PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Demographic Characterization of Patients
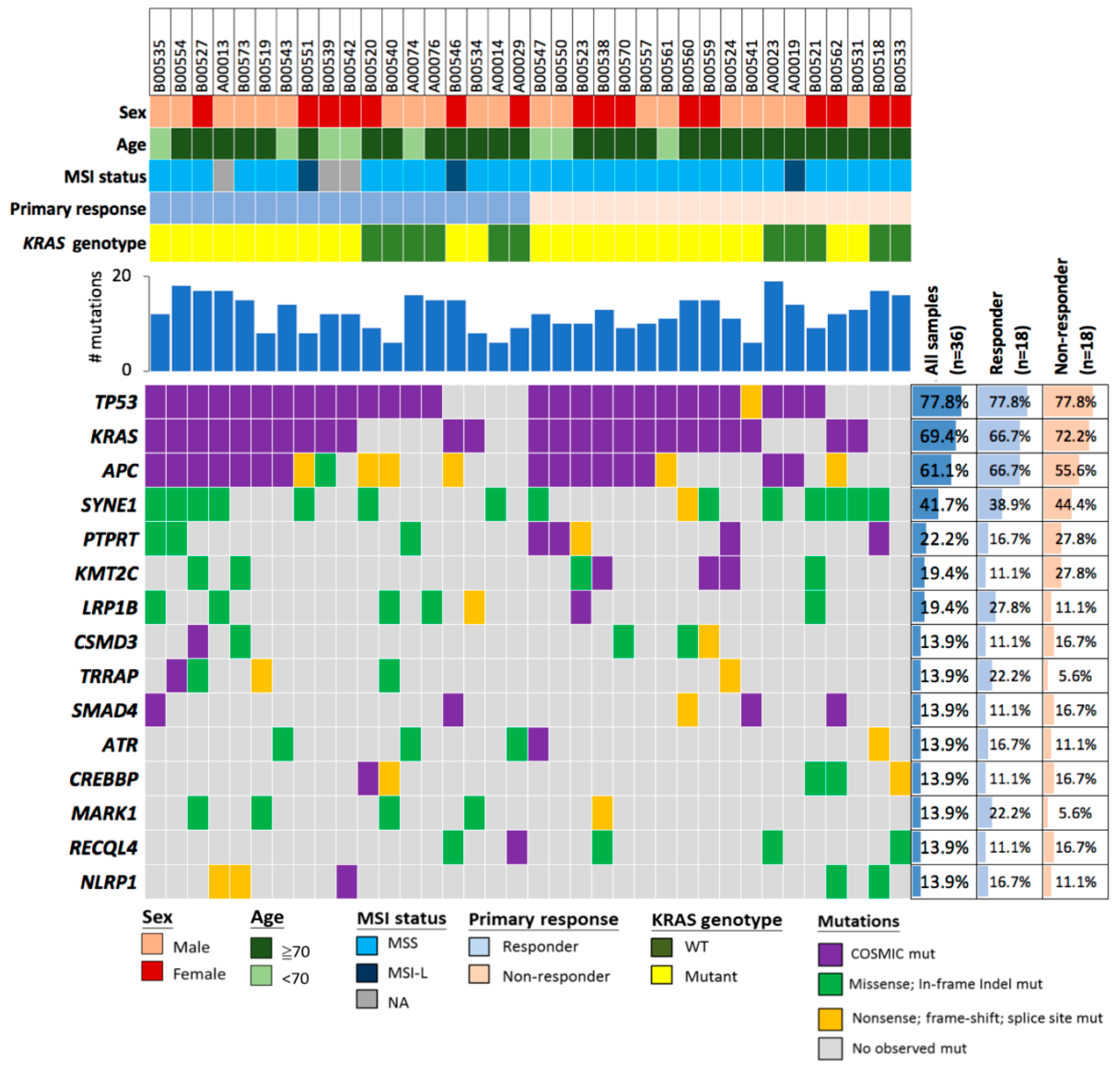
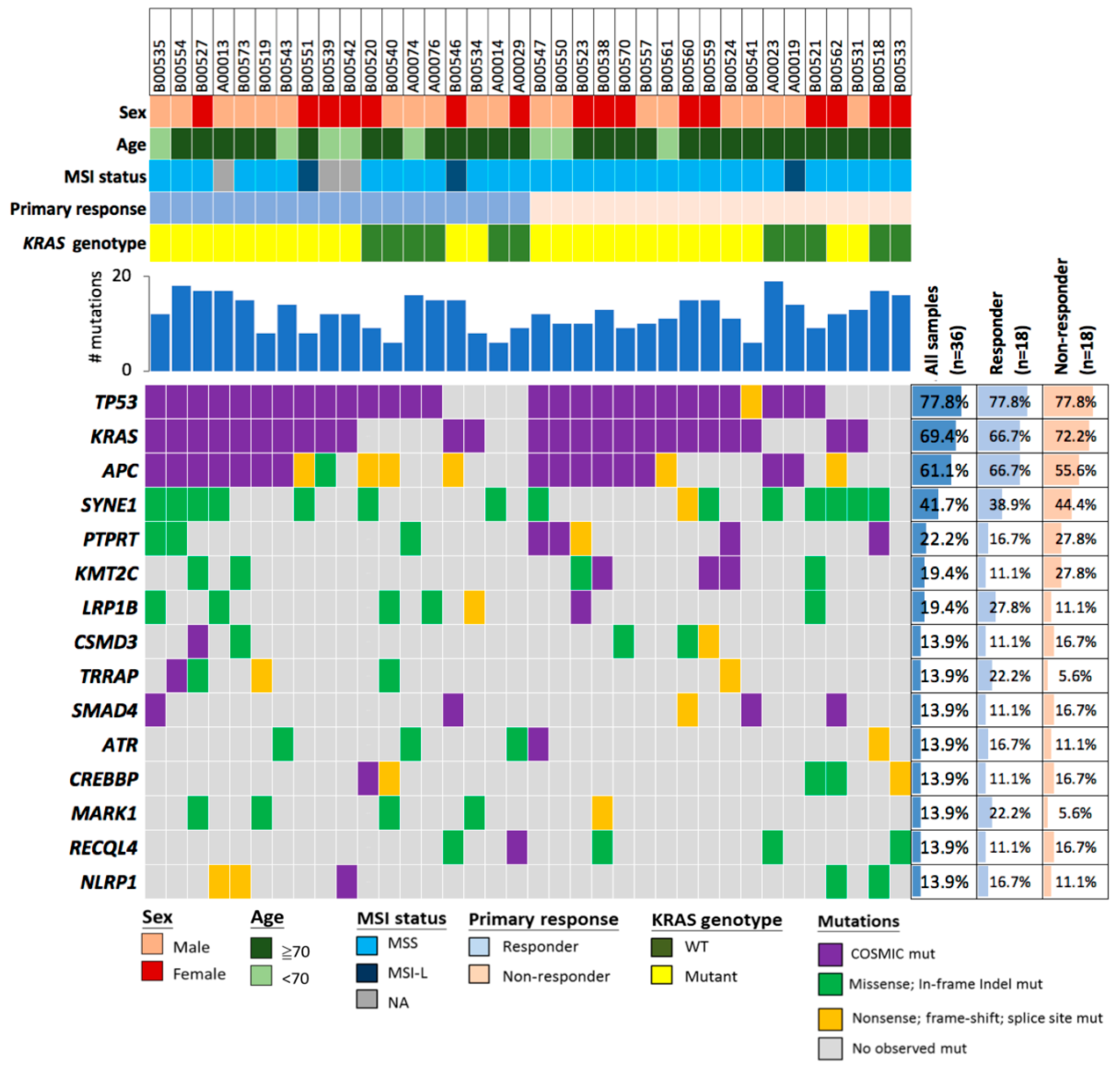
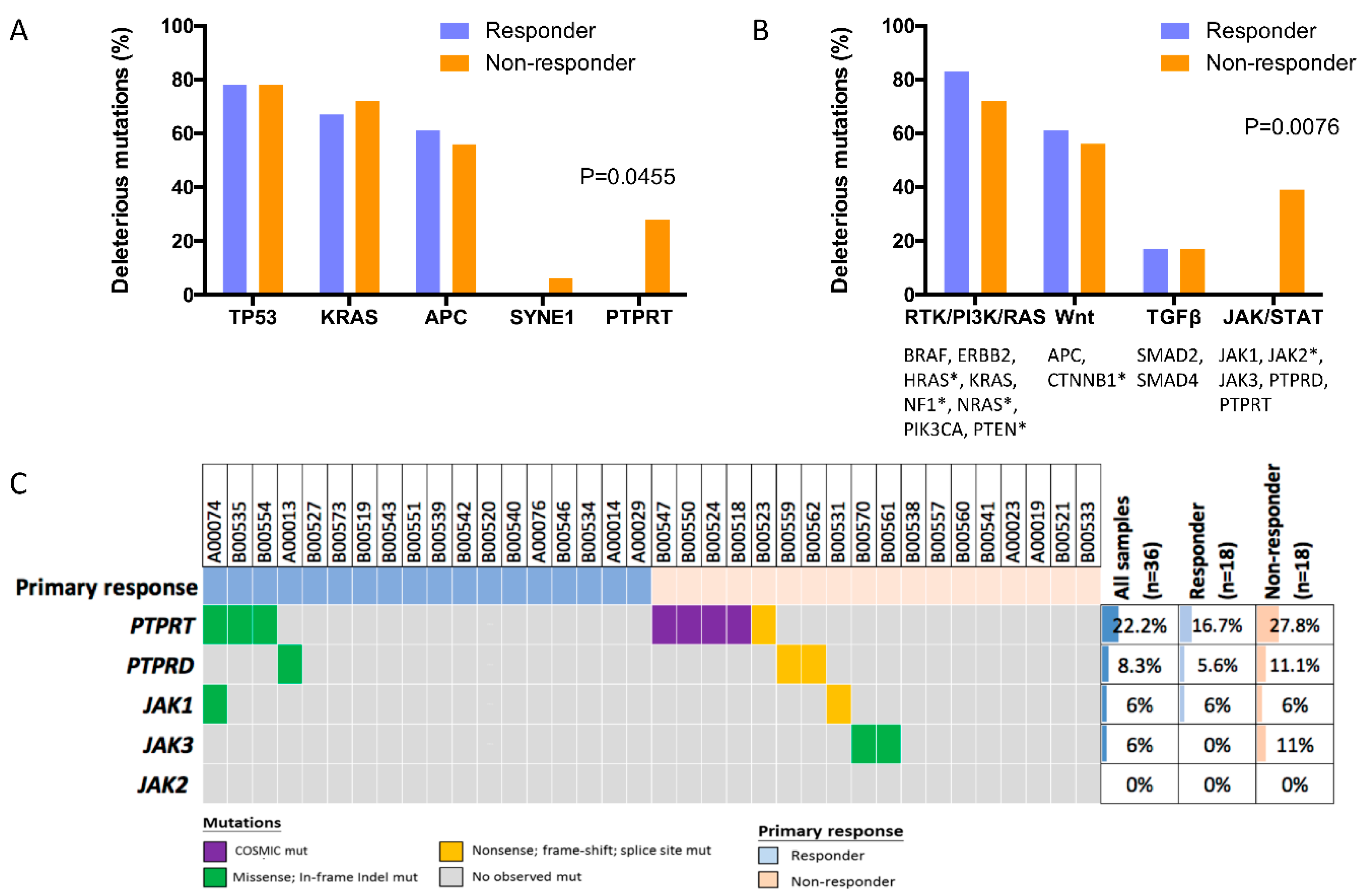
2.2. Deleterious Mutations in the Phosphatase Gene PTPRT Are Associated with Bevacizumab Response Status
2.3. Deleterious Mutations in PTPRT and PTPRD, Phosphatases of the JAK/STAT Pathway, Predict Bevacizumab Response
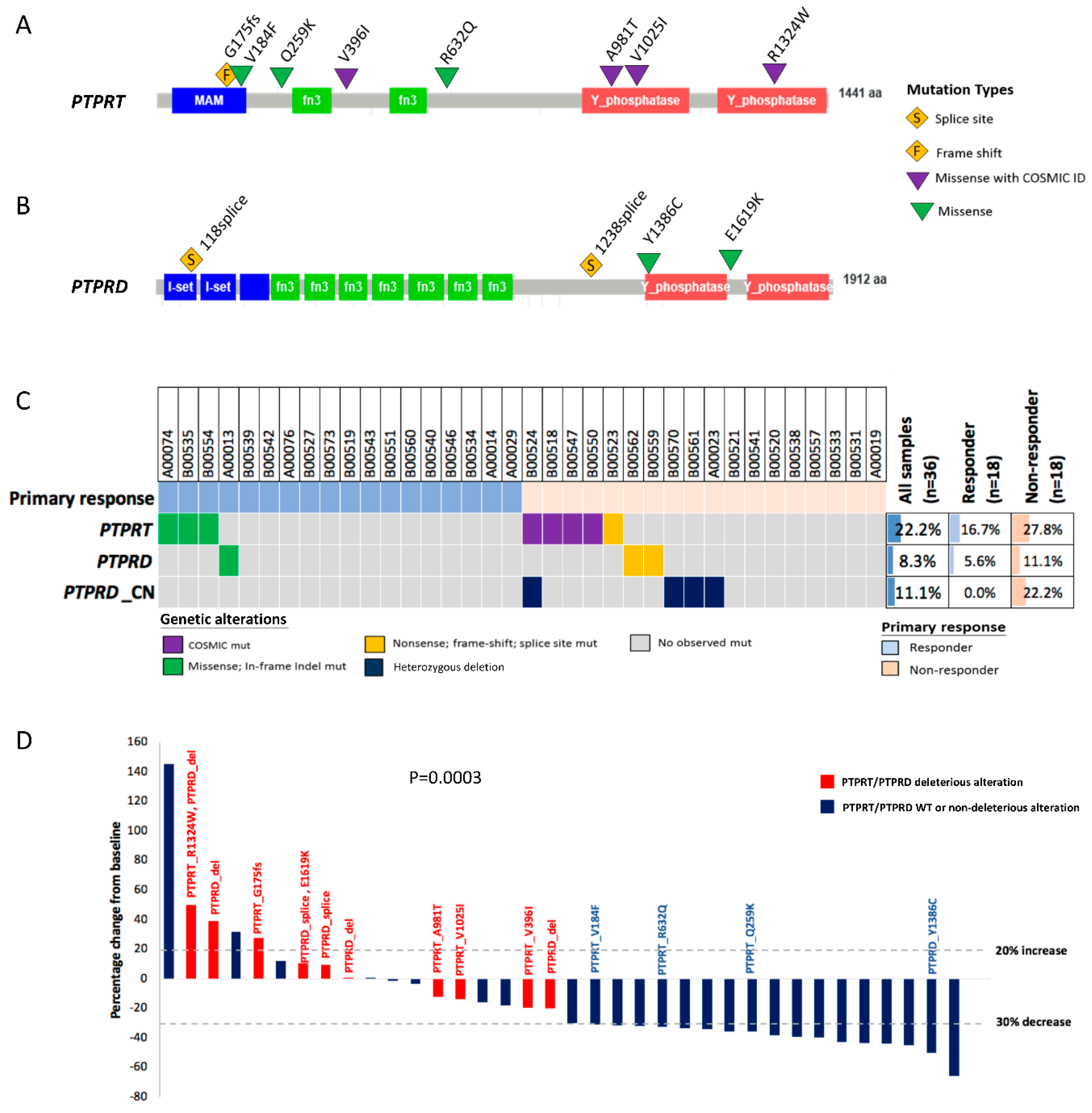
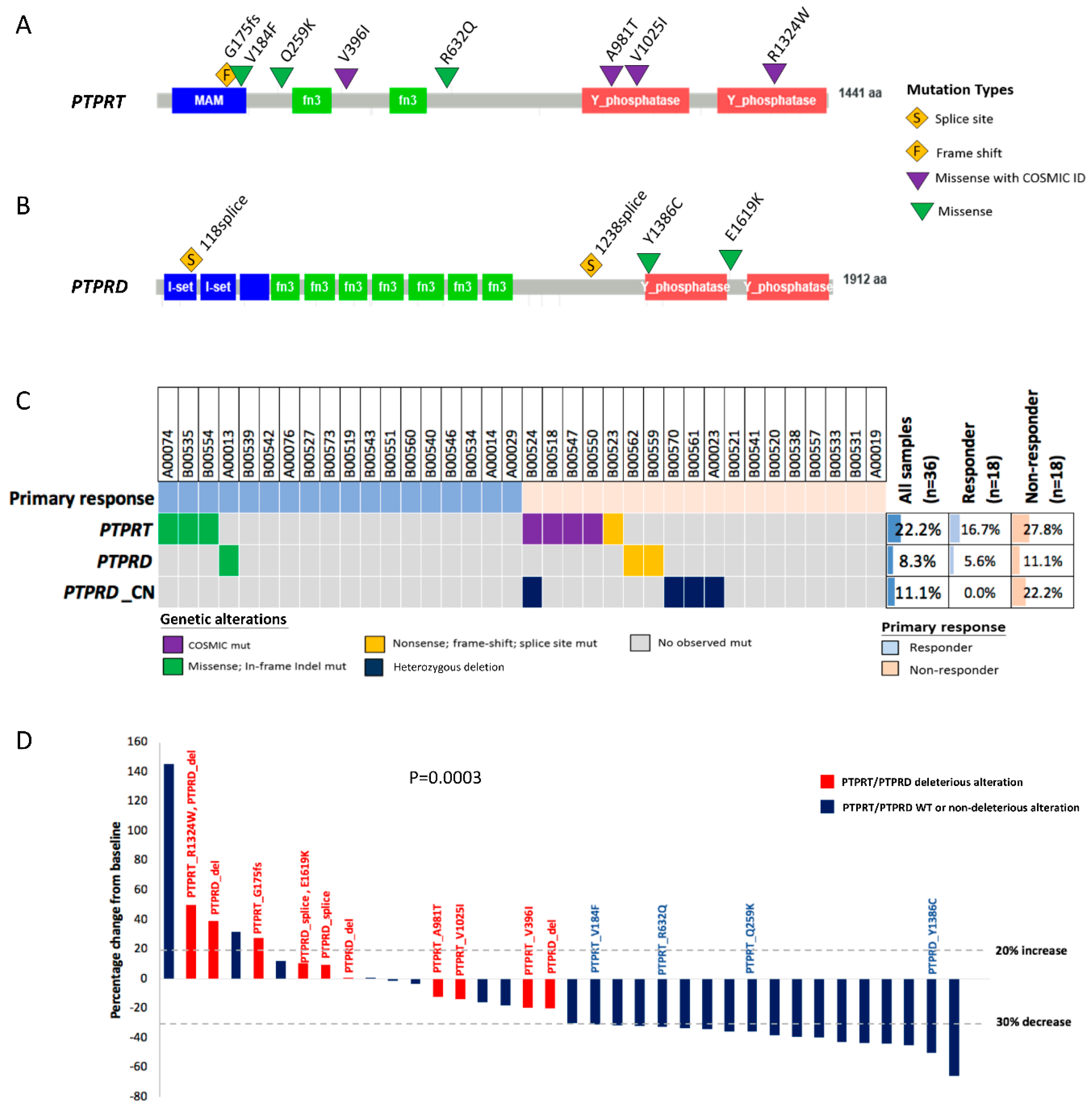
2.4. PTPRT Phosphatase Domain Missense Variants and PTPRT/PTPRD Truncating Variants Are Characteristic for Bevacizumab Non-Responders
2.5. Combining Copy Number Loss and Deleterious Mutations Improves the Predictive Power of PTPRT/PTPRD for Bevacizumab Response
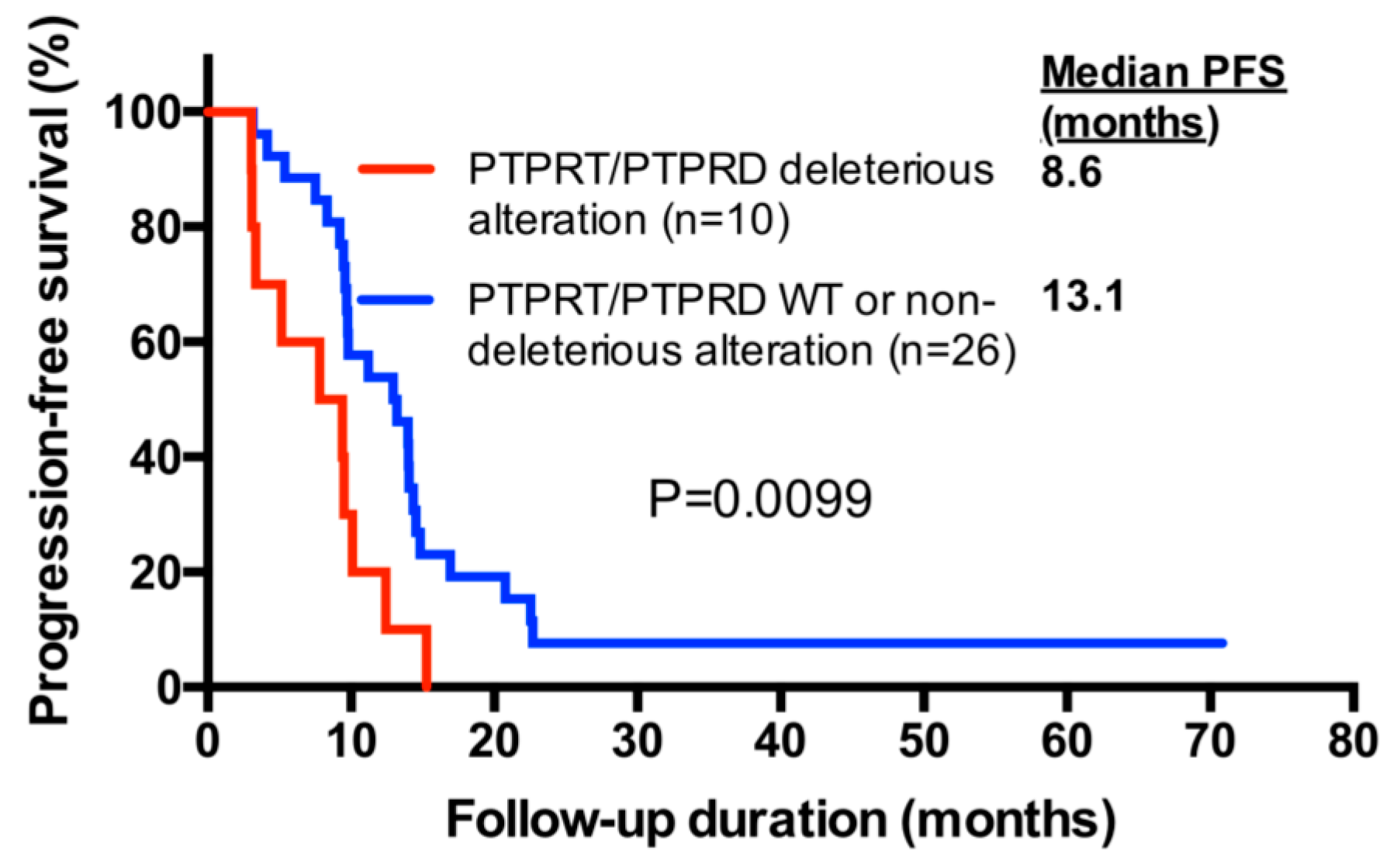
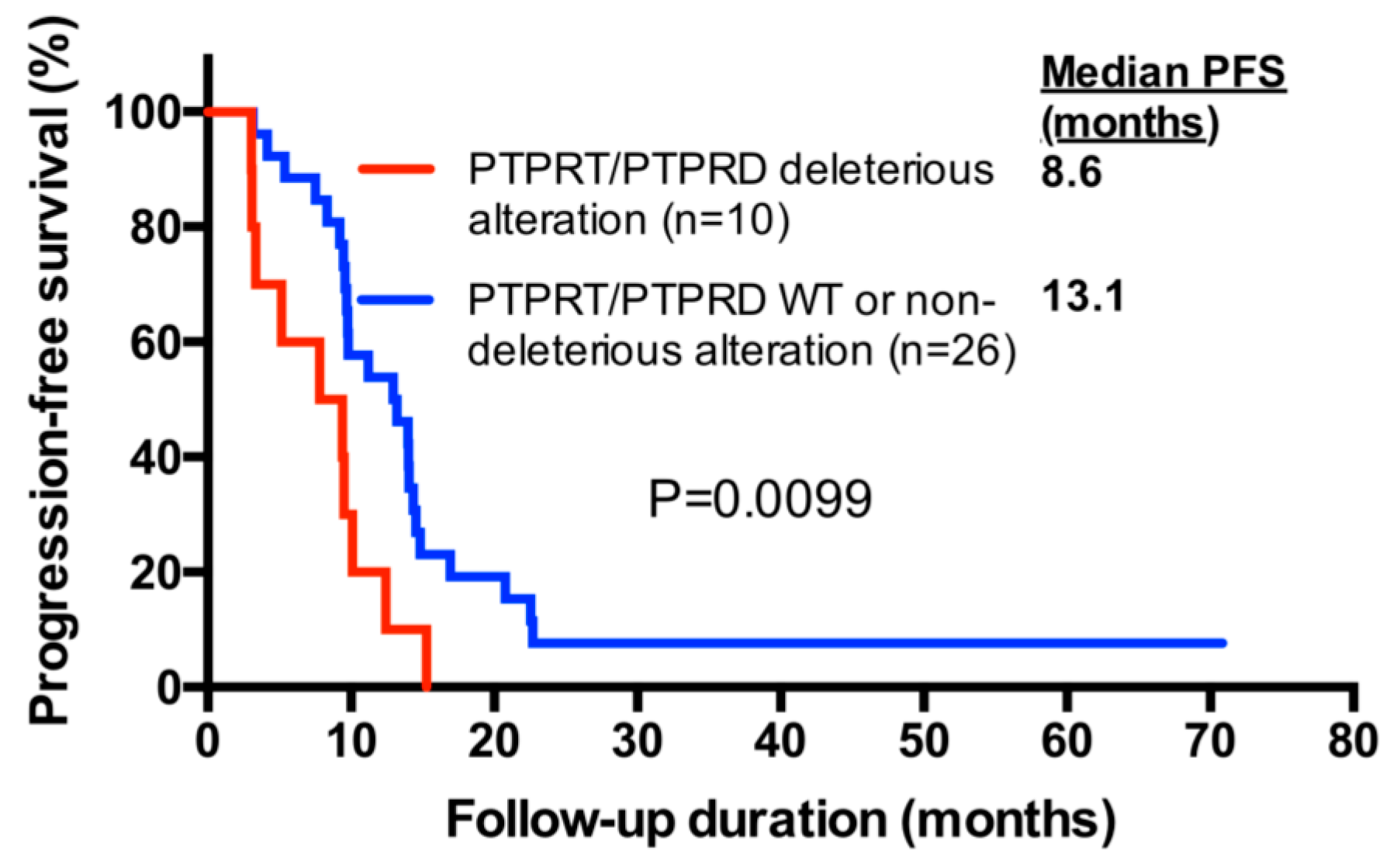
2.6. PTPRT/PTPRD Alterations and Progression-Free Survival
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Treatment Regimens
4.3. Tumor Sequencing and Analysis of Genetic Alterations
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer WHO. GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. World Fact Sheet. 2012. (Last Update 2012). Available online: http://globocan.iarc.fr/Pages/fact_sheets_population.aspx (accessed on 8 September 2016).
- National Cancer Institute. SEER Cancer Statistics Factsheets: Colon and Rectum Cancer. 2016; (Last Update 2016). Available online: http://seer.cancer.gov/ statfacts/html/colorect.html (accessed on 23 August 2016).
- Kasi, P.M.; Hubbard, J.M.; Grothey, A. Selection of biologics for patients with metastatic colorectal cancer: The role of predictive markers. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.C.; Wilson, K.; Gebski, V.J.; Cummins, M.M.; Zannino, D.; Van Hazel, G.A.; Robinson, B.; Broad, A.; Ganju, V.; Ackland, S.P.; et al. Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: Results of the Australasian Gastrointestinal Trials Group Randomized Phase III MAX Study. J. Clin. Oncol. 2010, 28, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Kabbinavar, F.; Irl, C.; Zurlo, A.; Hurwitz, H. Bevacizumab improves the overall and progression-free survival of patients with metastatic colorectal cancer treated with 5-fluorouracil-based regimens irrespective of baseline risk. Oncology 2008, 75, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Lang, I.; Marcuello, E.; Lorusso, V.; Ocvirk, J.; Shin, D.B.; Jonker, D.; Osborne, S.; Andre, N.; Waterkamp, D.; et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): An open-label, randomised phase 3 trial. Lancet Oncol. 2013, 14, 1077–1085. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii1–iii9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. Colon Cancer. Version 2. 2015. Available online: www.nccn.org (accessed on 11 June 2015).
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). European Public Assessment Reports. Avastin: EPAR—Product Information. Available online: http://www. ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000582/WC500029271.pdf (accessed on 11 June 2015).
- Loupakis, F.; Ruzzo, A.; Salvatore, L.; Cremolini, C.; Masi, G.; Frumento, P.; Schirripa, M.; Catalano, V.; Galluccio, N.; Canestrari, E.; et al. Retrospective exploratory analysis of VEGF polymorphisms in the prediction of benefit from first-line FOLFIRI plus bevacizumab in metastatic colorectal cancer. BMC Cancer 2011, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- Sibertin-Blanc, C.; Mancini, J.; Fabre, A.; Lagarde, A.; Del Grande, J.; Levy, N.; Seitz, J.-F.; Olschwang, S.; Dahan, L. Vascular Endothelial Growth Factor A c.*237C>T polymorphism is associated with bevacizumab efficacy and related hypertension in metastatic colorectal cancer. Dig. Liver Dis. 2015, 47, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Yang, D.; Salvatore, L.; Zhang, W.; Wakatsuki, T.; Bohanes, P.; Schirripa, M.; Benhaim, L.; Lonardi, S.; et al. Prospective validation of candidate SNPs of VEGF/VEGFR pathway in metastatic colorectal cancer patients treated with first-line FOLFIRI plus bevacizumab. PLoS ONE 2013, 8, e66774. [Google Scholar] [CrossRef] [PubMed]
- Sohn, B.S.; Park, S.J.; Kim, J.E.; Kim, K.P.; Hong, Y.S.; Suh, C.; Kim, Y.S.; Kim, S.Y.; Im, S.-A.; Kim, S.Y.; et al. Single-nucleotide polymorphisms in the vascular endothelial growth factor pathway and outcomes of patients treated with first-line cytotoxic chemotherapy combined with bevacizumab for advanced colorectal cancer. Oncology 2014, 87, 280–292. [Google Scholar] [CrossRef] [PubMed]
- AWeickhardt, J.A.; Williams, D.; Lee, C.; Simes, J.; Murone, C.; Cummins, M.; Asadi, K.; Price, T.J.; Mariadason, J.; Tebbutt, N.C.; et al. Vascular endothelial growth factors (VEGF) and VEGF receptor expression as predictive biomarkers for benefit with bevacizumab in metastatic colorectal cancer (mCRC): Analysis of the phase III MAX study. J. Clin. Oncol. 2011, 29 (Suppl. 15), 3531. [Google Scholar] [CrossRef]
- Guan, K.L.; Haun, R.S.; Watson, S.J.; Geahlen, R.L.; Dixon, J.E. Cloning and expression of a protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Czernilofsky, A.P.; Levinson, A.D.; Varmus, H.E.; Bishop, J.M.; Tischer, E.; Goodman, H.M. Nucleotide sequence of an avian sarcoma virus oncogene (src) and proposed amino acid sequence for gene product. Nature 1980, 287, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, H.; Tonks, N.K.; Kumar, S.; Diltz, C.D.; Harrylock, M.; Cool, D.E.; Krebs, E.G.; Fischer, E.H.; Walsh, K.A. Human placenta protein-tyrosine-phosphatase: Amino acid sequence and relationship to a family of receptor-like proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5252–5256. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuwanita, I.; Barnes, D.; Monterey, M.D.; O’Reilly, S.; Andrechek, E.R. Increased metastasis with loss of E2F2 in Myc-driven tumors. Oncotarget 2015, 6, 38210–38224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, X.; Guda, K.; Lawrence, E.; Sun, Q.; Watanabe, T.; Iwakura, Y.; Asano, M.; Wei, L.; Yang, Z.; et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc. Natl. Acad. Sci. USA 2010, 107, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; Van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, H.; Yoshida, K.; Blagitko-Dorfs, N.; Claus, R.; Pantic, M.; Abdelkarim, M.; Niemöller, C.; Greil, C.; Hackanson, B.; Shiraishi, Y.; et al. Tracing the development of acute myeloid leukemia in CBL syndrome. Blood 2014, 123, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.A.; Woo, M.S.; Getz, G.; Perner, S.; Ding, L.; Beroukhim, R.; Lin, W.M.; Province, M.A.; Kraja, A.; Johnson, L.A.; et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007, 450, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singchat, W.; Hitakomate, E.; Rerkarmnuaychoke, B.; Suntronpong, A.; Fu, B.; Bodhisuwan, W.; Peyachoknagul, S.; Yang, F.; Koontongkaew, S.; Srikulnath, K. Genomic Alteration in Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines Inferred from Karyotyping, Molecular Cytogenetics, and Array Comparative Genomic Hybridization. PLoS ONE 2016, 11, e0160901. [Google Scholar] [CrossRef] [PubMed]
- Beothe, T.; Zubakov, D.; Kovacs, G. Homozygous losses detected by array comparative genomic hybridization in multiplex urothelial carcinomas of the bladder. Cancer Genet. 2015, 208, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Acun, T.; Demir, K.; Oztas, E.; Arango, D.; Yakicier, M.C. PTPRD is homozygously deleted and epigenetically downregulated in human hepatocellular carcinomas. OMICS 2015, 19, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, K.; Yamazumi, Y.; Oda, T.; Akiyama, T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp. Ther. Med. 2011, 2, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becka, S.; Zhang, P.; Craig, S.E.; Lodowski, D.T.; Wang, Z.; Brady-Kalnay, S.M. Characterization of the adhesive properties of the type IIb subfamily receptor protein tyrosine phosphatases. Cell Commun. Adhes. 2010, 17, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Besco, J.A.; Hooft van Huijsduijnen, R.; Frostholm, A.; Rotter, A. Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPrho/PTPRT). Brain Res. 2006, 1116, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, B.; Fabius, A.W.; Wu, W.H.; Pedraza, A.; Brennan, C.W.; Schultz, N.; Pitter, K.L.; Bromberg, J.F.; Huse, J.T.; Holland, E.C.; et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 8149–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Niu, N.; Wei, T.; Tozawa, H.; Chen, X.; Zhang, C.; Zhang, J.; Wada, Y.; Kapron, C.M.; Liu, J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 2017, 8, 69139–69161. [Google Scholar] [CrossRef] [PubMed]
- Mesange, P.; Poindessous, V.; Sabbah, M.; Escargueil, A.E.; de Gramont, A.; Larsen, A.K. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget 2014, 5, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Videira, P.A.; Piteira, A.R.; Cabral, M.G.; Martins, C.; Correia, M.; Severino, P.; Gouveia, H.; Carrascal, M.; Almeida, J.F.; Trindade, H.; et al. Effects of bevacizumab on autocrine VEGF stimulation in bladder cancer cell lines. Urol. Int. 2011, 86, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Marisi, G.; Scarpi, E.; Passardi, A.; Nanni, O.; Ragazzini, A.; Valgiusti, M.; Gardini, A.C.; Neri, L.M.; Frassineti, G.L.; Amadori, D.; et al. Circulating VEGF and eNOS variations as predictors of outcome in metastatic colorectal cancer patients receiving bevacizumab. Sci. Rep. 2017, 7, 1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Jubb, A.M.; Chen, D.; Li, N.F.; Meng, Y.G.; Bernaards, C.; Elliott, R.; Scherer, S.J.; Chen, D.S. Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab. Clin. Cancer Res. 2013, 19, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.F.; Christensen, R.; Andersen, R.F.; Garm Spindler, K.L.; Johnsson, A.; Jakobsen, A. The predictive value of single nucleotide polymorphisms in the VEGF system to the efficacy of first-line treatment with bevacizumab plus chemotherapy in patients with metastatic colorectal cancer: Results from the Nordic ACT trial. Int. J. Colorectal Dis. 2012, 27, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, F.; Yuan, Q.; Chen, G.; Chen, C.; Yu, B. Role of VEGFA gene polymorphisms in colorectal cancer patients who treated with bevacizumab. Oncotarget 2017, 8, 105472–105478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; McCrudden, C.M.; Meng, C.; Lin, Y.; Kwok, H.F. The significance of combining VEGFA, FLT1, and KDR expressions in colon cancer patient prognosis and predicting response to bevacizumab. OncoTargets Ther. 2015, 8, 835–843. [Google Scholar] [CrossRef]
- Boisen, M.K.; Johansen, J.S.; Dehlendorff, C.; Larsen, J.S.; Osterlind, K.; Hansen, J.; Nielsen, S.E.; Pfeiffer, P.; Tarpgaard, L.S.; Holländer, N.H.; et al. Primary tumor location and bevacizumab effectiveness in patients with metastatic colorectal cancer. Ann. Oncol. 2013, 24, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, D.M.; Laudanna, C.; Migliozzi, S.; Zoppoli, P.; Santamaria, G.; Grillone, K.; Elia, L.; Mignogna, C.; Biamonte, F.; Sacco, R.; et al. Identification of different mutational profiles in cancers arising in specific colon segments by next generation sequencing. Oncotarget 2018, 9, 23960–23974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.S.; Marshall, J.; Mitchell, E.; Wierzbicki, R.; Ganju, V.; Jeffery, M.; Schulz, J.; Richards, D.; Soufi-Mahjoubi, R.; Wang, B.; et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: Results from the BICC-C Study. J. Clin. Oncol. 2007, 25, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Maspero, F.; Sauta, M.G.; Beretta, G.D.; Barni, S. FOLFIRI-bevacizumab as first-line chemotherapy in 3500 patients with advanced colorectal cancer: A pooled analysis of 29 published trials. Clin. Colorectal Cancer 2013, 12, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Lienard, M.; Toffoli, S.; Kamal, M.; Le Tourneau, C.; Gentien, D.; Servant, N.; Gestraud, P.; Rio Frio, T.; et al. Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics 2014, 30, 3443–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobson, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobson, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | Responders | Non-Responders | p Value | |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | ||
| Total number | 36 (100) | 18 (50) | 18 (50) | |
| Sex | 0.500 | |||
| Male | 21 (58.3) | 12 (67.7) | 9 (50.0) | |
| Female | 15 (41.7) | 6 (33.3) | 9 (50.0) | |
| Age | 0.740 | |||
| <60 | 18 (50.0) | 10 (55.6) | 8 (44.4) | |
| ≥60 | 18 (50.0) | 8 (44.4) | 10 (55.6) | |
| Median (range) | 59.5 (33–87) | 57 (40–84) | 61.5 (33–87) | |
| Histologic grade | 1.000 | |||
| low grade 1 | 33 (91.7) | 16 (88.9) | 17 (94.4) | |
| high grade 2 | 3 (8.3) | 2 (11.1) | 1 (5.6) | |
| Metastatic pattern | 1.000 | |||
| metachronous | 17 (47.2) | 8 (44.4) | 9 (50.0) | |
| synchronous | 19 (52.8) | 10 (55.6) | 9 (50.0) | |
| Primary tumor site | 0.500 | |||
| Colon | 21 (58.3) | 12 (66.7) | 9 (50.0) | |
| Rectum | 15 (41.7) | 6 (33.3) | 9 (50.0) | |
| Metastatic site | ||||
| Liver | 21 (58.3) | 8 (44.4) | 13 (72.2) | |
| Lung | 15 (41,7) | 9 (50.0) | 6 (33.3) | |
| Other | 15 (41.7) | 9 (50.0) | 6 (5.6) | |
| Number of metastatic sites | 1.000 | |||
| 1 | 21 (58.3) | 10 (55.6) | 11 (61.1) | |
| >1 | 15 (41.7) | 8 (44.4) | 7 (38.9) | |
| Treatment regimen | 0.603 | |||
| 1st line | 32 (88.9) | 15 (83.3) | 17 (94.4) | |
| 2nd line | 4 (11.1) | 3 (16.7) | 1 (5.6) | |
| PFS (months) | 0.001 | |||
| Median (range) | 9.8 (3.0–70.9) | 14.0 (7.5–70.9) | 9.2 (3.0–22.5) |
| Sample ID | Response | Gene | cDNA Change | Position/AA Change | COSMIC ID | Protein Domain | Variant Type | Deleterious |
|---|---|---|---|---|---|---|---|---|
| B00524 | PD | PTPRT | c.3970C>T | p.R1324W | COSM577363 | Phosphatase | Missense | Yes |
| B00524 | PD | PTPRD | Heterozygous deletion | Yes | ||||
| B00523 | PD | PTPRT | c.524delG | p.G175fs | Frameshift | Yes | ||
| A00023 | PD | PTPRD | Heterozygous deletion | Yes | ||||
| B00547 | SD | PTPRT | c.2941G>A | p.A981T | COSM1318833 | Phosphatase | Missense | Yes |
| B00550 | SD | PTPRT | c.3073G>A | p.V1025I | COSM3546449 | Phosphatase | Missense | Yes |
| B00559 | SD | PTPRD | c.3715-8T>C | Splice region | Splice region | Splice region | Yes | |
| B00561 | SD | PTPRD | Heterozygous deletion | Yes | ||||
| B00570 | SD | PTPRD | Heterozygous deletion | Yes | ||||
| B00518 | SD | PTPRT | c.1186G>A | p.V396I | COSM1411875 | MAM | Missense | Yes |
| B00562 | SD | PTPRD | c.353-4G>C | Splice region | Splice region | Splice region | Yes | |
| B00562 | SD | PTPRD | c.4855G>A | p.E1619K | Inter-domain | Missense | No | |
| A00074 | PR | PTPRT | c.774_775 delCCinsAA | p.Q259K | Inter-domain | Missense | No | |
| B00535 | PR | PTPRT | c.1895G>A | p.R632Q | Inter-domain | Missense | No | |
| B00554 | PR | PTPRT | c.550G>T | p.V184F | MAM | Missense | No | |
| A00013 | PR | PTPRD | c.4157A>G | p.Y1386C | Missense | No |
| Factors | n | HR 1 | 95% CI 1 | p Value 1 | HR 2 | 95% CI 2 | p Value 2 |
|---|---|---|---|---|---|---|---|
| Clinical | |||||||
| Sex (male/female) | 21/15 | 1.34 | 0.66–2.69 | 0.4164 | |||
| Age (≥60/<60) | 18/18 | 1.13 | 0.57–2.25 | 0.7191 | |||
| Histologic Grade (high/low) | 3/33 | 1.61 | 0.48–5.46 | 0.4422 | |||
| Metastatic pattern (meta/syn) | 17/19 | 1.19 | 0.60–2.35 | 0.6144 | |||
| Primary Tumor site (rectum/colon) | 15/21 | 2.11 | 1.04–4.26 | 0.0377 | 2.56 | 1.23–5.32 | 0.0118 |
| Number of metastatic sites (>1/1) | 15/21 | 1.01 | 0.51–2.01 | 0.9757 | |||
| Treatment regimen (2nd line/1st line) | 4/32 | 0.77 | 0.27–2.20 | 0.6274 | |||
| Genetic | |||||||
| PTPRT/PTPRD deleterious alteration (yes/no) | 10/26 | 2.67 | 1.23–5.80 | 0.0130 | 3.33 | 1.47–7.54 | 0.0038 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-C.; Lapke, N.; Chen, S.-J.; Lu, Y.-J.; Jhou, R.-S.; Yeh, C.-Y.; Tsai, W.-S.; Hung, H.-Y.; Hsieh, J.C.-H.; Yang, T.-S.; et al. PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients. Cancers 2018, 10, 314. https://doi.org/10.3390/cancers10090314
Hsu H-C, Lapke N, Chen S-J, Lu Y-J, Jhou R-S, Yeh C-Y, Tsai W-S, Hung H-Y, Hsieh JC-H, Yang T-S, et al. PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients. Cancers. 2018; 10(9):314. https://doi.org/10.3390/cancers10090314
Chicago/Turabian StyleHsu, Hung-Chih, Nina Lapke, Shu-Jen Chen, Yen-Jung Lu, Ren-Shiang Jhou, Chien-Yuh Yeh, Wen-Sy Tsai, Hsin-Yuan Hung, Jason Chia-Hsun Hsieh, Tsai-Sheng Yang, and et al. 2018. "PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients" Cancers 10, no. 9: 314. https://doi.org/10.3390/cancers10090314