Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review


 , and
, and
Abstract
:1. Introduction
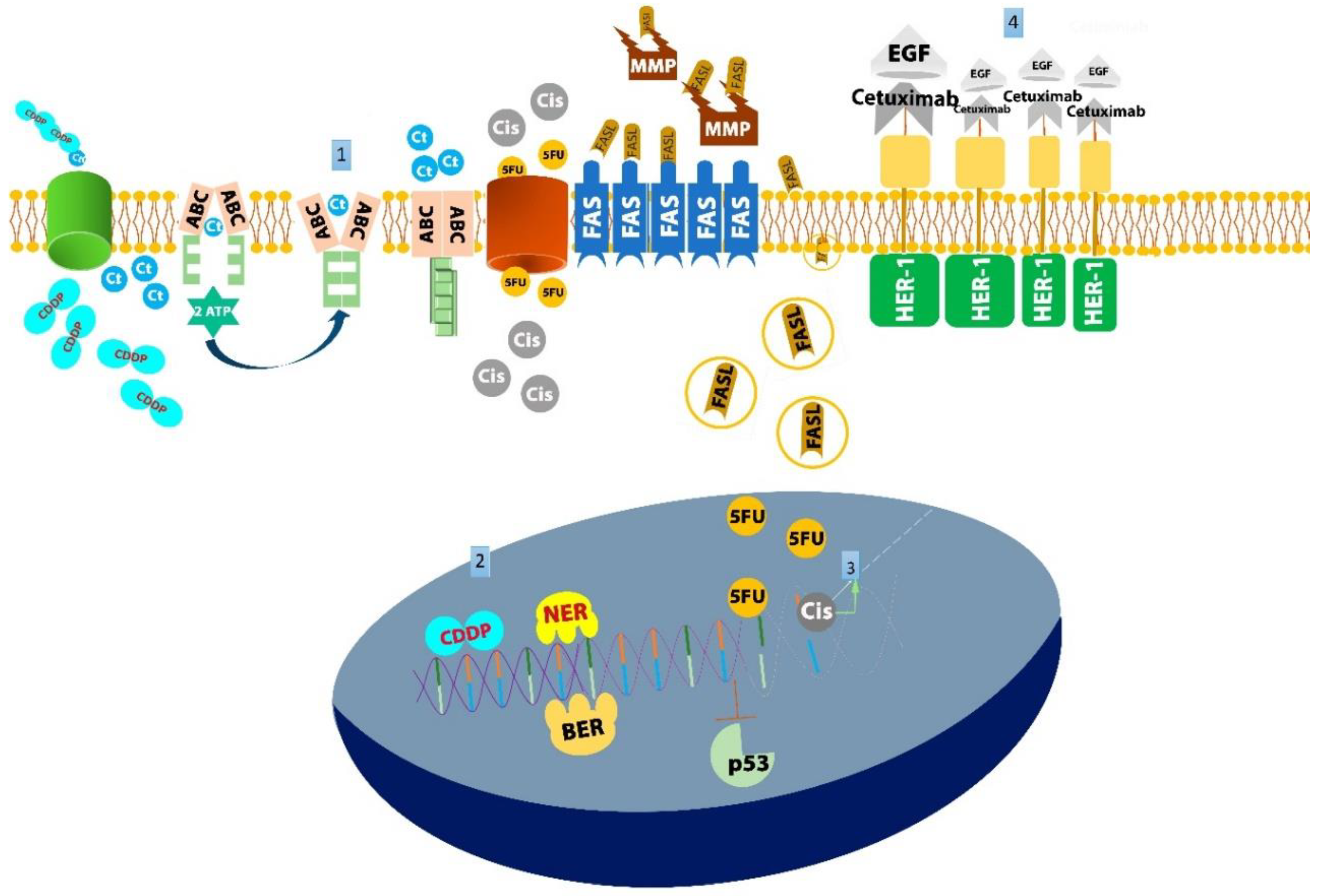
2. Reduced Concentration of Antineoplastic Drugs in Cancerous Cells
ATP-Binding Cassette Transporter (ABC)
3. Increased DNA Reparation Ability of Tumor Cells
Nucleotide Excision Repair (NER)/Base Excision Repair (BER)
4. Enhanced Tumor Survival and Routes of Dissemination
4.1. TP53
4.2. Fas/FasL
4.3. Complement System
5. Inactivation of Antineoplastic Drugs
5.1. HER1
5.2. Aurora Kinase A and B
6. EMT-CSC Involved in Treatment Resistance
7. Independent Molecular Markers of the Resistance Mechanism of Antineoplastic Drugs
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 13, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.P.; Saintigny, P.; Goudot, P.; Schouman, T.; Bertolus, C. The promising impact of molecular profiling on treatment strategies in oral cancers. J. Stomatol. Oral Maxillofac. Surg. 2017, 118, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Ciocan, C.; Gulei, D.; Mehterov, N.; Atanasov, A.G.; Dudea, D.; Berindan-Neagoe, I. Current Insights into oral cancer epigenetics. Int. J. Mol. Sci. 2018, 19, 670. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Komatsubara, H.; Ojima, Y.; Minamikawa, T.; Shigeta, T.; Shibuya, Y.; Yokoo, S.; Komori, T. Lack of survival advantage in patients with advanced, resectable squamous cell carcinoma of the oral cavity receiving induction chemotherapy with cisplatin (CDDP), docetaxel (TXT) and 5-fluorouracil (5FU). Kobe J. Med. Sci. 2004, 50, 189–196. [Google Scholar] [PubMed]
- Katori, H.; Tsukuda, M.; Mochimatu, I.; Ishitoya, J.; Kawai, S.; Mikami, Y.; Matsuda, H.; Tanigaki, Y.; Horiuchi, C.; Ikeda, Y.; et al. Phase I trial of concurrent chemoradiotherapy with docetaxel, cisplatin and 5-fluorouracil (TPF) in patients with locally advanced squamous cell carcinoma of the head and neck (SCCHN). Br. J. Cancer 2004, 90, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, A.; Jedlinski, A.; Johansson, A.C.; Roberg, K. Epidermal growth factor is a potential biomarker for poor cetuximab response in tongue cancer cells. J. Oral Pathol. Med. 2016, 45, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Cognelti, F.; Pinnaro, P.; Carlini, P.; Ruggeri, E.M.; Ambesi Impiombato, F.; Del Vecchio, M.R.; Giannarelli, D.; Perrino, A. Neoadjuvant chemotherapy in previously untreated patients with advanced head and neck squamous cell cancer. Cancer 1988, 62, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Decker, D.A.; Drelichman, A.; Jacobs, J.; Hoschner, J.; Kinzie, J.; Loh, J.J.; Weaver, A.; Al-Sarraf, M. Adjuvant chemotherapy with cisdiamminedichloroplatinum and 120-hour infusion 5-fluorouracil in stage III and IV squamous cell carcinoma of the head and neck. Cancer 1983, 51, 1353–1355. [Google Scholar] [CrossRef]
- Ervin, T.J.; Clark, J.R.; Weichselbaum, R.R.; Fallon, B.G.; Miller, D.; Fabian, R.L.; Posner, M.R.; Norris, C.M., Jr.; Tuttle, S.A.; Schoenfeld, D.A. An analysis of induction and adjuvant chemotherapy in the multidisciplinary treatment of squamous cell carcinoma of the head and neck. J. Clin. Oncol. 1987, 5, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, E.; Oridate, N.; Furuta, Y.; Suzuki, S.; Hatakeyama, H.; Sawa, H.; Sunayashiki-Kusuzaki, K.; Yamazaki, K.; Inuyama, Y.; Fukuda, S. Differentially expressed genes associated with ClS-diamminedichloroplatinum (II) resistance in head and neck cancer using differential display and cDNA microarray. Head Neck 2003, 25, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Ishikawa, H.; Tanaka, A.; Mataga, I. Heterogeneity of anticancer drug sensitivity in squamous cell carcinoma of the tongue. Hum. Cell 2011, 24, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.P.; Divakar, D.D.; Al Kheraif, A.A.; Ramakrishnaiah, R.; Mustafa, S.M.; Durgesh, B.H.; Basavarajappa, S.; Khan, A.A. Outcome of Palliative and Radical Radiotherapy in Patients with Oral Squamous Cell Carcinoma—A Retrospective Study. Asian Pac. J. Cancer Prev. 2015, 16, 6919–6922. [Google Scholar] [CrossRef] [PubMed]
- Tobias, J.S. Has chemotherapy proved itself in head and neck cancer? Br. J. Cancer 1990, 6, 649–651. [Google Scholar] [CrossRef]
- Becker, M.; Levy, D. Modeling the Transfer of Drug Resistance in Solid Tumors. Bull. Math. Biol. 2017, 79, 2394–2412. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Rational Treatment of Cancer in the Biology of Cancer, 2nd ed.; Weinberg, R., Ed.; Garland Science: New York, NY, USA, 2013; pp. 833–834. [Google Scholar]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Jaffrezou, J.P.; Fleming, W.H.; Durán, G.E.; Sikic, B.I. Prevalence of multidrug resistance related to activation of the mdr1 gene in human sarcoma mutants derived by single-step doxorubicin selection. Cancer Res. 1994, 54, 4980–4987. [Google Scholar] [PubMed]
- Ng, I.O.; Lam, K.Y.; Ng, M.; Kwong, D.L.; Sham, J.S. Expression of p-glycoprotein, a multidrug-resistance gene product, is induced by radiotherapy in patients with oral squamous cell carcinoma. Cancer 1998, 83, 851–857. [Google Scholar] [CrossRef]
- Rabkin, D.; Chieng, D.C.; Miller, M.B.; Jennings, T.; Feustel, P.; Steiniger, J.; Parnes, S.M. P-glycoprotein expression in the squamous cell carcinoma of the tongue base. Laryngoscope 1995, 105, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Das, S.N.; Luthra, K.; Shukla, N.K.; Ralhan, R. Differential expression of multidrug resistance gene product, P-glycoprotein, in normal, dysplastic and malignant oral mucosa in India. Int. J. Cancer 1997, 74, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, P.M.; Roninson, I.B. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J. Natl. Cancer Inst. 1993, 85, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Brock, I.; Hipfner, D.R.; Nielsen, B.S.; Jensen, P.B.; Deeley, R.G.; Cole, S.P.; Sehested, M. Sequential coexpression of the multidrug resistance genes MRP and mdr1 and their products in VP-16 (etoposide)-selected H69 small cell lung cancer cells. Cancer Res. 1995, 55, 459–462. [Google Scholar] [PubMed]
- Abe, Y.; Ohnishi, Y.; Yoshimura, M.; Ota, E.; Ozeki, Y.; Oshika, Y.; Tokunaga, T.; Yamazaki, H.; Ueyema, Y.; Ogata, T.; et al. P-glycoprotein-mediated acquired multidrug resistance of human lung cancer cells in vivo. Br. J. Cancer 1996, 74, 1929–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.F.; Slater, A.; Wall, D.M.; Kantharidis, P.; Parkin, J.D.; Cowman, A.; Zalcberg, J.R. Rapid up-regulation of mdr1 expression by anthracyclines in a classical multidrug-resistant cell line. Br. J. Cancer 1995, 71, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, K.; Kikuchi, J.; Sato, S.; Takano, H.; Saburi, Y.; Asoh, K.; Kuwano, M. Vincristine-resistant human cancer KB cell line and increased expression of multidrug-resistance gene. Jpn. J. Cancer Res. 1988, 79, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A.; Rybalkina, E.Y. Recent Advances in the Studies of Molecular Mechanisms Regulating Multidrug Resistance in Cancer Cells. Biochemistry (Mosc) 2018, 83, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.E.; Punke, C.; Reymann, A. Expression of multi-drug resistance genes (mdr1, mrp1, bcrp) in primary oral squamous cell carcinoma. In Vivo 2004, 18, 133–147. [Google Scholar] [PubMed]
- Nakamura, M.; Nakatani, K.; Uzawa, K.; Ono, K.; Uesugi, H.; Ogawara, K.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Wada, T.; et al. Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R. Oncol. Rep. 2005, 14, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, G.A.S.; Costa, E.F.D.; Lopes-Aguiar, L.; Lima, T.R.P.; Visacri, M.B.; Pincinato, E.C.; Lourenco, G.J.; Calonga, L.; Mariano, F.V.; Altemani, A.M.A.M.; et al. Polymorphism in DNA mismatch repair pathway genes predict toxicity and response to cisplatin chemoradiation in head and neck squamous cell carcinoma patients. Oncotarget 2018, 9, 29538–29547. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Spitz, M.R.; Lee, J.J.; Huang, M.; Lippman, S.M.; Wu, X. Nucleotide Excision Repair Pathway Genes and Oral Premalignant Lesions. Clin. Cancer Res. 2007, 13, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Grube, M.; Hanna, B.M.F.; Benitez-Buelga, C.; Cázares-Körner, A.; Helleday, T. Targeting BER enzymes in cancer therapy. DNA Repair 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Quintela-Fandino, M.; Hitt, R.; Medina, P.P.; Gamarra, S.; Manso, L.; Cortés-Funes, H.; Sanchez-Cespedes, M. DNA-Repair gene polymorphisms predict favorable clinical outcome among patients with advanced squamous cell carcinoma of the head and neck treated with cisplatin-based induction chemotherapy. J. Clin. Oncol. 2006, 24, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- Vaezi, A.; Feldman, C.H.; Niedernhofer, L.J. ERCC1 and XRCC1 as biomarkers for lung and head and neck cancer. Pharmgenomics. Pers. Med. 2011, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Ameri, A.; Mortazavi, N.; Ahmadi, H.K.; Novin, K. ERCC1 Expression can predict response to platinum-based induction chemotherapy in head and neck cancer cases. Asian Pac. J. Can. Prev. 2016, 17, 87–91. [Google Scholar] [CrossRef]
- Bozec, A.; Sudaka, A.; Etienne-Grimaldi, M.C.; Brunstein, M.C.; Fischel, J.L.; Milano, G. Antitumor activity of cetuximab associated with the taxotere-cisplatin-fluorouracil (TPF) combination on an orthotopic head and neck cancer model. Oral Oncol. 2011, 47, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.T.; Borges, G.A.; Rêgo, D.F.; E. Silva, L.F.; Avelino, S.; DE Matos Neto, J.N.; Simeoni, L.A.; Guerra, E.N. Combined paclitaxel, cisplatin and fluorouracil therapy enhances ionizing radiation effects, inhibits migration and induces G0/G1 cell cycle arrest and apoptosis in oral carcinoma cell lines. Oncol. Lett. 2015, 10, 1721–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabelguenne, A.; Blons, H.; de Waziers, I.; Carnot, F.; Houllier, A.M.; Soussi, T.; Brasnu, D.; Beaune, P.; Laccourreye, O.; Laurent-Puig, P. p53 alterations predict tumor response to neoadjuvant chemotherapy in head and neck squamous cell carcinoma: a prospective series. J. Clin. Oncol. 2000, 18, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Cutilli, T.; Leocata, P.; Dolo, V.; Altobelli, E. Evaluation of p53 as a prognostic factor for oral cancer surgery. Br. J. Oral Maxillofac Surg. 2013, 51, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Poulaki, V.; Mitsiades, C.S.; Mitsiades, N. The role of Fas and FasL as mediators of anticancer chemotherapy. Drug Resist. Updat. 2001, 4, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Sun, L.; Hu, F.F.; Chen, Q.E. Effects of FasL expression in oral squamous cell cancer. Asian Pac. J. Cancer Prev. 2013, 14, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.H.; Chang, W.M.; Lee, W.J.; Chang, Y.C.; Lai, T.C.; Chan, D.V.; Sharma, R.; Lin, Y.F.; Hsiao, M. A Fas Ligand (FasL)-Fused Humanized Antibody Against Tumor-Associated Glycoprotein 72 Selectively Exhibits the Cytotoxic Effect Against Oral Cancer Cells with a Low FasL/Fas Ratio. Mol. Cancer Ther. 2017, 16, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Blons, H.; Gad, S.; Zinzindohoue, F.; Maniere, I.; Beauregard, J.; Tregouet, D.; Brasnu, D.; Beaune, P.; Laccourreye, O.; Laurent-Puig, P. Matrix Metalloproteinase 3 polymorphism: A predictive factor of response to noadjuvant chemotherapy in head and neck squamous cell carcinoma. Clin. Cancer Res. 2004, 10, 2594–2599. [Google Scholar] [CrossRef] [PubMed]
- Thielen, A.J.F.; van Baarsen, I.M.; Jongsma, M.L.; Zeerleder, S.; Spaapen, R.M.; Wouters, D. CRISPR/Cas9 generated human CD46, CD55 and CD59 knockout cell lines as a tool for complement research. J. Immunol. Methods. 2018, 456, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Bellosillo, B.; Villamor, N.; López-Guillermo, A.; Marcé, S.; Esteve, J.; Campo, E.; Colomer, D.; Montserrat, E. Complement-mediated cell death induced by rituximab in B-cell lymphoproliferative disorders is mediated in vitro by a caspase-independent mechanism involving the generation of reactive oxygen species. Blood 2001, 98, 2771–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dho, S.H.; Lim, J.C.; Kim, L.K. Beyond the Role of CD55 as a Complement Component. Immune Netw. 2018, 18, e11. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Concha-Benavente, F.; Santegoets, S.J.A.M.; Welters, M.J.P.; Ehsan, I.; Ferris, R.L.; van der Burg, S.H. EGFR signaling suppresses type 1 cytokine-induced T-cell attracting chemokine secretion in head and neck cancer. PLoS ONE 2018, 13, e0203402. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Minamino, Y.; Kakudo, K.; Nozaki, M. Resistance of oral squamous cell carcinoma cells to cetuximab is associated with EGFR insensitivity and enhanced stem cell-like potency. Oncol. Rep. 2014, 32, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Merlano, M.; Occelli, M. Review of cetuximab in the treatment of squamous cell carcinoma of the head and neck. Ther. Clin. Risk. Manag. 2007, 3, 871–876. [Google Scholar] [PubMed]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 2001, 8, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, F.A.; Noguti, J.; Oshima, C.T.; Ribeiro, D.A. Effective targeting of the epidermal growth factor receptor (EGFR) for treating oral cancer: A promising approach. Anticancer Res. 2014, 34, 1547–1552. [Google Scholar] [PubMed]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Favrot, M.C.; Coll, J.L.; Hurbin, A. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol. Ther. 2010, 18, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Klinghammer, K.; Weichert, W.; Knödler, M.; Stenzinger, A.; Gauler, T. Expression of amphiregulin and EGFRvIII outcome of patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin. Cancer Res. 2011, 17, 5197–5204. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Cheng, H.; Wirth, P.; Counsell, A.; Marcrom, S.R.; Wood, C.B.; Pohlmann, P.R.; Gilbert, J.; Murphy, B.; Yarbrough, W.G.; et al. Regulation of heparin-binding EGF-like growth factor by miR-212 and acquired cetuximab-resistance in head and neck squamous cell carcinoma. PLoS ONE 2010, 5, e12702. [Google Scholar] [CrossRef] [PubMed]
- Boeckx, C.; Baay, M.; Wouters, A.; Specenier, P.; Vermorken, J.B.; Peeters, M.; Lardon, F. Anti-epidermal growth factor receptor therapy in head and neck squamous cell carcinoma: Focus on potential molecular mechanisms of drug resistance. Oncologist 2013, 18, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Jeong, J.H.; Kim, H.S.; Kim, J.H. The role of anti-EGFR agents in patients with locoregionally advanced head and neck cancer: A meta-analysis of randomized trials. Oncotarget 2017, 8, 102371–102380. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; van-Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.D. Aurora kinases: shining lights on the therapeutic horizon? Oncogene 2005, 24, 5005–5015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraizer, G.C.; Díaz, M.F.; Lee, I.L.; Grossman, H.B.; Sen, S. Aurora-A/STK15/BTAK enhances chromosomal instability in bladder cancer cells. Int. J. Oncol. 2004, 25, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.J.; Kang, H.F.; Wang, X.J.; Shao, Y.P.; Lin, S.; Zhao, Y.; Ren, H.T.; Min, W.L.; Wang, M.; Liu, X.X. Association between genetic polymorphism in AURKA (rs2273535 and rs1047972) and breast cancer risk: a meta-analysis involving 37,221 subjects. Cancer Cell Int. 2014, 14, e91. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Chou, Y.E.; Chuang, C.Y.; Yang, S.F.; Lin, C.W. Combined effect of genetic polymorphisms of AURKA and environmental factors on oral cancer development in Taiwan. PLoS ONE 2017, 12, e0171583. [Google Scholar] [CrossRef] [PubMed]
- Hoellein, A.; Pickhard, A.; von Keitz, F.; Schoeffmann, S.; Piontek, G.; Rudelius, M. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Pickhard, A.; Siegl, M.; Baumann, A.; Huhn, M.; Wirth, M.; Reiter, R. The response of head and neck squamous cell carcinoma to cetuximab treatment depend on Aurora kinase A polymorphism. Oncotarget. 2014, 5, 5428–5438. [Google Scholar] [CrossRef] [PubMed]
- Masui, T.; Ota, I.; Yook, J.I.; Mikami, S.; Yane, K.; Yamanaka, T.; Hosoi, H. Snail-induced epithelial-mesenchymal transition promotes cancer stem cell-like phenotype in head and neck cancer cells. Int. J. Oncol. 2014, 44, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Setúbal Destro Rodrigues, M.F.; Gammon, L.; Rahman, M.M.; Biddle, A.; Nunes, F.D.; Mackenzie, I.C. Effects of Cetuximab and Erlotinib on the behaviour of cancer stem cells in head and neck squamous cell carcinoma. Oncotarget 2018, 9, 13488–13500. [Google Scholar]
- La Fleur, L.; Johansson, A.C.; Roberg, K. A CD44high/EGFRlow subpopulation within head and neck cancer cell lines shows an epithelial-mesenchymal transition phenotype and resistance to treatment. PLoS ONE 2012, 7, e44071. [Google Scholar] [CrossRef] [PubMed]
- Fullár, A.; Kovalszky, I.; Bitsche, M.; Romani, A.; Schartinger, V.H.; Sprinzl, G.M.; Riechelmann, H.; Dudás, J. Tumor cell and carcinoma-associated fibroblast interaction regulates matrix metalloproteinases and their inhibitors in oral squamous cell carcinoma. Exp. Cell Res. 2012, 318, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.N.; Xu, B.N.; Cai, J.; Yang, J.B.; Lin, N. Tumor-associated fibroblast-conditioned medium promotes tumor cell proliferation and angiogenesis. Genet Mol. Res. 2013, 12, 5863–5871. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Herchenhorn, D.; Dias, F.L.; Viegas, C.M.; Federico, M.H.; Araújo, C.M.; Small, I.; Bezerra, M.; Fontão, K.; Knust, R.E.; Ferreira, C.G.; et al. Phase I/II study of erlotinib combined with cisplatin and radiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzidis, E.; Gammon, L.; Biddle, A.; Emich, H.; Mackenzie, I.C. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget 2015, 6, 43964–43977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfaki, I.; Mir, R.; Almutairi, F.M.; Abu Duhier, F.M. Cytochrome P450: Polymorphisms and roles in Cancer, Diabetes and Atherosclerosis. Asian Pac. J. Cancer Prev. 2018, 19, 2057–2070. [Google Scholar] [PubMed]
- Sim, S.C.; Ingelman-Sundberg, M. Update on allele nomenclature for human cytochromes P450 and the human cytochrome P450 allele (CYP-allele) nomenclature database. Methods Mol. Biol. 2013, 987, 251–259. [Google Scholar] [PubMed]
- Yadav, S.S.; Seth, S.; Khan, A.J.; Maurya, S.S.; Dhawan, A.; Pant, S.; Pant, M.C.; Parmar, D. Association of polymorphism in cytochrome P450 2C9 with susceptibility to head and neck cancer and treatment outcome. Appl. Transl. Genom. 2014, 3, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, A.; Mascitti, M.; Lo-Russo, L.; Sartini, D.; Troiano, G.; Emanuelli, M.; Lo-Muzio, L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, e971. [Google Scholar] [CrossRef] [PubMed]
- Hang, W.; Yin, Z.X.; Liu, G.; Zeng, Q.; Shen, X.F.; Sun, Q.H.; Li, D.D.; Jian, Y.P.; Zhang, Y.H.; Wang, Y.S.; et al. Piperlongumine and p53-reactivator APR-246 selectively induce cell death in HNSCC by targeting GSTP1. Oncogene 2018, 37, 3384–3398. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Year | Genes | Methodology | Conclusions |
|---|---|---|---|---|
| Reduced concentration of antineoplastic drugs in cancerous cells. | ||||
| Friedrich | 2004 | MDR1, MRP1 and BCRP | Gene expression in primary SCC using IH and PCR. | MDR1 and MRP1 are co-expressed; MDR1 and BCRP are not co-dependent. Patient survival can be influenced by the altered expression of at least one of the genes implicated in chemotherapeutic resistance. |
| Nakamura | 2005 | MDR1, MRP1 | Expression levels in CDDP-resistant/sensitive cell lines using in-house cDNA microarray (2021 genes originated from normal oral tissue, primary oral cancer, and oral cancer cell lines) and PCR. | Resistant cells have high MDR1 and low MRP1 expression. |
| Suzuki | 2010 | MDR1, MRP1 and MRP2 | Gene expression analysis of single cell clones dissociated from primary tumors using PCR. | MDR1 was not expressed in any single cell clone from primary SCC tumor, although MRP1 and MRP2 were expressed. |
| Genes involved in DNA repair | ||||
| Quintela | 2006 | XPD, ERCC1 and XRCC1 | SNP detected using RFLP in DNA from peripheral lymphocytes of HNSCC patients. | The accumulation of polymorphic variants increases the probability of achieving a complete response. |
| Ameri | 2016 | ERCC1 | Expression status determined using PCR in tumor samples. | Tumor samples with high ERCC1 expression showed no response to induction chemotherapy. |
| Enhanced tumor survival and routes of dissemination | ||||
| Cabelguenne | 2000 | TP53 | Gene status (mutations, allele loss) detected using PCR amplification in tumor samples. | P53 status may be a useful indicator of responding to neoadjuvant chemotherapy in HNSCC. |
| Blons | 2004 | MMP3 | MMP1, MMP3, and MMP7 polymorphisms detected using PCR in tumor samples and blood. | A significant correlation between MMP3 polymorphism and response to chemotherapy. |
| Nakamura | 2005 | CD55 | Expression levels in CDDP-resistant/sensitive cell lines using in-house cDNA microarray (2021 genes originated from normal oral tissue, primary oral cancer, and oral cancer cell lines) and PCR. | CD55 was overexpressed in the H-1R colony. |
| Inactivation of antineoplastic drugs | ||||
| Ansell | 2016 | AR, EPR and EGF | Response was evaluated by adding recombinant human proteins or siRNA-mediated downregulation of endogenous ligand production. | The amount of EGF strongly influences the tumor cell proliferation rate and response to cetuximab treatment. Proposed EGF as a potential predictive biomarker |
| Pickhard | 2014 | AurkA and AurkB | IH in tissue samples. | Provide evidence that AurkA genotypically homozygous HNSCC cells respond to cetuximab monotreatment, whereas heterozygous cells do not. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Verdín, S.; Lavalle-Carrasco, J.; Carreón-Burciaga, R.G.; Serafín-Higuera, N.; Molina-Frechero, N.; González-González, R.; Bologna-Molina, R. Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review. Cancers 2018, 10, 376. https://doi.org/10.3390/cancers10100376
López-Verdín S, Lavalle-Carrasco J, Carreón-Burciaga RG, Serafín-Higuera N, Molina-Frechero N, González-González R, Bologna-Molina R. Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review. Cancers. 2018; 10(10):376. https://doi.org/10.3390/cancers10100376
Chicago/Turabian StyleLópez-Verdín, Sandra, Jesús Lavalle-Carrasco, Ramón G. Carreón-Burciaga, Nicolás Serafín-Higuera, Nelly Molina-Frechero, Rogelio González-González, and Ronell Bologna-Molina. 2018. "Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review" Cancers 10, no. 10: 376. https://doi.org/10.3390/cancers10100376