Structure–Antioxidant–Antiproliferative Activity Relationships of Natural C7 and C7–C8 Hydroxylated Flavones and Flavanones

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
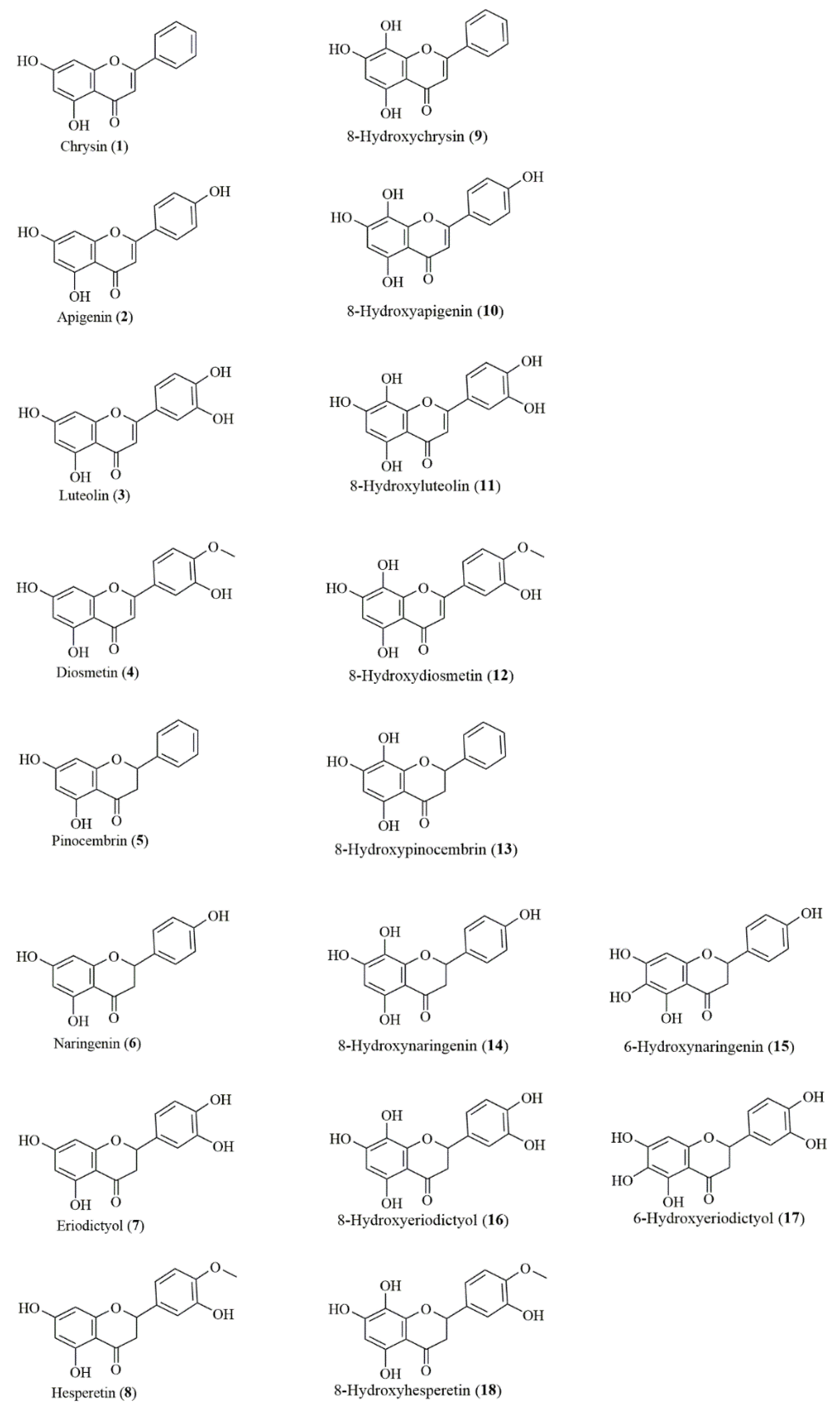
2.1. Compounds
2.2. Analytical Methods
2.3. Antioxidant Activity
2.3.1. ABTS Radical Cation Decolorization Assay
2.3.2. DPPH Free Radical Scavenging Activity Assay
2.3.3. FRAP Assay
2.4. Antiproliferative Activity In Vitro
2.4.1. MTT Assay
2.4.2. SRB Assay
2.5. Stability of Flavonoids under Antiproliferative Test Conditions
2.5.1. Stability of Flavonoids before Administration to the Cell Cultures
2.5.2. Stability of Flavonoids in the Cell Cultures
3. Results
3.1. Biotransformation
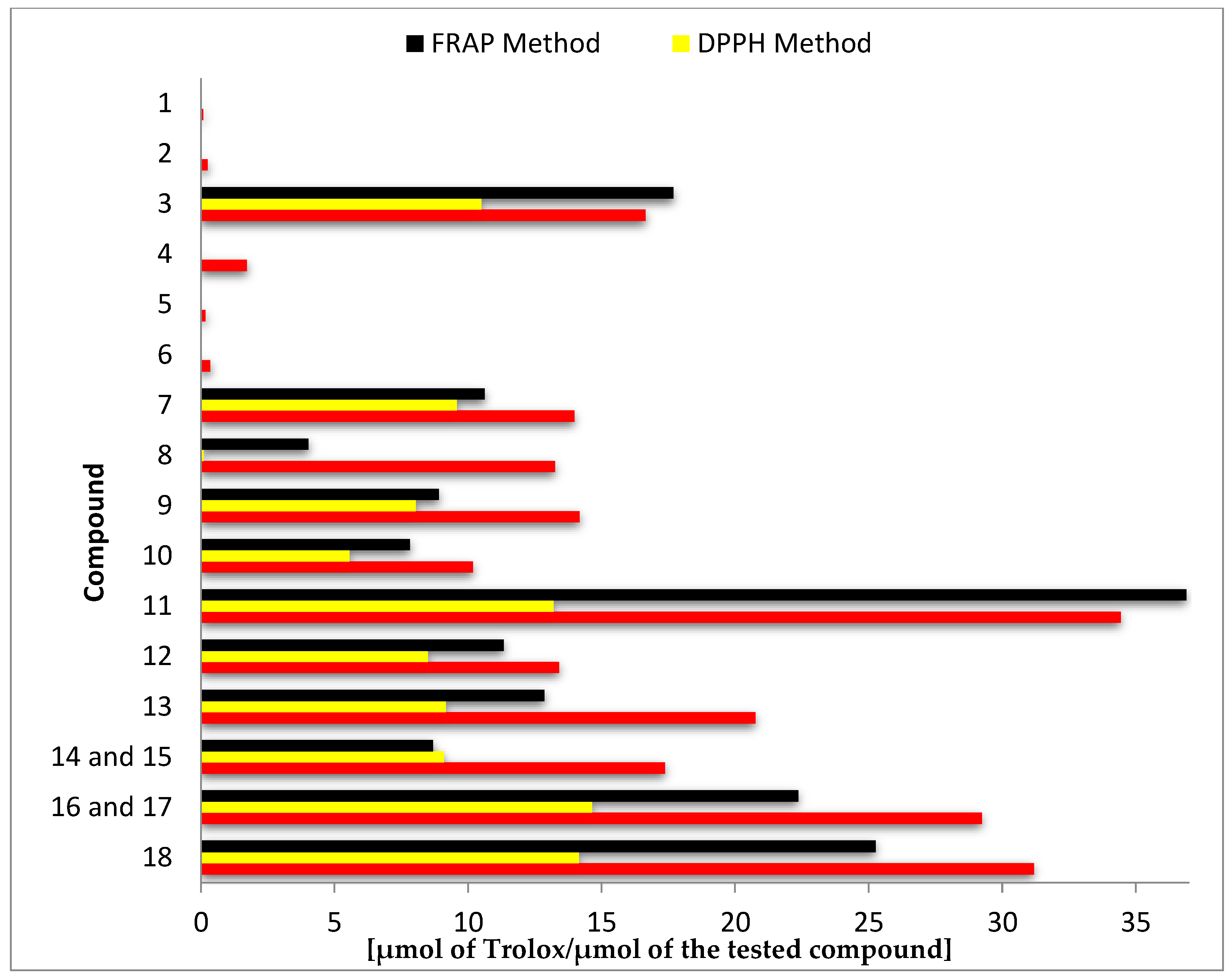
3.2. Antioxidant Activity
3.3. Antiproliferative Activity In Vitro
4. Discussion
4.1. Biotransformation
4.2. Antioxidant Activity
4.3. Antiproliferative Activity In Vitro
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Usman, H.; Abdulrahman, F.; Kaita, H.A.; Khan, I.R.; Tijjani, M.A. Flavonoids: The bioactive phytochemical agent—A review. Chem. Res. J. 2017, 2, 59–72. [Google Scholar]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Roleira, F.M.F.; Tavares-da-Silva, E.J.; Varela, C.L.; Costa, S.C.; Silva, T.; Garrido, J.; Borges, F. Plant derived and dietary phenolic antioxidants: Anticancer properties. Food Chem. 2015, 183, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R. Role of plant polyphenols in genomic stability. Mutat. Res. 2001, 475, 89–111. [Google Scholar] [CrossRef]
- Banjarnahor, S.D.S.; Artanti, N. Antioxidant properties of flavonoids. Med. J. Indones. 2014, 23, 239–244. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Lin, C.Z.; Zhu, C.C.; Hu, M.; Wu, A.Z.; Bairu, Z.D.; Kangsa, S.Q. Structure-activity relationships of antioxidant activity in vitro about flavonoids isolated from Pyrethrum tatsienense. J. Intercult. Ethnopharmacol. 2014, 3, 123–127. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Burda, S.; Oleszek, W. Antioxidant and antiradical activities of flavonoids. J. Agric. Food Chem. 2001, 49, 2774–2779. [Google Scholar] [CrossRef]
- Hyun, J.; Woo, Y.; Hwang, D.S.; Jo, G.; Eom, S.; Lee, Y.; Park, J.C.; Lim, Y. Relationships between structures of hydroxyflavones and their antioxidative effects. Bioorg. Med. Chem. Lett. 2010, 20, 5510–5513. [Google Scholar] [CrossRef] [PubMed]
- Grigalius, I.; Petrikaite, V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules 2017, 22, 2169. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Nair, M.G.; Strasburg, G.M. Structure-activity relationships for antioxidant activities of a series of flavonoids in a liposomal system. Free Radic. Biol. Med. 1998, 24, 1355–1363. [Google Scholar] [CrossRef]
- Chang, H.; Mi, M.; Ling, W.; Zhu, J.; Zhang, Q.; Wei, N.; Zhou, Y.; Tang, Y.; Yuan, J. Structurally related cytotoxic effects of flavonoids on human cancer cells in vitro. Arch. Pharm. Res. 2008, 31, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Li, Q.; Bi, K.S. Bioactive flavonoids in medicinal plants: Structure, activity and biological fate. AJPS 2018, 13, 12–23. [Google Scholar] [CrossRef]
- Roh, C. Biotransformation for multiple regio-selective hydroxylation of isoflavonoid. Biocatal. Agric. Biotechnol. 2013, 2, 403–408. [Google Scholar] [CrossRef]
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Regioselective O-glycosylation of flavonoids by fungi Beauveria bassiana, Absidia coerulea and Absidia glauca. Bioorg. Chem. 2019. [Google Scholar] [CrossRef]
- Sordon, S.; Madej, A.; Popłoński, J.; Bartmańska, A.; Tronina, T.; Brzezowska, E.; Juszczyk, P.; Huszcza, E. Regioselective ortho-hydroxylation of flavonoids by yeast. J. Agric. Food Chem. 2016, 64, 5525–5530. [Google Scholar] [CrossRef]
- Alam, M.N.; Bristi, N.J.; Rafiquzzaman, M. Review on in vivo and in vitro methods evaluation of antioxidant activity. Saudi Pharm. J. 2013, 21, 143–152. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying and improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Yen, G.C.; Chen, H.Y. Antioxidant activity of various tea extracts in relation to their antimutagenicity. J. Agric. Food Chem. 1995, 43, 27–32. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “Anti-oxidant Power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Venturella, P.; Bellino, A.; Marino, M.L. Three acylated flavone glycosides from Sideritis syriaca. Phytochemistry 1995, 38, 527–530. [Google Scholar] [CrossRef]
- Hu, B.H.; Liu, Y.L. Studies on the structures of new flavonoids from the root of Scutellaria amoena. Yao Xue Xue Bao Acta Pharm. Sin. 1989, 24, 200–206. [Google Scholar]
- Bilia, A.R.; Ciampi, L.; Mendez, J.; Morelli, I. Phytochemical investigation of Licania genus. Flavonoids Lic. Pyrifolia. Pharm. Acta Helv. 1996, 71, 199–204. [Google Scholar] [CrossRef]
- van Acker, S.A.; de Groot, M.J.; van den Berg, D.J.; Tromp, M.N.; Donné-Op den Kelder, G.; van der Vijgh, W.J.; Bast, A. A quantum chemical explanation of the antioxidant activity of flavonoids. Chem. Res. Toxicol. 1996, 9, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Yuan, S.; Cong, X.D. Biotransformation of puerarin into 3’-hydroxypuerarin by Trichoderma harzianum NJ01. Enzyme Microb. Technol. 2007, 40, 594–597. [Google Scholar] [CrossRef]
- Foti, M.; Piattelli, M.; Baratta, M.T.; Ruberto, G. Flavonoids, coumarins, and cinnamic acids as antioxidants in a micellar system. Structure—activity relationship. J. Agric. Food Chem. 1996, 44, 497–501. [Google Scholar] [CrossRef]
- Yokozawa, T.; Chen, C.P.; Dong, E.; Tanaka, T.; Nonaka, G.I.; Nishioka, I. Study on the inhibitory effect of tannins and flavonoids against the 1,1-diphenyl-2 picrylhydrazyl radical. Biochem. Pharm. 1998, 56, 213–222. [Google Scholar] [CrossRef]
- Long, L.H.; Hoi, A.; Halliwell, B. Instability of, and generation of hydrogen peroxide by, phenolic compounds in cell culture media. Arch. Biochem. Biophys. 2010, 501, 162. [Google Scholar] [CrossRef]
- Xiao, J.; Hoegger, P. Stability of dietary polyphenols under the cell culture conditions avoiding erroneous conclusions. J. Agric. Food Chem. 2015, 63, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J. Stability of dietary polyphenols: It's never too late to mend? Food Chem. Toxicol. 2018, 119, 3–5. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | IC50 ± SD (µM) | ||||||
|---|---|---|---|---|---|---|---|
| MV-4-11 | MCF7 | LoVo | LoVo/DX | DU 145 | MCF 10A | HLMEC | |
| 1 | 15.14 ± 1.99 | 74.97 ± 26.08 | 28.01 ± 8.17 | 12.98 ± 1.29 | ND | 26.98 ± 11.96 | 22.85 ± 2.02 |
| 2 | 8.18 ± 2.76 | 22.50 ± 1.29 | 13.80 ± 1.38 | 11.58 ± 1.35 | 28.86 ± 8.4 | 35.67 ± 11.93 | 14.06 ± 0.91 |
| 3 | 8.73 ± 2.23 | 25.50 ± 6.33 | 13.45 ± 0.65 | 13.69 ± 1.65 | 21.94 ± 6.11 | 33.12 ± 10.31 | 9.22 ± 0.64 |
| 4 | 8.39 ± 3.53 | 122.73 ± 37.36 | 14.99 ± 2.2 | 13.39 ± 3.02 | ND | ND | 14.39 ± 1.95 |
| 5 | 99.12 ± 34.11 | 147.63 ± 6.57 | 116.10 ± 10.63 | 67.04 ± 15.81 | 118.60 ± 22.45 | 125.19 ± 5.77 | 96.59 ± 7.23 |
| 6 | 146.19 ± 19.09 | 215.35 ± 32.28 | 120.84 ± 4.3 | 128.67 ± 9.74 | 241.65 ± 15.61 | 185.16 ± 23.19 | 113.98 ± 18.55 |
| 7 | ND | 150.70 ± 9.31 | 100.68 ± 10.86 | 99.22 ± 12.46 | 120.66 ± 18.45 | 146.61 ± 36.21 | 25.81 ± 6.71 |
| 8 | 140.47 ± 12.93 | 167.33 ± 13.12 | 103.81 ± 5.55 | 77.25 ± 19.39 | 214.54 ± 32.09 | 206.97 ± 23.42 | 98.29 ± 7.62 |
| 9 | ND | ND | ND | ND | ND | ND | 119.60 ± 2.24 |
| 10 | ND | ND | 235.29 ± 57.07 | ND | ND | ND | 122.21 ± 14.38 |
| 11 | ND | ND | 237.76 ± 35.36 | ND | ND | ND | 105.98 ± 1.72 |
| 12 | ND | ND | 223.65 ± 15.29 | ND | ND | ND | 107.70 ± 15.4 |
| 13 | ND | ND | ND | ND | ND | ND | 148.50 ± 13.38 |
| 14 and 15 * | ND | 121.80 ± 4.11 | ND | ND | ND | ND | 110.22 ± 0.18 |
| 16 and 17 ** | ND | 115.53 ± 7.1 | 179.49 ± 31.97 | ND | ND | 202.93 ± 93.02 | 90.68 ± 18.85 |
| 18 | ND | 135.45 ± 20.25 | ND | ND | ND | ND | 97.06 ± 5.15 |
| Cisplatin | 1.60 ± 0.33 | 7.50 ± 1.32 | 5.87 ± 1.72 | 3.23 ± 0.77 | 1.47 ± 0.12 | 10.63 ± 1.17 | 1.07 ± 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sordon, S.; Popłoński, J.; Milczarek, M.; Stachowicz, M.; Tronina, T.; Kucharska, A.Z.; Wietrzyk, J.; Huszcza, E. Structure–Antioxidant–Antiproliferative Activity Relationships of Natural C7 and C7–C8 Hydroxylated Flavones and Flavanones. Antioxidants 2019, 8, 210. https://doi.org/10.3390/antiox8070210
Sordon S, Popłoński J, Milczarek M, Stachowicz M, Tronina T, Kucharska AZ, Wietrzyk J, Huszcza E. Structure–Antioxidant–Antiproliferative Activity Relationships of Natural C7 and C7–C8 Hydroxylated Flavones and Flavanones. Antioxidants. 2019; 8(7):210. https://doi.org/10.3390/antiox8070210
Chicago/Turabian StyleSordon, Sandra, Jarosław Popłoński, Magdalena Milczarek, Martyna Stachowicz, Tomasz Tronina, Alicja Z. Kucharska, Joanna Wietrzyk, and Ewa Huszcza. 2019. "Structure–Antioxidant–Antiproliferative Activity Relationships of Natural C7 and C7–C8 Hydroxylated Flavones and Flavanones" Antioxidants 8, no. 7: 210. https://doi.org/10.3390/antiox8070210