Machine Learning-Guided Prediction of Antigen-Reactive In Silico Clonotypes Based on Changes in Clonal Abundance through Bio-Panning

 ,
, 
Abstract
:1. Introduction
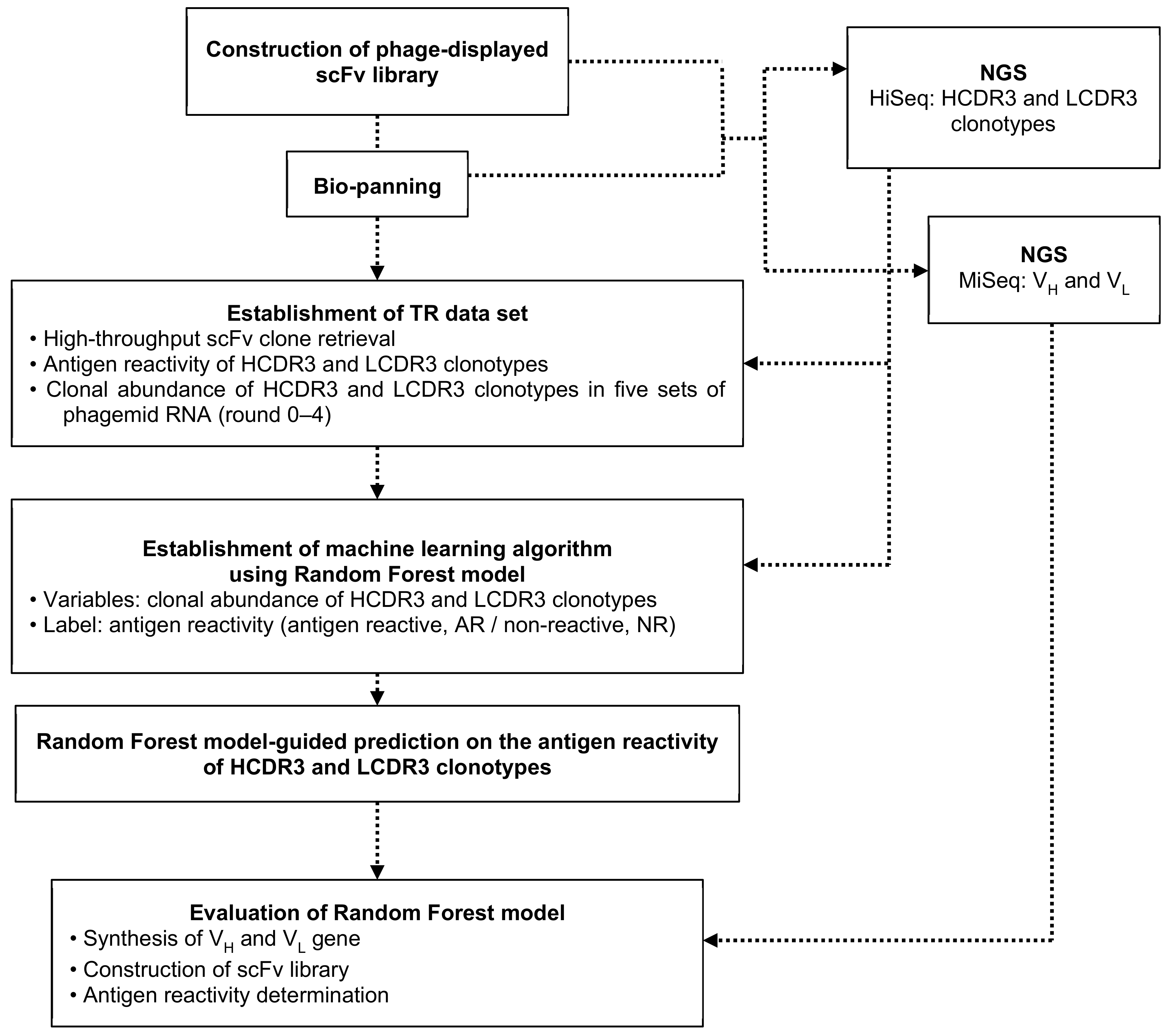
2. Materials and Methods
2.1. Immunization, Construction of Phage-Displayed scFv Library, and Bio-Panning
2.2. Next-Generation Sequencing (NGS)
2.3. High-Throughput Clone Retrieval and Phage ELISA
2.4. Establishment of the Random forest (RF) Models
2.5. Construction of Antigen-Reactive (AR) and Non-Reactive (NR) Phage-Displayed scFv Library and Phage ELISA
3. Results
3.1. Construction of Phage-Displayed scFv Library, Biopanning, Selection of Positive Clones, Next-Generation Sequencing (NGS), And Establishment of TR Data Set
3.2. Establishing Random Forest (RF) Binding Reactivity Prediction Model
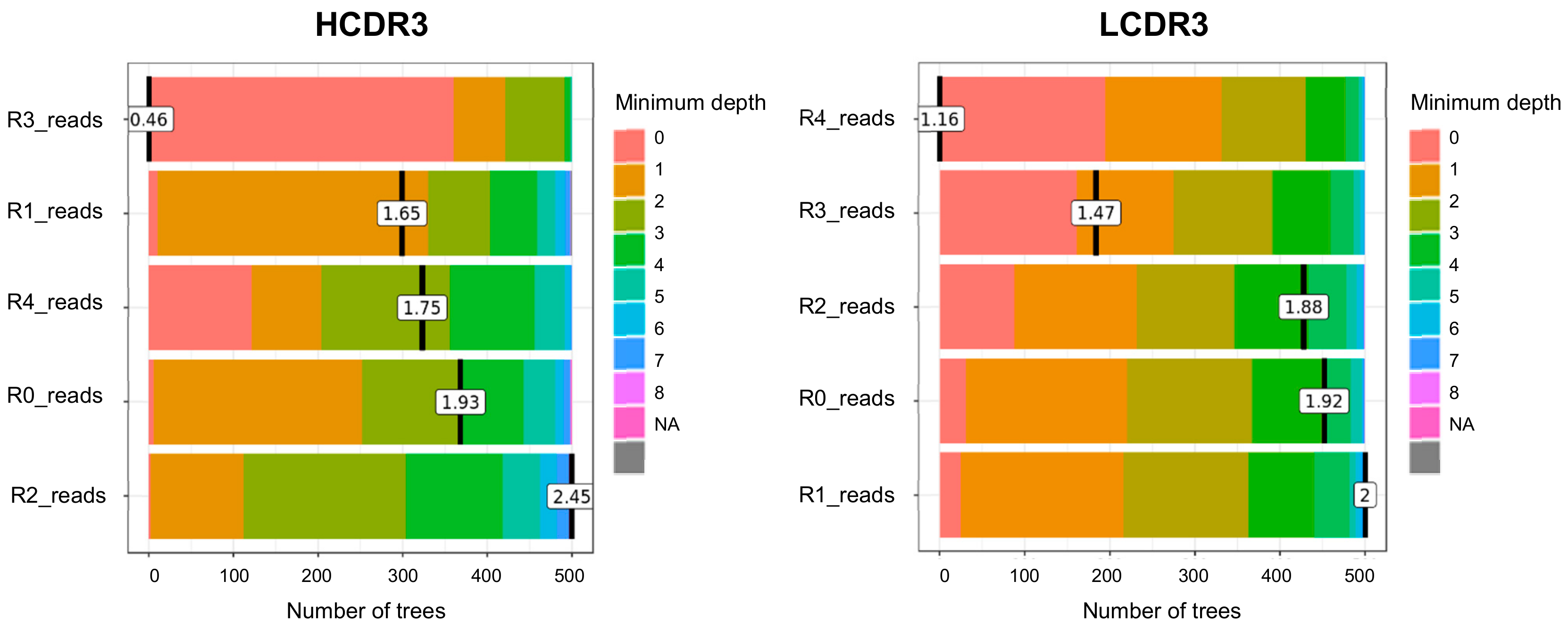
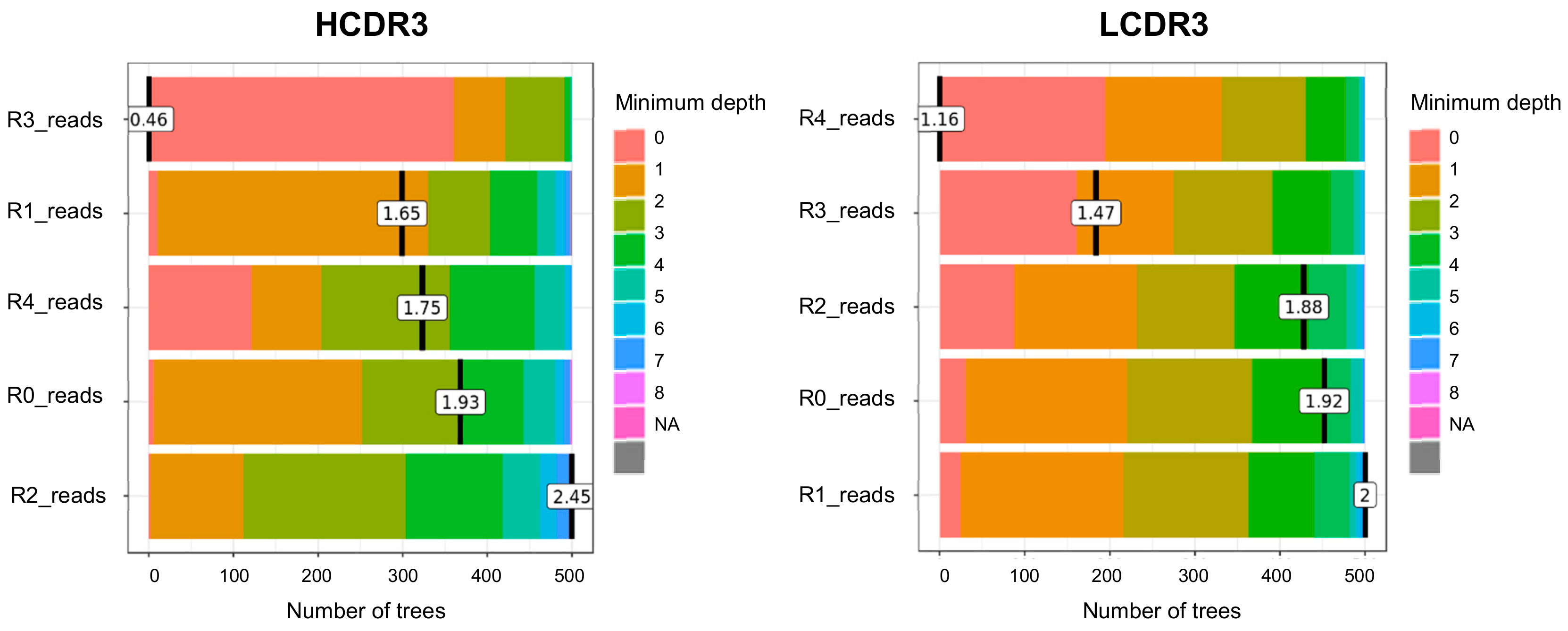
3.3. Measurement of the Minumum Depth Value of a Predictor Variable
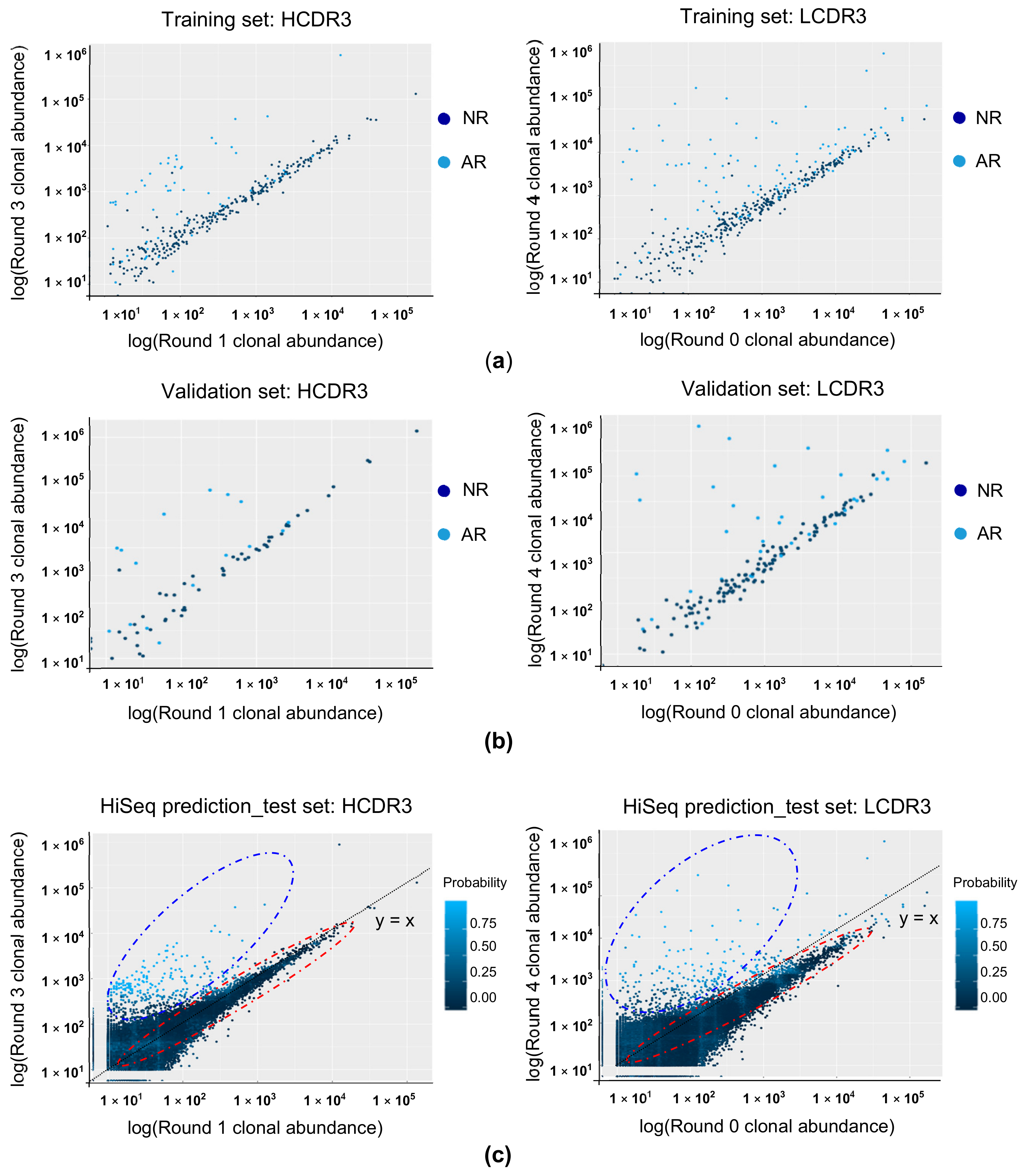
3.4. Predicting the Binding Property of the CDR3 Clonotype Using RF Modeling
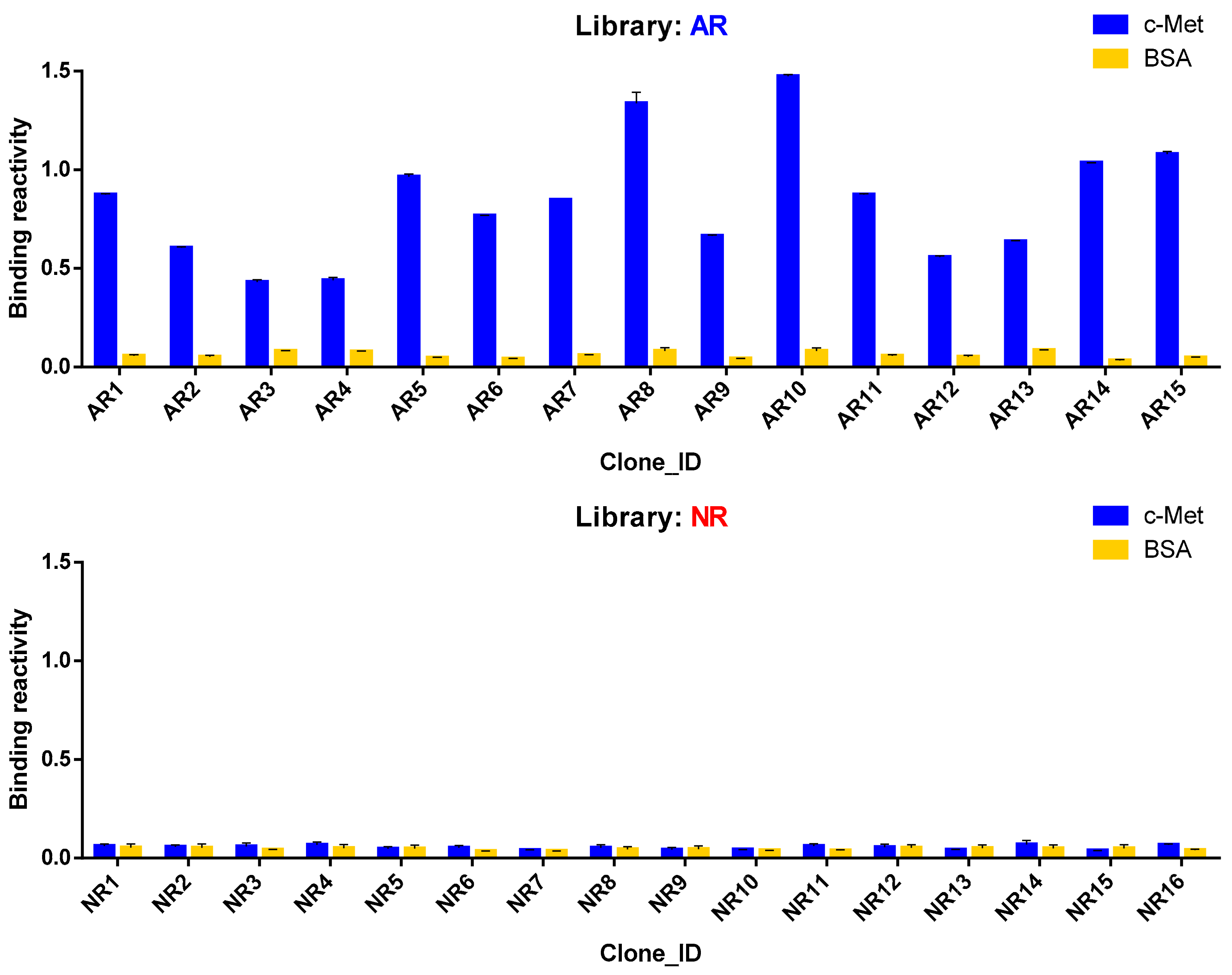
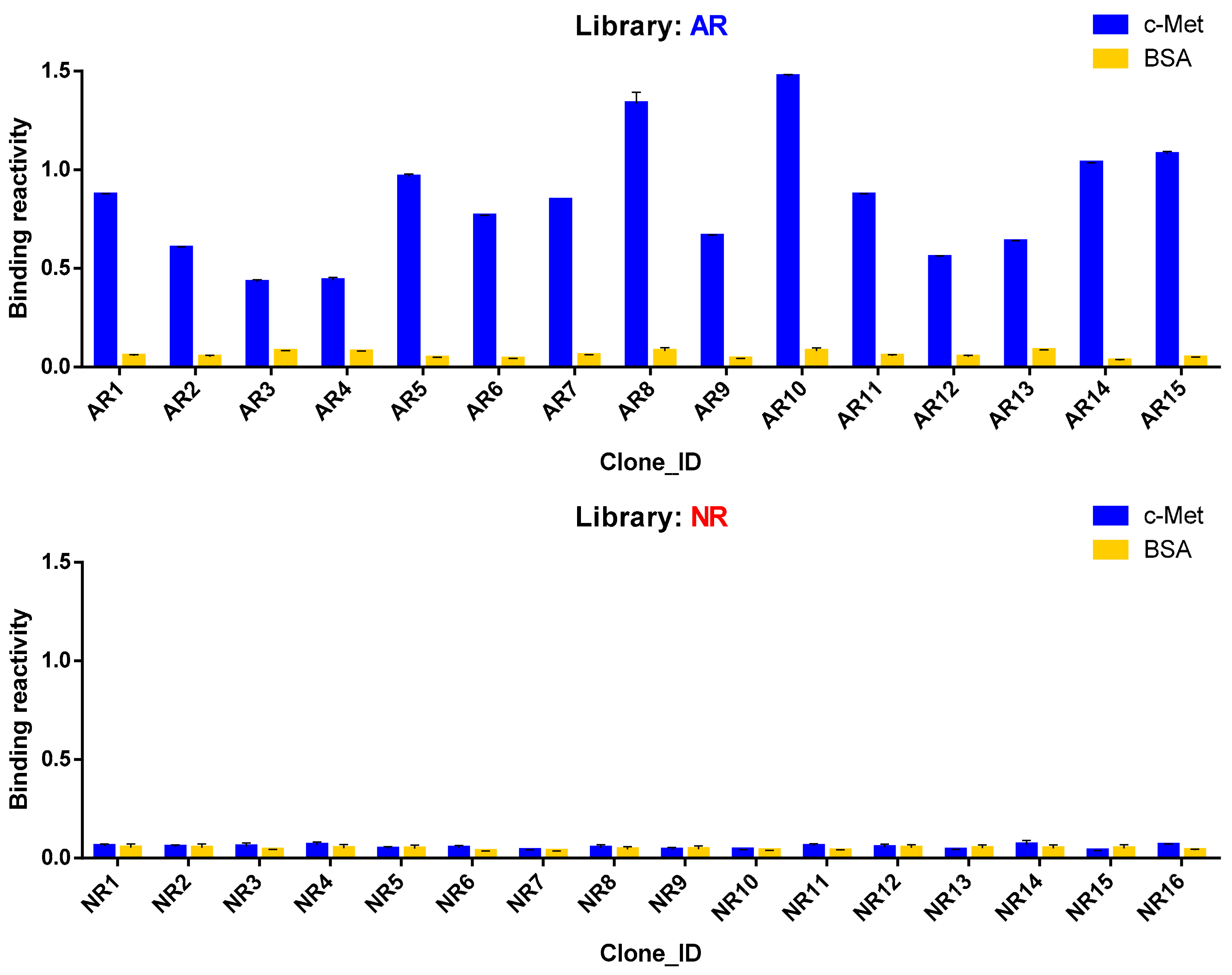
3.5. Antigen Reactivity Validation of In Silico CDR3 Clonotypes in Phage ELISA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Comoglio, P.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.G.; Lim, T.H.; Lim, J.; Liew, P.J.; Kwang, X.L.; Nahar, R.; Aung, Z.W.; Takano, A.; Lee, Y.Y.; Lau, D.P.; et al. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor–Mutant Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, Y.; Ko, Y.S.; Kim, Y.; Pyo, J.-S.; Jang, B.G.; A Kim, M.; Lee, J.-S.; Chang, M.S.; Park, J.-W.; et al. FOXO1 Suppression is a Determinant of Acquired Lapatinib-Resistance in HER2-Positive Gastric Cancer Cells Through MET Upregulation. Cancer Res. Treat. 2018, 50, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Fucà, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of Primary Resistance to Trastuzumab in HER2-Positive Metastatic Gastric Cancer Patients: the AMNESIA Case-Control Study. Clin. Cancer Res. 2017, 24, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Chiriaco, C.; Modica, C.; Acquadro, A.; Cortese, M.; Galimi, F.; Perera, T.; Gammaitoni, L.; Aglietta, M.; Comoglio, P.M.; et al. Met inhibition revokes IFNγ-induction of PD-1 ligands in MET-amplified tumours. Br. J. Cancer 2019, 120, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Saigí, M.; Alburquerque-Bejar, J.J.; Mc Leer-Florin, A.; Pereira, C.; Pros, E.; Romero, O.A.; Baixeras, N.; Esteve-Codina, A.; Nadal, E.; Brambilla, E.; et al. MET-Oncogenic andJAK2-Inactivating Alterations Are Independent Factors That Affect Regulation of PD-L1 Expression in Lung Cancer. Clin. Cancer Res. 2018, 24, 4579–4587. [Google Scholar] [CrossRef] [Green Version]
- Glodde, N.; Bald, T.; Boorn-Konijnenberg, D.V.D.; Nakamura, K.; O’Donnell, J.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e9. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, S.; Nam, D.-H.; Lee, S.J.; Park, S.H.; Park, J.O.; Lee, J.-W.; Kim, K.-M.; Lee, H.; Park, N.; et al. First-in-human phase I trial of anti-hepatocyte growth factor (HGF) antibody (YYB101) in refractory solid tumor patients: Integrative pathologic-genomic analysis and the final results. J. Clin. Oncol. 2019, 37, 3104. [Google Scholar] [CrossRef]
- Rolfo, C.; Van Der Steen, N.; Pauwels, P.; Cappuzzo, F. Onartuzumab in lung cancer: the fall of Icarus? Expert Rev. Anticancer. Ther. 2015, 15, 487–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parola, C.; Neumeier, D.; Reddy, S. Integrating high-throughput screening and sequencing for monoclonal antibody discovery and engineering. Immunology 2017, 153, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Snapkov, I.; Akbar, R.; Pavlović, M.; Miho, E.; Sandve, G.K.; Greiff, V. Augmenting adaptive immunity: progress and challenges in the quantitative engineering and analysis of adaptive immune receptor repertoires. Mol. Syst. Des. Eng. 2019, 4, 701–736. [Google Scholar] [CrossRef]
- A Norman, R.; Ambrosetti, F.; Bonvin, A.M.J.J.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Briefings Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 9–118. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yoon, A.; Lee, S.; Kim, S.; Han, J.; Chung, J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp. Mol. Med. 2017, 49, e308. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.; Kim, O.; Jung, Y.; Han, H.; Kim, J.-E.; Kim, S.; Lee, S.; Park, J.; Jung, R.H.; Kim, S.I.; et al. High-throughput retrieval of physical DNA for NGS-identifiable clones in phage display library. mAbs 2019, 11, 532–545. [Google Scholar] [CrossRef]
- Singh, A.; Thakur, N.; Sharma, A. A review of supervised machine learning algorithms. In Proceedings of the 2016 3rd International Conference on Computing for Sustainable Global Development (INDIACom), New Delhi, India, 16–18 March 2016; pp. 1310–1315. [Google Scholar]
- Kulkarni, V.Y.; Sinha, P.K. Effective Learning and Classification using Random Forest Algorithm. Int. J. Eng. Innov. Technolgy 2014, 3, 267–273. [Google Scholar]
- Andris-Widhopf, J.; Rader, C.; Steinberger, P.; Fuller, R.; Barbas, C.F. Methods for the generation of chicken monoclonal antibody fragments by phage display. J. Immunol. Methods 2000, 242, 159–181. [Google Scholar] [CrossRef]
- Barbas, C.F., III; Burton, D.R.; Scott, J.K.; Silverman, G.J. Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Suffolk County, NY, USA, 2001. [Google Scholar]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 5. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- Wright, M.N.; Ziegler, A. ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77, 77. [Google Scholar] [CrossRef] [Green Version]
- Rempała, G.A.; Seweryn, M.; Seweryn, M. Methods for diversity and overlap analysis in T-cell receptor populations. J. Math. Boil. 2012, 67, 1339–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, V.S.; Siemann, D.W. Have Clinical Trials Properly Assessed c-Met Inhibitors? Trends Cancer 2018, 4, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, F.; Giovannetti, E.; Saso, L.; Firuzi, O. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers. Crit. Rev. Clin. Lab. Sci. 2019, 56, 533–566. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 2017, 49, e307. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.M.; Song, Y.-J.; Lee, S.B.; Jeong, Y.; Kim, B.; Kim, G.W.; Kim, K.E.; Lee, J.M.; Cho, M.-Y.; Choi, J.; et al. A New Anti-c-Met Antibody Selected by a Mechanism-Based Dual-Screening Method: Therapeutic Potential in Cancer. Mol. Cells 2012, 34, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Gordon, M.; Tsai, F.; Papadopoulos, K.P.; Rasco, D.; Beeram, M.; Fu, S.; Janku, S.; Hynes, S.M.; Gundala, S.R.; et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Ahn, M.; Lee, S.; Kim, H.; Hong, M.; Millington, D.; Curtis, M.; Triantos, S.; Chaplan, S.; Haddish-Berhane, N.; et al. OA10.06 A First-in-Human Phase 1 Trial of the EGFR-cMET Bispecific Antibody JNJ-61186372 in Patients with Advanced Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2018, 13, S344–S345. [Google Scholar] [CrossRef] [Green Version]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab—The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 8. [Google Scholar] [CrossRef]
- Zahavi, D.; Aldeghaither, D.; O’Connell, A.; Weiner, L.M. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib. Ther. 2018, 1, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Boil. 2019, 29, 635–646. [Google Scholar] [CrossRef]
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef] [Green Version]
- Romero, D. Haematological cancer: Blinatumomab facilitates complete responses. Nat. Rev. Clin. Oncol. 2018, 15, 200. [Google Scholar]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The Structural Basis of Antibody-Antigen Recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Pena, E.; Barderas, R.; Bondi, M.C.M. Phage Display in the Quest for New Selective Recognition Elements for Biosensors. ACS Omega 2019, 4, 11569–11580. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2019, 12, 219–238. [Google Scholar] [CrossRef]
- Ravn, U.; Gueneau, F.; Baerlocher, L.; Osteras, M.; Desmurs, M.; Malinge, P.; Magistrelli, G.; Farinelli, L.; Kosco-Vilbois, M.H.; Fischer, N. By-passing in vitro screening--next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010, 38, e193. [Google Scholar] [CrossRef] [PubMed]
- Ravn, U.; Didelot, G.; Venet, S.; Ng, K.-T.; Gueneau, F.; Rousseau, F.; Calloud, S.; Kosco-Vilbois, M.; Fischer, N. Deep sequencing of phage display libraries to support antibody discovery. Methods 2013, 60, 99–110. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.; Kumar, S.; Naranjo, L.; Ferrara, F.; Kiss, C.; Bradbury, A. From deep sequencing to actual clones. Protein Eng. Des. Sel. 2014, 27, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.; Hu, S.; Wan, W.; Xu, M.; Du, R.; Zhao, W.; Gao, X.; Liu, J.; Liu, H.; Hong, J. Effective Optimization of Antibody Affinity by Phage Display Integrated with High-Throughput DNA Synthesis and Sequencing Technologies. PLOS ONE 2015, 10, e0129125. [Google Scholar] [CrossRef] [Green Version]
- Spiliotopoulos, A.; Owen, J.P.; Maddison, B.C.; Dreveny, I.; Rees, H.; Gough, K. Sensitive recovery of recombinant antibody clones after their in silico identification within NGS datasets. J. Immunol. Methods 2015, 420, 50–55. [Google Scholar] [CrossRef]
- Miyazaki, N.; Kiyose, N.; Akazawa, Y.; Takashima, M.; Hagihara, Y.; Inoue, N.; Matsuda, T.; Ogawa, R.; Inoue, S.; Ito, Y.; et al. Isolation and characterization of antigen-specific alpaca (Lama pacos) VHH antibodies by biopanning followed by high-throughput sequencing. J. Biochem. 2015, 158, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Lövgren, J.; Pursiheimo, J.-P.; Pyykkö, M.; Salmi, J.; Lamminmäki, U. Next generation sequencing of all variable loops of synthetic single framework scFv—Application in anti-HDL antibody selections. New Biotechnol. 2016, 33, 790–796. [Google Scholar] [CrossRef]
- Lowden, M.J.; Henry, K.A. Oxford nanopore sequencing enables rapid discovery of single-domain antibodies from phage display libraries. Biotech niques 2018, 65, 351–356. [Google Scholar] [CrossRef]
- Barreto, K.; Maruthachalam, B.V.; Hill, W.; Hogan, D.; Sutherland, A.R.; Kusalik, A.; Fonge, H.; DeCoteau, J.F.; Geyer, C. Next-generation sequencing-guided identification and reconstruction of antibody CDR combinations from phage selection outputs. Nucleic Acids Res. 2019, 47, e50. [Google Scholar] [CrossRef]
- Mei, M.; Zhou, Y.; Peng, W.; Yu, C.; Ma, L.; Zhang, G.; Yi, L. Application of modified yeast surface display technologies for non-Antibody protein engineering. Microbiol. Res. 2017, 196, 118–128. [Google Scholar] [CrossRef]
- Sha, S.; Agarabi, C.; Brorson, K.; Lee, N.-Y.; Yoon, S. N-Glycosylation Design and Control of Therapeutic Monoclonal Antibodies. Trends Biotechnol. 2016, 34, 835–846. [Google Scholar] [CrossRef]
- Sydow, J.F.; Lipsmeier, F.; Larraillet, V.; Hilger, M.; Mautz, B.; Mølhøj, M.; Kuentzer, J.; Klostermann, S.; Schoch, J.; Voelger, H.R.; et al. Structure-Based Prediction of Asparagine and Aspartate Degradation Sites in Antibody Variable Regions. PLOS ONE 2014, 9, e100736. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.S.; Li, L.; Broulidakis, M.P.; Luksha, N.G.; Burns, C.T.; Singh, S.K.; Kumar, S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. mAbs 2017, 9, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Obrezanova, O.; Arnell, A.; De La Cuesta, R.G.; E Berthelot, M.; Gallagher, T.R.; Zurdo, J.; Stallwood, Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. mAbs 2015, 7, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Sankar, K.; Hoi, K.H.; Yin, Y.; Ramachandran, P.; Andersen, N.; Hilderbrand, A.; McDonald, P.; Spiess, C.; Zhang, Q. Prediction of methionine oxidation risk in monoclonal antibodies using a machine learning method. mAbs 2018, 10, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Mason, D.M.; Friedensohn, S.; Weber, C.; Jordi, C.; Wagner, B.; Meng, S.; Gainza, P.; E Correia, B.; Reddy, S. Deep learning enables therapeutic antibody optimization in mammalian cells by deciphering high-dimensional protein sequence space. bioRxiv 2019. [Google Scholar]
- Liu, G.; Zeng, H.; Mueller, J.; Carter, B.; Wang, Z.; Schilz, J.; Horny, G.; Birnbaum, M.E.; Ewert, S.; Gifford, D.K. Antibody Complementarity Determining Region Design Using High-Capacity Machine Learning. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Bujotzek, A.; Lipsmeier, F.; Harris, S.F.; Benz, J.; Kuglstatter, A.; Georges, G. VH-VL orientation prediction for antibody humanization candidate selection: A case study. Platf. Dev. Expr. Purif. Stable Isot. Labeled Monoclon. Antibodies Escherichia Coli 2015, 8, 288–305. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clonotypes | Round 0 | Round 1 | Round 2 | Round 3 | Round 4 | Total |
|---|---|---|---|---|---|---|
| HCDR3 | 390,814 | 395,459 | 402,854 | 311,678 | 308,547 | 860,207 |
| LCDR3 | 272,317 | 253,899 | 250,630 | 187,314 | 117,239 | 443,292 |
| Clone ID | HCDR3 AA * Sequence | LCDR3 AA * Sequence |
|---|---|---|
| AR1 | GSGGVDSIDA | GSYDNTYAGI |
| AR2 | SADGYGWDTAGNMDA | GSIDSNYDGI |
| AR3 | TAGTCTTSCNAGAYIDA | GGYDGSSAA |
| AR4 | TTCSGSYGWCADSIDA | GAYDSSYIGI |
| AR5 | SADSCATCATYPSEIDT | GSFDSSYVGM |
| AR6 | SADSCATCATYPSEIDT | GSFDSSYVGM |
| AR7 | SADSCATCATYPSEIDT | GSIDSNYDGI |
| AR8 | SADSCATCATYPSEIDT | GSYDSSYVGL |
| AR9 | SADSCATCATYPSEIDT | GSYDSSYDGV |
| AR10 | SADSCATCATYPSEIDT | GSFDSSYTGI |
| AR11 | SADSCATCATYPSEIDT | GSIDSRYVGI |
| AR12 | SADSCATCATYPSEIDT | GSYDSSYVGYVGV |
| AR13 | SADSCATCATYPSEIDT | GSYDNTYAGI |
| AR14 | SADSCATCATYPSEIDT | GGYDSSSGA |
| AR15 | SADSCATCATYPSEIDT | GAYDSSYIGI |
| Drug Name | Targets | FDA Approval Status | Approved Year |
|---|---|---|---|
| Tivantinib | c-Met, microtubule | None | N.A.* |
| Foretinib | c-Met, VEGFR-2 * | None | N.A. |
| Cabozantinib | c-Met, VEGFR, Axl | Medullary thyroid cancer Advanced renal cell carcinoma Hepatocellular carcinoma | 2012 2016 2019 |
| Crizotinib | c-Met, ALK *, ROS1, RON * | ALK or ROS-1 positive NSCLC * | 2011 |
| Capmatinib | c-Met, EGFR *, ErbB-3 | None | N.A. |
| AMG337 | c-Met | None | N.A. |
| AZD6094 | c-Met | None | N.A. |
| BMS777607/ASLAN002 | c-Met, Axl, Tyro3, RON | None | N.A. |
| Glesatinib | c-Met, Axl | None | N.A. |
| Tepotinib | c-Met | None | N.A. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, D.K.; Lee, S.R.; Jung, Y.; Han, H.; Lee, H.K.; Han, J.; Kim, S.; Chae, J.; Ryu, T.; Chung, J. Machine Learning-Guided Prediction of Antigen-Reactive In Silico Clonotypes Based on Changes in Clonal Abundance through Bio-Panning. Biomolecules 2020, 10, 421. https://doi.org/10.3390/biom10030421
Yoo DK, Lee SR, Jung Y, Han H, Lee HK, Han J, Kim S, Chae J, Ryu T, Chung J. Machine Learning-Guided Prediction of Antigen-Reactive In Silico Clonotypes Based on Changes in Clonal Abundance through Bio-Panning. Biomolecules. 2020; 10(3):421. https://doi.org/10.3390/biom10030421
Chicago/Turabian StyleYoo, Duck Kyun, Seung Ryul Lee, Yushin Jung, Haejun Han, Hwa Kyoung Lee, Jerome Han, Soohyun Kim, Jisu Chae, Taehoon Ryu, and Junho Chung. 2020. "Machine Learning-Guided Prediction of Antigen-Reactive In Silico Clonotypes Based on Changes in Clonal Abundance through Bio-Panning" Biomolecules 10, no. 3: 421. https://doi.org/10.3390/biom10030421