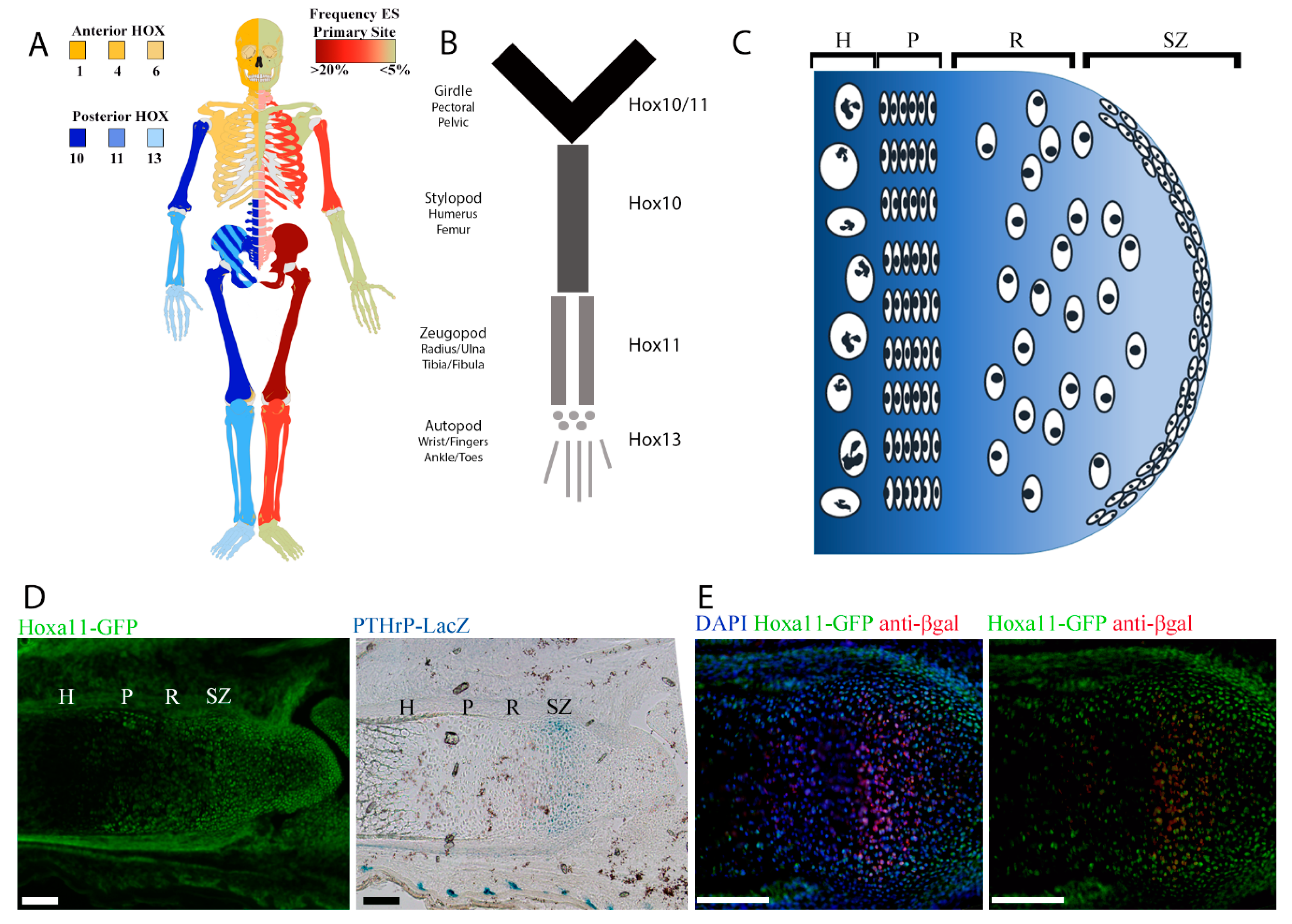
2.1. Regional Differences in Ewing sarcoma Presentation Mirror Regional Hox Compartmentalization
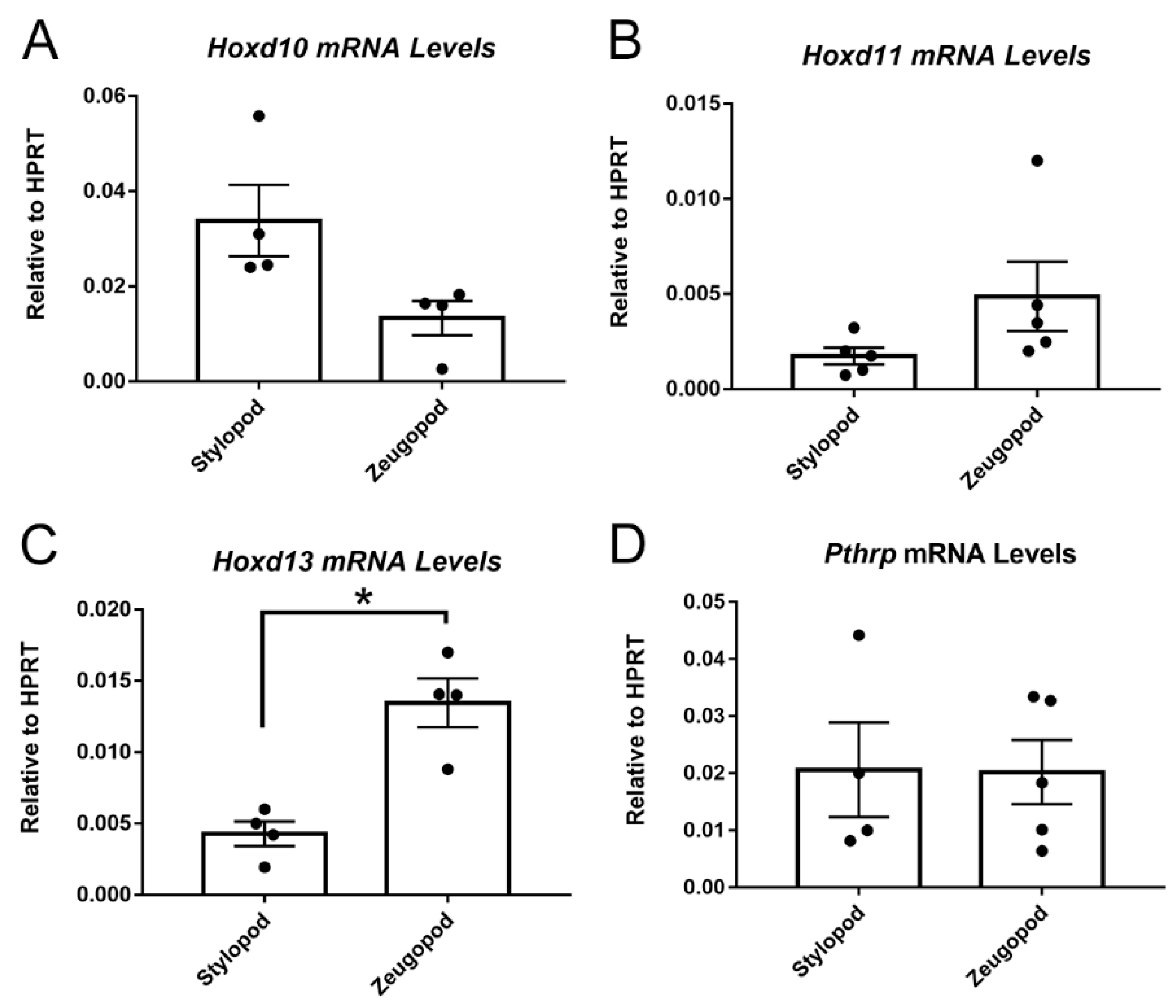
2.2. Posterior HoxD Genes Are Expressed by Isolated eSZ Cells
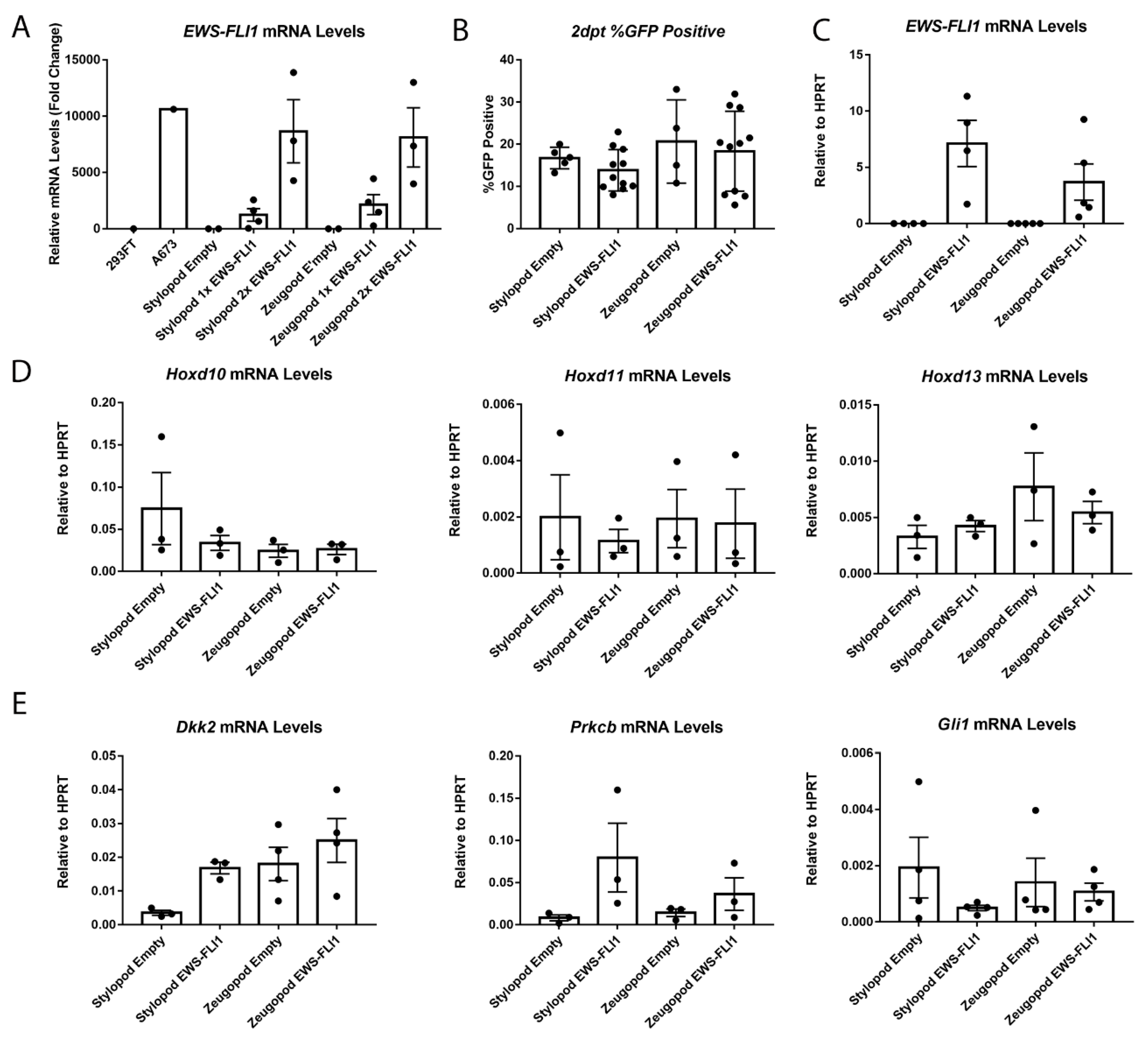
2.3. EWS-FLI1 Transduction of Stylopod and Zeugopod eSZ Cells
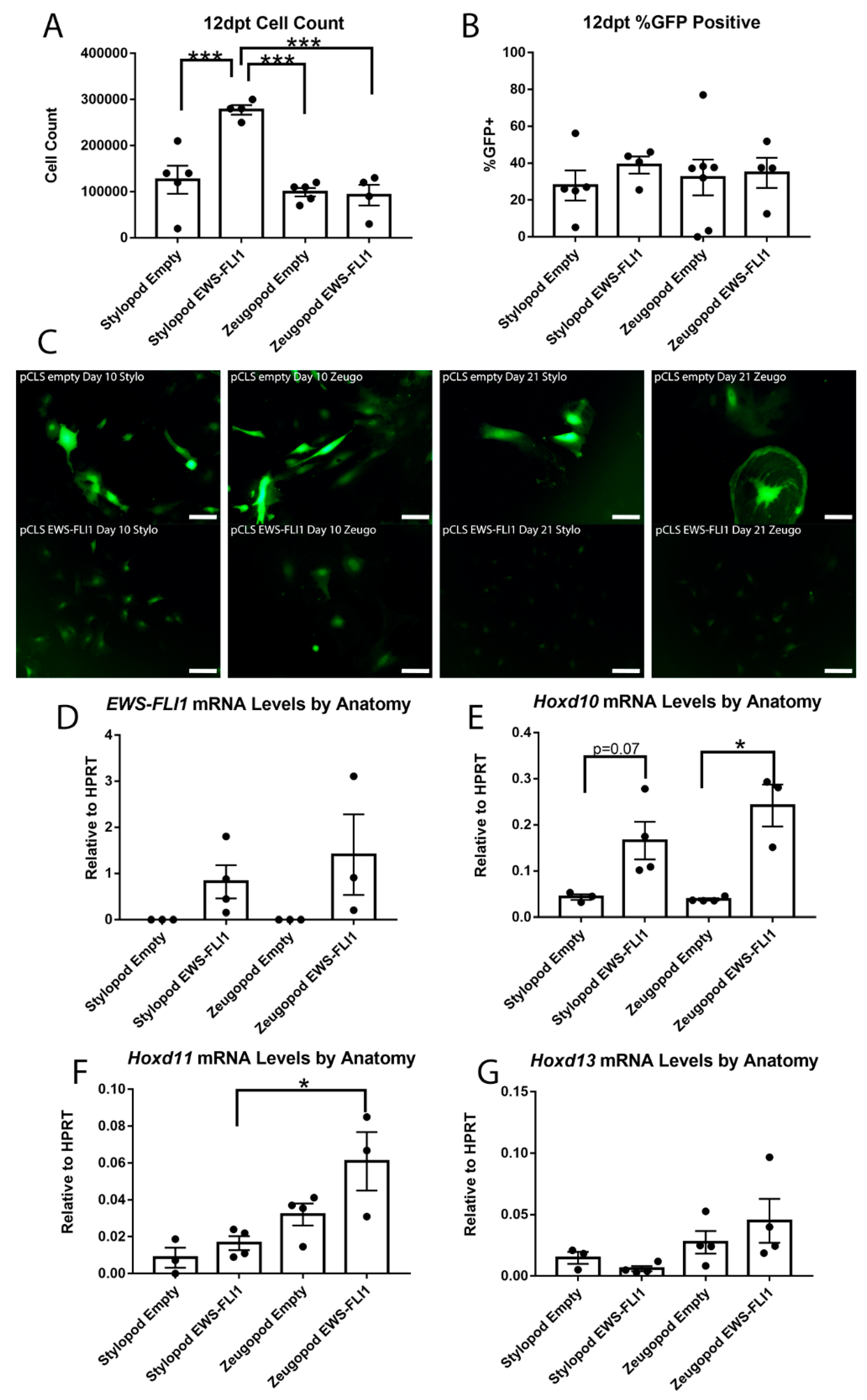
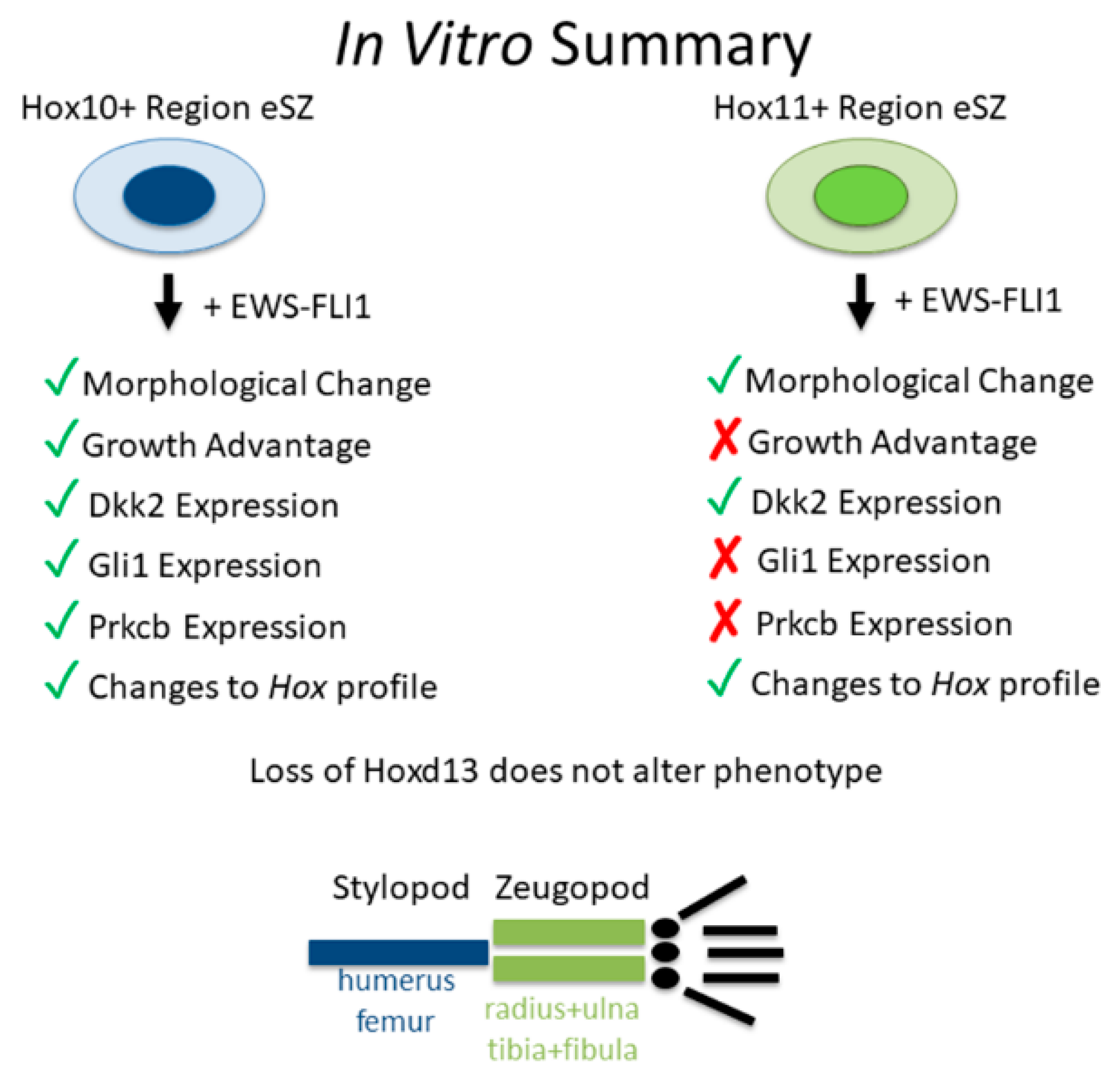
2.4. Impact of Anatomic Origin on EWS-FLI1-Induced Phenotype
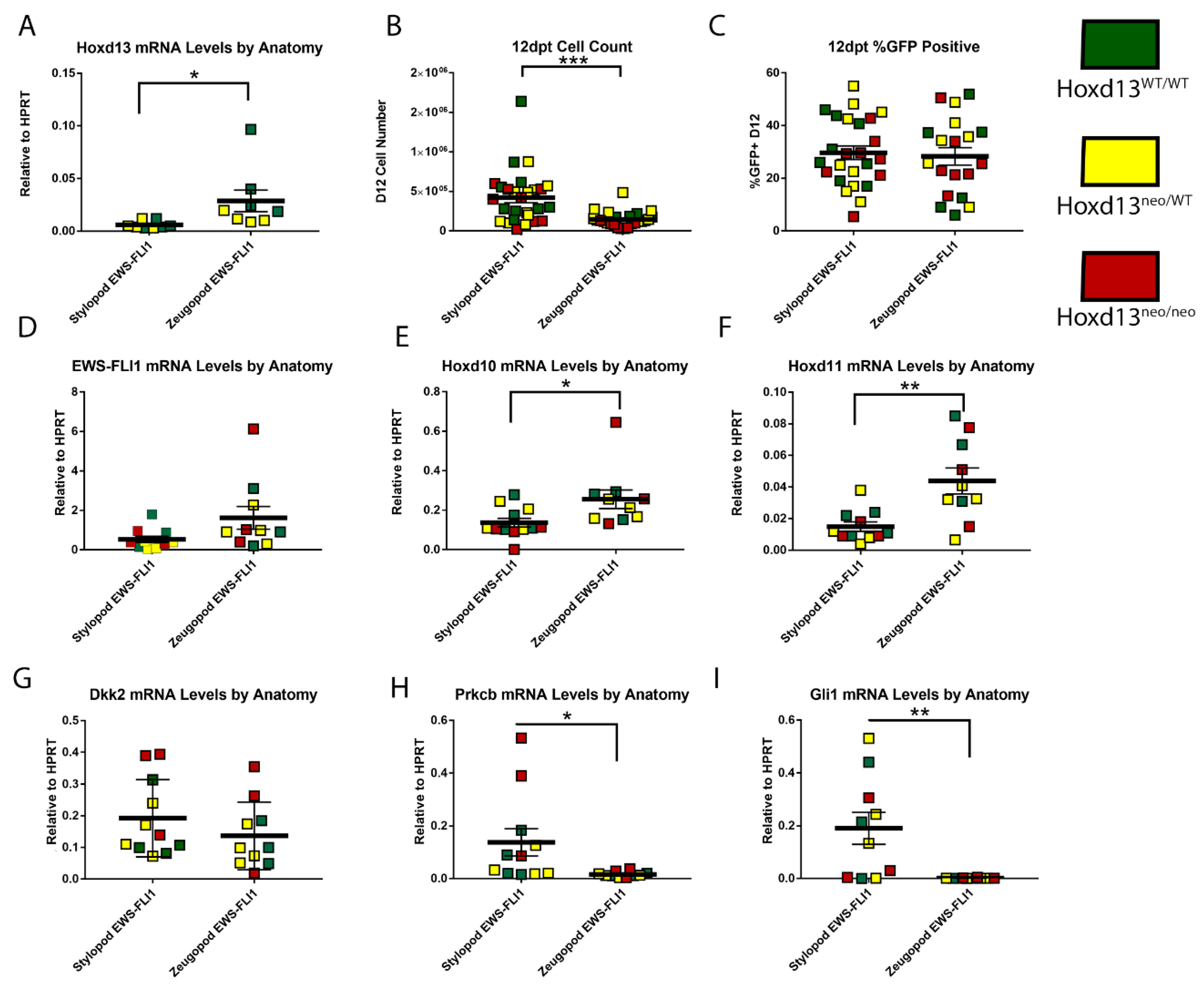
2.5. Impact of Hoxd13 Loss on EWS-FLI1-Induced Transformation
3. Discussion
4. Materials and Methods
4.1. Staining of E18.5 eSZ
4.2. Isolation and Transduction of eSZ
4.3. RNA Isolation
4.4. qPCR
4.5. Flow Cytometry
4.6. Imaging
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernstein, M.; Kovar, H.; Paulussen, M.; Randall, R.L.; Schuck, A.; Teot, L.A.; Juergens, H. Ewingv’s sarcoma family of tumors: Current management. Oncologist 2006, 11, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [PubMed]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Deneen, B.; Denny, C.T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene 2001, 20, 6731–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessnick, S.L.; Dacwag, C.S.; Golub, T.R. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 2002, 1, 393–401. [Google Scholar] [CrossRef]
- Riggi, N.; Suva, M.L.; Suva, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, Y.; Okita, H.; Nakaijima, H.; Horiuchi, Y.; Sato, B.; Taguchi, T.; Toyoda, M.; Katagiri, Y.U.; Fujimoto, J.; Hata, J.; et al. Inducible expression of chimeric EWS/ETS proteins confers Ewing’s family tumor-like phenotypes to human mesenchymal progenitor cells. Mol. Cell. Biol. 2008, 28, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.H.; Lawlor, E.R. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS ONE 2011, 6, e19305. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Suva, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suva, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minas, T.Z.; Surdez, D.; Javaheri, T.; Tanaka, M.; Howarth, M.; Kang, H.J.; Han, J.; Han, Z.Y.; Sax, B.; Kream, B.E.; et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017, 8, 34141–34163. [Google Scholar] [CrossRef] [PubMed]
- Castillero-Trejo, Y.; Eliazer, S.; Xiang, L.; Richardson, J.A.; Ilaria, R.L., Jr. Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1-dependent, ewing sarcoma-like tumors. Cancer Res. 2005, 65, 8698–8705. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Cironi, L.; Provero, P.; Suva, M.L.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yamazaki, Y.; Kanno, Y.; Igarashi, K.; Aisaki, K.; Kanno, J.; Nakamura, T. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J. Clin. Investig. 2014, 124, 3061–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javaheri, T.; Kazemi, Z.; Pencik, J.; Pham, H.T.; Kauer, M.; Noorizadeh, R.; Sax, B.; Nivarthi, H.; Schlederer, M.; Maurer, B.; et al. Increased survival and cell cycle progression pathways are required for EWS/FLI1-induced malignant transformation. Cell Death Dis. 2016, 7, e2419. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Harris, A.; Bailey, N.J.; Schwentner, R.; Tomazou, E.; von Levetzow, C.; Magnuson, B.; Ljungman, M.; Kovar, H.; Lawlor, E.R. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014, 9, 1613–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Heyking, K.; Roth, L.; Ertl, M.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Lawlor, E.R.; Burdach, S.; Richter, G.H. The posterior HOXD locus: Its contribution to phenotype and malignancy of Ewing sarcoma. Oncotarget 2016, 7, 41767–41780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rux, D.R.; Wellik, D.M. Hox genes in the adult skeleton: Novel functions beyond embryonic development. In Developmental Dynamics: An. Official Publication of the American Association of Anatomists; Wiley-Liss Inc.: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Wellik, D.M.; Capecchi, M.R. Hox10 and Hox11 genes are required to globally pattern the mammalian skeleton. Science 2003, 301, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Rux, D.R.; Song, J.Y.; Swinehart, I.T.; Pineault, K.M.; Schlientz, A.J.; Trulik, K.G.; Goldstein, S.A.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Regionally Restricted Hox Function in Adult Bone Marrow Multipotent Mesenchymal Stem/Stromal Cells. Dev. Cell 2016, 39, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Fabre, P.J.; Leleu, M.; Mascrez, B.; Lo Giudice, Q.; Cobb, J.; Duboule, D. Heterogeneous combinatorial expression of Hoxd genes in single cells during limb development. BMC Biol. 2018, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, Y.; Okita, H.; Itagaki, M.; Toyoda, M.; Katagiri, Y.U.; Fujimoto, J.; Hata, J.; Umezawa, A.; Kiyokawa, N. EWS/ETS regulates the expression of the Dickkopf family in Ewing family tumor cells. PLoS ONE 2009, 4, e4634. [Google Scholar] [CrossRef] [PubMed]
- Surdez, D.; Benetkiewicz, M.; Perrin, V.; Han, Z.Y.; Pierron, G.; Ballet, S.; Lamoureux, F.; Redini, F.; Decouvelaere, A.V.; Daudigeos-Dubus, E.; et al. Targeting the EWSR1-FLI1 oncogene-induced protein kinase PKC-beta abolishes ewing sarcoma growth. Cancer Res. 2012, 72, 4494–4503. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Bailey, N.; Van Noord, R.A.; Krook, M.A.; Harris, A.; Cramer, C.; Jasman, B.; Patel, R.M.; Thomas, D.; Borkin, D.; et al. Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget 2017, 8, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Beccari, L.; Yakushiji-Kaminatsui, N.; Woltering, J.M.; Necsulea, A.; Lonfat, N.; Rodriguez-Carballo, E.; Mascrez, B.; Yamamoto, S.; Kuroiwa, A.; Duboule, D. A role for HOX13 proteins in the regulatory switch between TADs at the HoxD locus. Genes Dev. 2016, 30, 1172–1186. [Google Scholar] [PubMed]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, E.R.; Sorensen, P.H. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit. Rev. Oncog. 2015, 20, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Pineault, K.M.; Wellik, D.M. Hox genes and limb musculoskeletal development. Curr. Osteoporos Rep. 2014, 12, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Volorio, A.; Iyer, S.; Broye, L.C.; Stamenkovic, I.; Riggi, N.; Rivera, M.N. Epigenome editing of microsatellite repeats defines tumor-specific enhancer functions and dependencies. Genes Dev. 2018, 32, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Tanaka, M.; Tsutsumi, S.; Aburatani, H.; Yamazaki, Y.; Homme, M.; Kitagawa, Y.; Nakamura, T. EWS-FLI1 regulates a transcriptional program in cooperation with Foxq1 in mouse Ewing sarcoma. Cancer Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Douglas, D.; Hsu, J.H.; Hung, L.; Cooper, A.; Abdueva, D.; van Doorninck, J.; Peng, G.; Shimada, H.; Triche, T.J.; Lawlor, E.R. BMI-1 promotes Ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008, 68, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.T.; Brignola, E.; Chen, L.; Guo, Y.; Zhao, S.; Wang, Q.; Li, B.; Correa, H.; Ermilov, A.N.; Dlugosz, A.A.; et al. Insight into the etiology of undifferentiated soft tissue sarcomas from a novel mouse model. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Capecchi, M.R. A mutational analysis of the 5′ HoxD genes: Dissection of genetic interactions during limb development in the mouse. Development 1996, 122, 1175–1185. [Google Scholar] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfaltzgraff, E.R.; Apfelbaum, A.; Kassa, A.P.; Song, J.Y.; Jiang, W.; Suhan, T.K.; Wellik, D.M.; Lawlor, E.R. Anatomic Origin of Osteochondrogenic Progenitors Impacts Sensitivity to EWS-FLI1-Induced Transformation. Cancers 2019, 11, 313. https://doi.org/10.3390/cancers11030313
Pfaltzgraff ER, Apfelbaum A, Kassa AP, Song JY, Jiang W, Suhan TK, Wellik DM, Lawlor ER. Anatomic Origin of Osteochondrogenic Progenitors Impacts Sensitivity to EWS-FLI1-Induced Transformation. Cancers. 2019; 11(3):313. https://doi.org/10.3390/cancers11030313
Chicago/Turabian StylePfaltzgraff, Elise R., April Apfelbaum, Andrew P. Kassa, Jane Y. Song, Wei Jiang, Tahra K. Suhan, Deneen M. Wellik, and Elizabeth R. Lawlor. 2019. "Anatomic Origin of Osteochondrogenic Progenitors Impacts Sensitivity to EWS-FLI1-Induced Transformation" Cancers 11, no. 3: 313. https://doi.org/10.3390/cancers11030313