HER2-Targeted Tyrosine Kinase Inhibitors Cause Therapy-Induced-Senescence in Breast Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
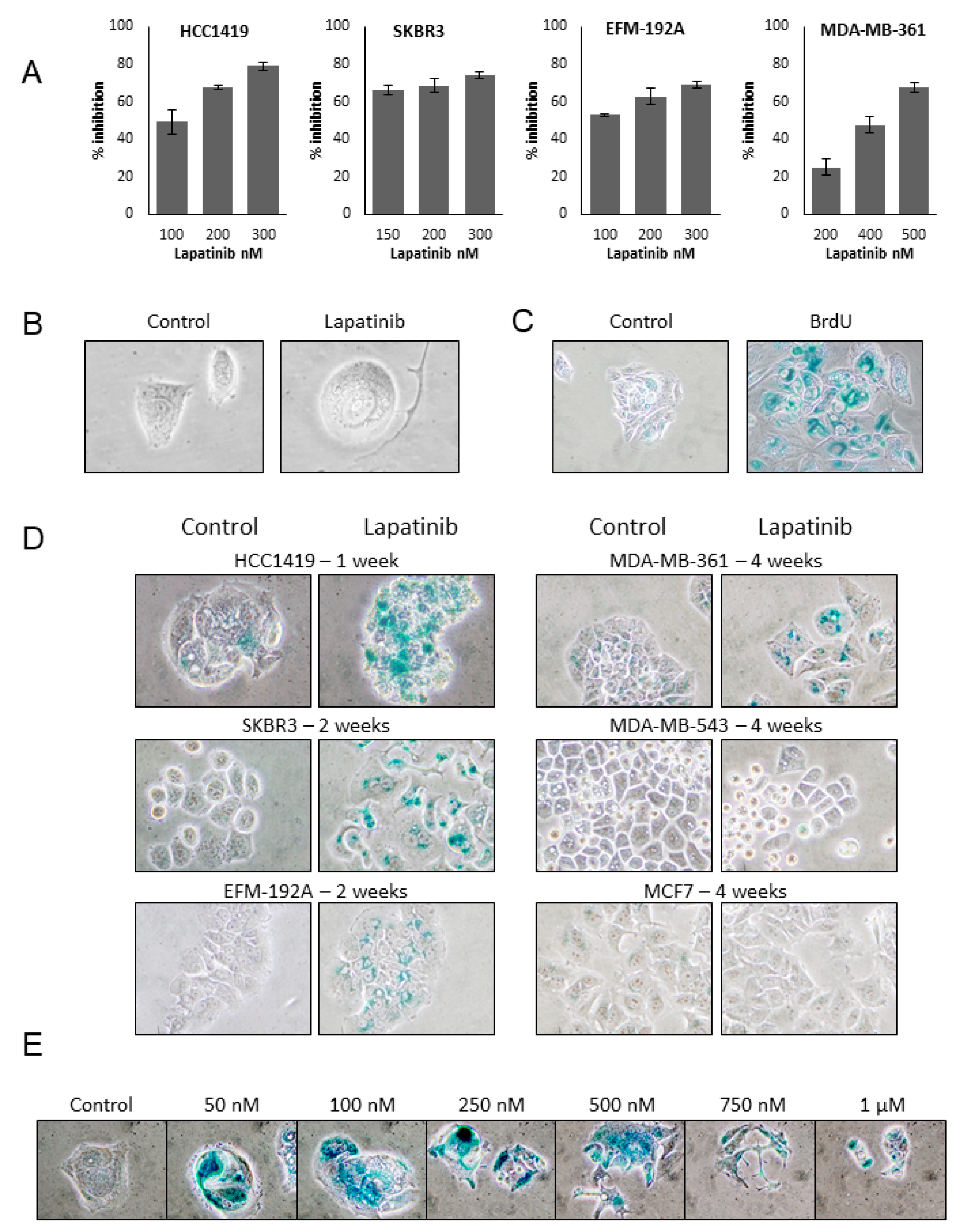
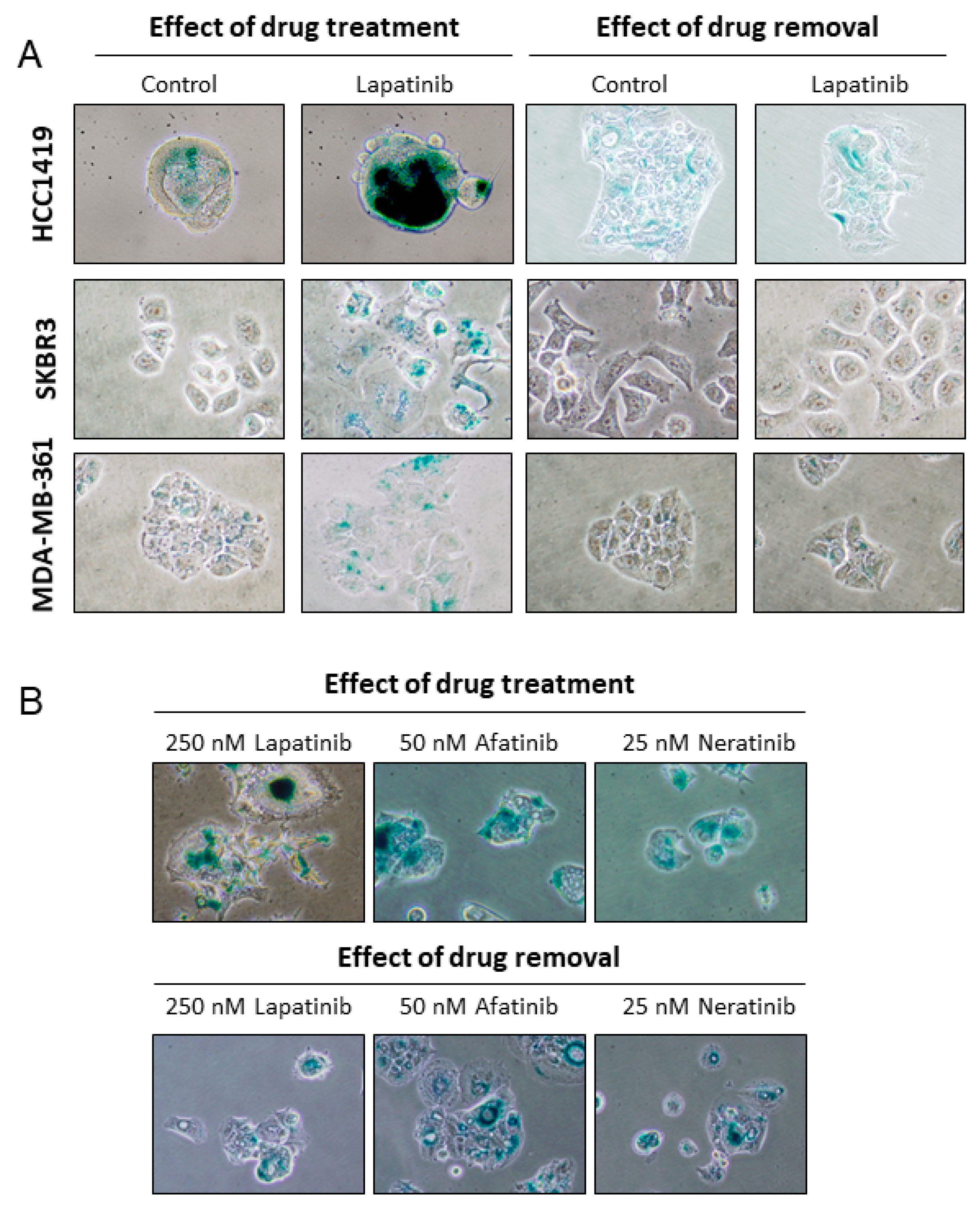
2.1. Lapatinib Induces Senescence in HER2 Positive Breast Cancer Cell Lines
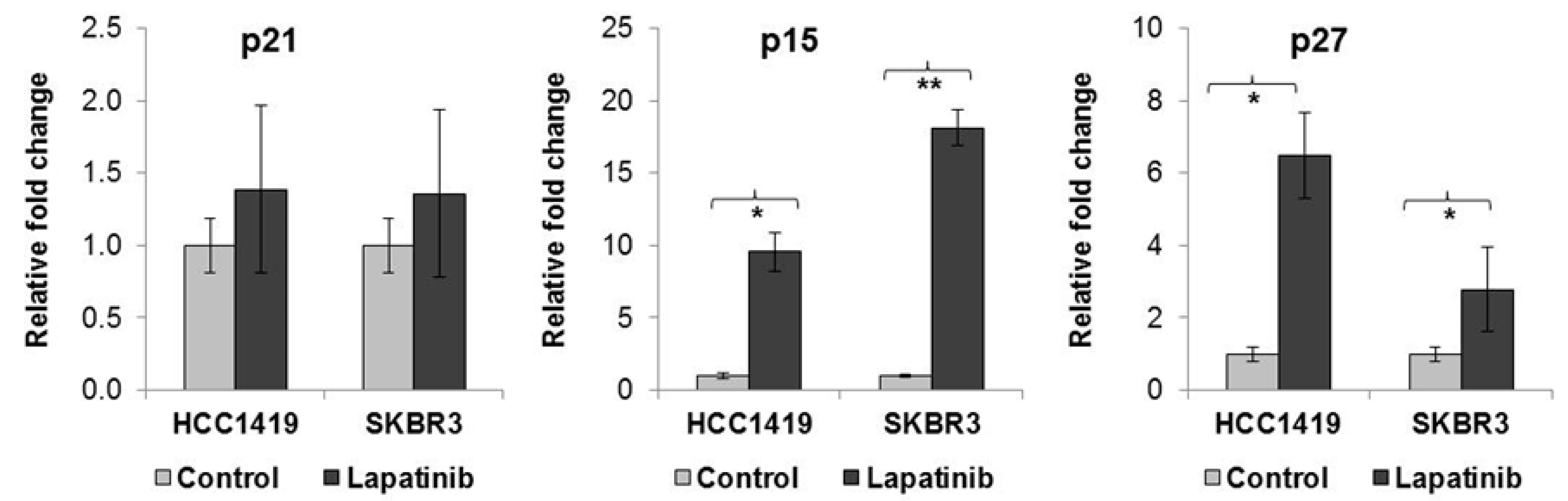
2.2. Lapatinib-Induced Senescence Is Associated with Increased p15 and p27 Expression
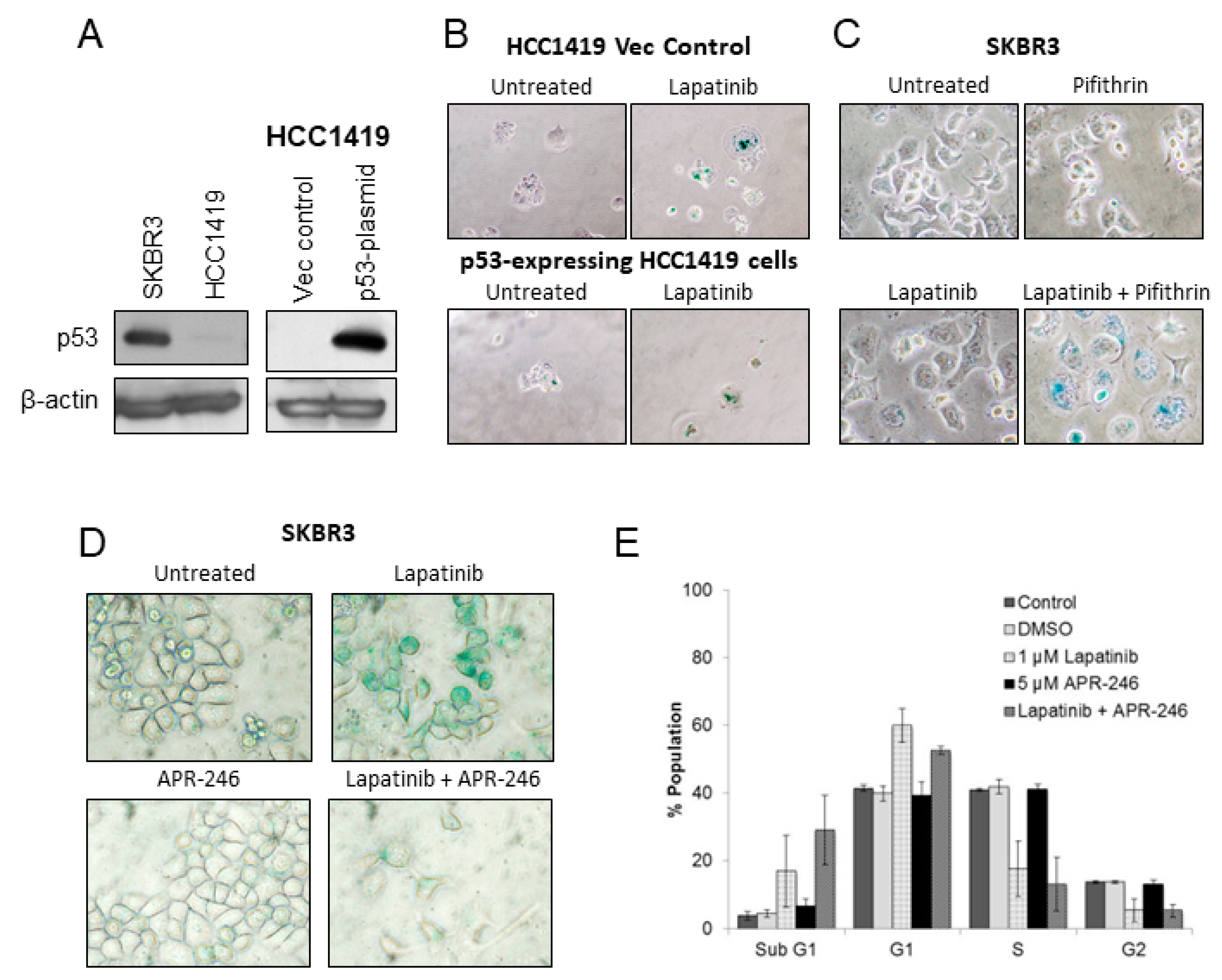
2.3. p53 in Lapatinib-Induced Senescence
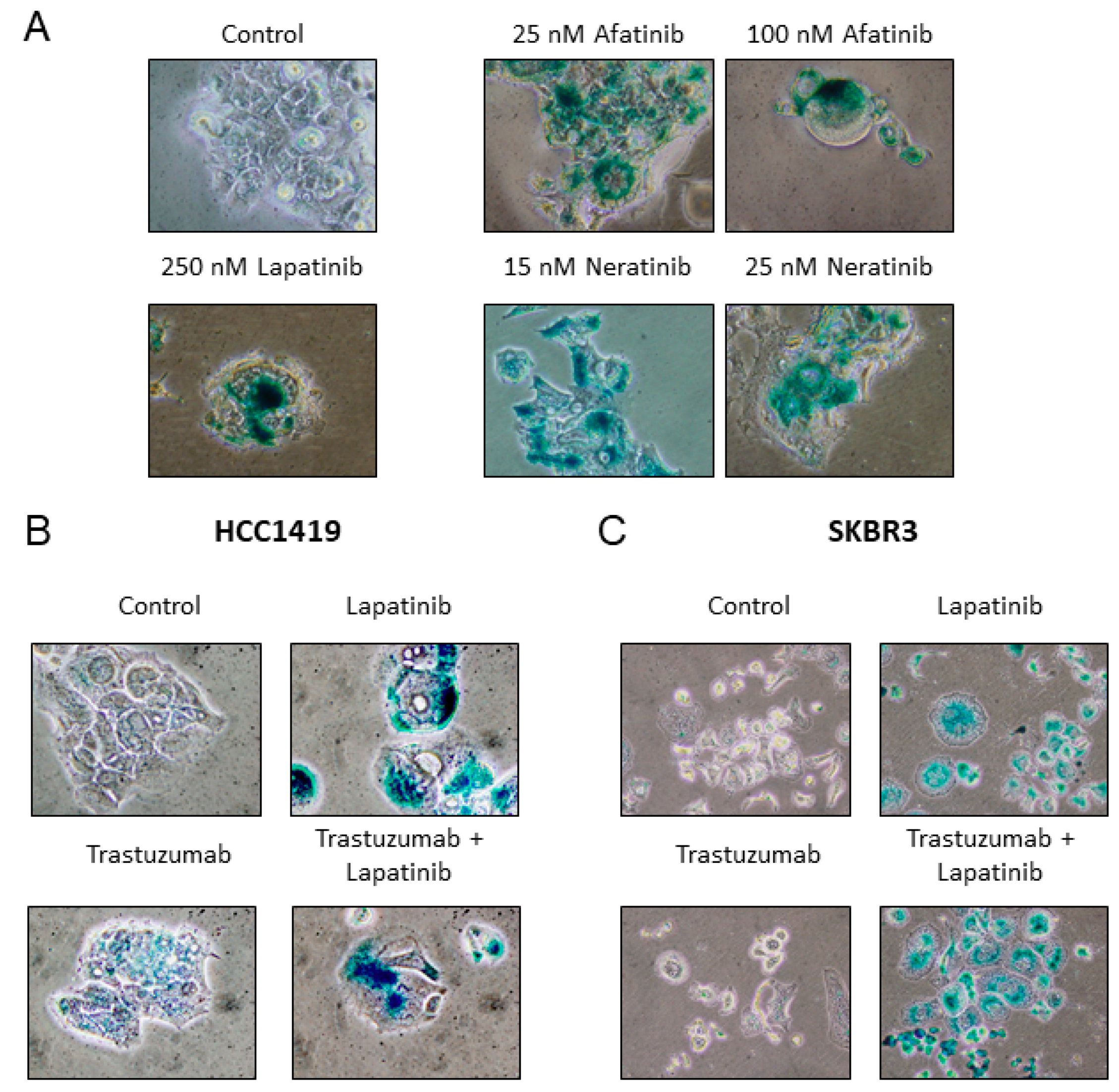
2.4. Senescence is Induced by Anti-HER2 TKIs but not by Trastuzumab
2.5. Lapatinib-Induced Senescence Is Reversible
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Growth Inhibition Assays
4.3. Senescence-Associated β-Galactosidase Activity Assay
4.4. Cell Cycle Assay
4.5. RNA Extraction and qRT-PCR Analysis
4.6. Western Blot Analysis
4.7. p53 Overexpression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.S.; Franco, S.X.; Florance, A.; O’Rourke, L.; Maltzman, J.; Johnston, S. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist 2010, 15, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar] [PubMed]
- Zhu, X.; Wu, L.; Qiao, H.; Han, T.; Chen, S.; Liu, X.; Jiang, R.; Wei, Y.; Feng, D.; Zhang, Y.; et al. Autophagy stimulates apoptosis in HER2-overexpressing breast cancers treated by lapatinib. J. Cell Biochem. 2013, 114, 2643–2653. [Google Scholar] [CrossRef]
- Yu, T.; Cho, B.J.; Choi, E.J.; Park, J.M.; Kim, D.H.; Kim, I.A. Radiosensitizing effect of lapatinib in human epidermal growth factor receptor 2-positive breast cancer cells. Oncotarget 2016, 7, 79089–79100. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.M. The impact of cellular senescence in cancer therapy: Is it true or not? Acta Pharmacol. Sin. 2011, 32, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef]
- McDermott, M.S.; Browne, B.C.; Conlon, N.T.; O’Brien, N.A.; Slamon, D.J.; Henry, M.; Meleady, P.; Clynes, M.; Dowling, P.; Crown, J.; et al. PP2A inhibition overcomes acquired resistance to HER2 targeted therapy. Mol. Cancer 2014, 13, 157. [Google Scholar] [CrossRef]
- Xia, W.; Bacus, S.; Hegde, P.; Husain, I.; Strum, J.; Liu, L.; Paulazzo, G.; Lyass, L.; Trusk, P.; Hill, J.; et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 7795–7800. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Atadja, P.; Wong, H.; Garkavtsev, I.; Veillette, C.; Riabowol, K. Increased activity of p53 in senescing fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 8348–8352. [Google Scholar] [CrossRef]
- Ewald, J.; Desotelle, J.; Almassi, N.; Jarrard, D. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br. J. Cancer 2008, 98, 1244–1249. [Google Scholar] [CrossRef]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Hurwitz, H.I.; Dees, E.C.; Dowlati, A.; Blackwell, K.L.; O’Neil, B.; Marcom, P.K.; Ellis, M.J.; Overmoyer, B.; Jones, S.F.; et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J. Clin. Oncol. 2005, 23, 5305–5313. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, J.; Akusjarvi, G.; Goike, H.M.; Roos, G.; Collins, V.P.; Pettersson, R.F. Adenovirus-mediated overexpression of p15INK4B inhibits human glioma cell growth, induces replicative senescence, and inhibits telomerase activity similarly to p16INK4A. Cell Growth Differ. 2000, 11, 373–384. [Google Scholar] [PubMed]
- Majumder, P.K.; Grisanzio, C.; O’Connell, F.; Barry, M.; Brito, J.M.; Xu, Q.; Guney, I.; Berger, R.; Herman, P.; Bikoff, R.; et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 2008, 14, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Bringold, F.; Serrano, M. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 2000, 35, 317–329. [Google Scholar] [CrossRef]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.M. p53 and p16(INK4A) independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef]
- Huck, J.J.; Zhang, M.; McDonald, A.; Bowman, D.; Hoar, K.M.; Stringer, B.; Ecsedy, J.; Manfredi, M.G.; Hyer, M.L. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol. Cancer Res. 2010, 8, 373–384. [Google Scholar] [CrossRef]
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, P.; Altieri, P.; Aloi, C.; Garibaldi, S.; Barisione, C.; Ghigliotti, G.; Fugazza, G.; Barsotti, A.; Brunelli, C. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2169–H2181. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Tabata, M.; Kiura, K.; Kozuki, T.; Hisamoto, A.; Katayama, H.; Takigawa, N.; Fujimoto, N.; Fujiwara, K.; Ueoka, H.; et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol. Rep. 2007, 17, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Morsbach, F.; Sander, D.; Gheorghiu, L.; Nanda, A.; Benes, C.; Kriegs, M.; Krause, M.; Dikomey, E.; Baumann, M.; et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011, 71, 6261–6269. [Google Scholar] [CrossRef] [PubMed]
- Subik, K.; Lee, J.-F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.-C.; Bonfiglio, T.; Hicks, D.G.; et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer Basic Clin. Res. 2010, 4, 35–41. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Irreversible protein kinase inhibitors. Curr. Med. Chem. 2011, 18, 2981–2994. [Google Scholar] [CrossRef]
- Baselga, J.; Bradbury, I.; Eidtmann, H.; Di Cosimo, S.; de Azambuja, E.; Aura, C.; Gomez, H.; Dinh, P.; Fauria, K.; Van Dooren, V.; et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): A randomised, open-label, multicentre, phase 3 trial. Lancet 2012, 379, 633–640. [Google Scholar] [CrossRef]
- De Azambuja, E.; Holmes, A.P.; Piccart-Gebhart, M.; Holmes, E.; Di Cosimo, S.; Swaby, R.F.; Untch, M.; Jackisch, C.; Lang, I.; Smith, I.; et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): Survival outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. Lancet Oncol. 2014, 15, 1137–1146. [Google Scholar] [CrossRef]
- Piccart-Gebhart, M.J.; Holmes, A.P.; Baselga, J.; De Azambuja, E.; Dueck, A.C.; Viale, G.; Zujewski, J.A.; Goldhirsch, A.; Santillana, S.; Pritchard, K.I.; et al. First results from the phase III ALTTO trial (BIG 2-06; NCCTG [Alliance] N063D) comparing one year of anti-HER2 therapy with lapatinib alone (L), trastuzumab alone (T), their sequence (T→L), or their combination (T+L) in the adjuvant treatment of HER2-positive early breast cancer (EBC). J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Sargent, D.J.; Grothey, A.; Kerbel, R.S. Drug rechallenge and treatment beyond progression--implications for drug resistance. Nat. Rev. Clin. Oncol. 2013, 10, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Muss, H.B.; Smith, L.R.; Cooper, M.R. Tamoxifen rechallenge: Response to tamoxifen following relapse after adjuvant chemohormonal therapy for breast cancer. J. Clin. Oncol. 1987, 5, 1556–1558. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Krell, J.; James, C.R.; Harper-Wynne, C.; Misra, V.; Cleator, S.; Miles, D. Rechallenging with anthracyclines and taxanes in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Gore, M. Treatment of recurrent ovarian cancer relapsing 6–12 months post platinum-based chemotherapy. Crit. Rev. Oncol. Hematol. 2007, 64, 129–138. [Google Scholar] [CrossRef]
- Breen, L.; Heenan, M.; Amberger-Murphy, V.; Clynes, M. Investigation of the role of p53 in chemotherapy resistance of lung cancer cell lines. Anticancer Res. 2007, 27, 1361–1364. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDermott, M.S.J.; Conlon, N.; Browne, B.C.; Szabo, A.; Synnott, N.C.; O’Brien, N.A.; Duffy, M.J.; Crown, J.; O’Donovan, N. HER2-Targeted Tyrosine Kinase Inhibitors Cause Therapy-Induced-Senescence in Breast Cancer Cells. Cancers 2019, 11, 197. https://doi.org/10.3390/cancers11020197
McDermott MSJ, Conlon N, Browne BC, Szabo A, Synnott NC, O’Brien NA, Duffy MJ, Crown J, O’Donovan N. HER2-Targeted Tyrosine Kinase Inhibitors Cause Therapy-Induced-Senescence in Breast Cancer Cells. Cancers. 2019; 11(2):197. https://doi.org/10.3390/cancers11020197
Chicago/Turabian StyleMcDermott, Martina S. J., Neil Conlon, Brigid C. Browne, Adam Szabo, Naoise C. Synnott, Neil A. O’Brien, Michael J. Duffy, John Crown, and Norma O’Donovan. 2019. "HER2-Targeted Tyrosine Kinase Inhibitors Cause Therapy-Induced-Senescence in Breast Cancer Cells" Cancers 11, no. 2: 197. https://doi.org/10.3390/cancers11020197