Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations?


1
Predictive Medicine and Data Analytics Lab, Department of Signal Processing, Tampere University of Technology, 33720 Tampere, Finland
2
Institute of Biosciences and Medical Technology, 33520 Tampere, Finland
3
Department of Mechatronics and Biomedical Computer Science, University for Health Sciences, Medical Informatics and Technology (UMIT), 6060 Hall in Tyrol, Austria
4
College of Computer and Control Engineering, Nankai University, Tianjin 300071, China
5
Institute for Intelligent Production, Faculty for Management, University of Applied Sciences Upper Austria, Steyr Campus, 4400 Steyr, Austria
*
Author to whom correspondence should be addressed.
Mach. Learn. Knowl. Extr. 2019, 1(1), 138-148; https://doi.org/10.3390/make1010008
Submission received: 27 July 2018
/
Revised: 17 August 2018
/
Accepted: 20 August 2018
/
Published: 22 August 2018
(This article belongs to the Section Network)
Abstract
:Causal networks, e.g., gene regulatory networks (GRNs) inferred from gene expression data, contain a wealth of information but are defying simple, straightforward and low-budget experimental validations. In this paper, we elaborate on this problem and discuss distinctions between biological and clinical validations. As a result, validation differences for GRNs reflect known differences between basic biological and clinical research questions making the validations context specific. Hence, the meaning of biologically and clinically meaningful GRNs can be very different. For a concerted approach to a problem of this size, we suggest the establishment of the HUMAN GENE REGULATORY NETWORK PROJECT which provides the information required for biological and clinical validations alike.
1. Introduction
In recent years, there have been many new developments in machine learning and artificial intelligence [1,2,3]. One of these is in the causal inference of gene regulatory networks [4,5]. This approach is based on large-scale, high-throughput genomics data [6,7,8]. Gene regulatory networks (GRNs) provide a framework for representing condition and disease-specific interactions. For this reason, such networks are playing an increasingly important role in biology and medicine and translational applications derived from these [9,10,11,12,13,14,15,16,17,18]. Despite the importance of gene regulatory networks and their ample application opportunities, there are still many questions that are either unanswered or misunderstood that present serious obstacles to basic and translational research.
For instance, it is generally recognized that the validation of GRNs from high-throughput molecular data is a formidable challenge. Still, so far, it has received little attention from publications in the literature. Notable contributions to this topic were from Dougherty [19,20], who discussed the general theoretical aspects of this problem by assuming a reference network for comparison is available. However, particular biological measurements or the clinical utility of GRNs is not addressed. Another contribution was given by the paper of Walhout [21] that focuses on biological, but not clinical, aspects and the validation of individual interactions. Hence, it presents a reductionist approach without embedding the discussion in a wider holistic context, omitting, in this way, problems we encounter during this transition to genome-scale GRNs. Yet, other types of validations are discussed for simulated data, for which the true but artificial network is known by construction [22,23], skipping experimental validations entirely. This allows a standard statistical validation.
In this paper, we focus on a question that can be easily asked, but turns out to be difficult to answer practically. Specifically, we take a detailed look at the experimental validation of gene regulatory networks inferred from gene expression data. In order to clearly state the problem, we first discuss the size of such networks in terms of the number of inferred interactions and then describe the means for validating them experimentally. This leads us directly to the need to re-assess our approaches to deal with GRNs.
Before we start with our discussion, we would like to emphasize that here we consider gene regulatory networks to be causal networks [24,25,26]. That means whenever there is an interaction in a GRN between two genes, this is supposed to correspond to a biochemical interaction, e.g., a transcription regulation, a protein interaction, or a signaling event between gene products that can be experimentally validated. Hence, a gene regulatory network can, in principle, be fully experimentally validated. This is a strong requirement and goes far beyond the mere utility of gene regulatory networks, because it is possible to infer a network structure from gene expression data, and such a network may not correspond to a causal network in the above sense, but nevertheless, it can be a sensible biological auxiliary model [27]. In this paper, we make the assumption that gene regulatory networks are causal networks, and the following discussions are based on this and bound by this assumption.
2. How Large Are Gene Regulatory Networks?
In order to get a better appreciation for the validation problem of GRNs, we show, in Table 1, an overview of some inferred GRNs from the literature and the number of interactions they contain. It is clear that the size of a GRN depends on the number of genes in an organism and on the physiological and disease stages but also on the quality of the data. For this reason, the GRNs for E. coli and S. cerevisiae are smaller than those for human cancer networks. However, even for E. coli, we have a network with over 20,000 interactions. As an additional reference, we also added information about the size of transcriptional regulatory networks (TRN) and protein interaction networks (PIN) from S. cerevisiae and humans to Table 1 .
Although the true number of interactions in particular gene regulatory networks is largely unknown, from the comparison of the different types of networks in Table 1, it is not implausible to assume that the typical GRNs of humans contain more than 50,000 interactions. In the following discussion, the exact number is not crucial, but is only in the order of magnitude of interactions in a GRN.
3. Biological Validation of GRNs
Now, we turn to the important question of how the interactions, predicted by an inferred GRN, can be biologically validated. Specifically, by biologically validated, we mean experimental molecular biology techniques that provide us with direct measurements of the biochemical binding between gene products. In other words, these measurements provide us with information if a predicted interaction (X) is a true positive (TP) or a false positive (FP), e.g., TP53 binding to PFN1. In addition, depending on the experimental technique, biological validation will also obtain information about the types of interactions (see Table 2; second column).
Here, it is crucial to emphasize the important differences between the gene expression data used to infer a GRN and the validation experiments. Whereas gene expression data provide only indirect measurements of the potential binding or interactions between gene products, because they measure only the concentration of mRNAs, biological validation experiments need to be direct measurements of such binding events (see below). From an epistemological point of view, this means that a GRN is obtained in a data-driven manner, whereas the validation experiments are hypothesis-driven [33,34]; see also Figure 1.
Obviously, due to the inclusive nature of GRNs, the interactions between connected genes on such networks can occur at multiple levels. For the biological validation of such interactions, a number of basic laboratory techniques should help to decipher the biological processes involved. These include real-time polymerase chain reaction (PCR) and Western blot analyses following specific knockdown of nodal genes (using approaches such as short interfering RNAs) to verify if peripheral genes represent the transcriptional targets of GRN nodal genes. Publically available ChIP-seq information from sources such as ENCODE (Encyclopedia of DNA Elements) may help to confirm whether nodal genes have previously been localised to the genomic loci of putative target genes and may also provide valuable information about the likely site(s) of interaction within promoters or enhancer regions. ChIP analyses using DNA primers based on this information may then be used to confirm the localization to specific target loci as well to quantify the intensity of such interactions.
Co-immunoprecipitation (Co-IP) experiments are the logical first steps in the identification of protein–protein interactions. Other techniques, such as pull-down assays, Crosslinking Protein Interaction Analysis, Luminex and MALDI-TOF mass spectrometry, Label Transfer Protein Interaction Analysis, Far-Western Blot Analysis, and Bimolecular Fluorescence Complementation can also be used. Briefly, Crosslinking Protein Interaction Analysis enables transient protein interactions to be frozen in place while weakly interacting molecules can be localized in a complex that is stable enough for isolation and characterization [35]. Label Transfer Protein Interaction Analysis can be used to investigate the interface of an interacting protein of interest [36]. Far-Western Blot Analysis has been used to determine receptor–ligand interactions and screen libraries for interacting proteins [37]. Bimolecular fluorescence complementation (BiFC) enables the direct visualization of protein interactions in living cells [38]. Again, all of these techniques allow us to measure direct (biochemical) interactions.
We would like to note that there are many more experimental methods to detect molecular interactions. For instance, the INTACT database [39] contains information about interactions from dozens of different experimental methods, separated into nine subcategories: biochemical, biophysical, genetic interference, phenotype-based detection assay, imaging technique, post transcriptional interference, and protein complementation assay.
From this discussion, one can see that the validation of a GRN with 50,000 or more interactions is a new, large-scale experiment rather than a single small-scale experiment. To bring the order of magnitude of the number of interactions into a practical context, consider the following. Assuming (very optimistically) that, on average, a postdoc can validate 50 interactions in 3 years in the laboratory. Then, we need 1000 postdocs to validate 50,000 interactions in 3 years for one GRN.
If one places this discussion in a more general context, one sees that our problem is actually even bigger. Specifically, for humans, we distinguish between about 200 different cell types and over 1200 disorders [45]. That means there are at least 1400 different gene regulatory networks. However, it is likely that the number of different diseases will increase further in the near future when more and more genomic information becomes available and will be used to sub-classify disorders, as has been done, e.g., for breast cancer [46,47].
Specifically, in their seminal work, Perou and colleagues identified up to five molecular subtypes [48,49]. However, subsequently, it has been shown that a simple three-gene model can robustly classify breast tumors into four subtypes—including luminal A, luminal B, HER2-enriched, and basal-like tumors—calling into question the existence of the normal-like category [50]. More recently Curtis et al. jointly analyzed copy number alteration and gene expression profiles from the largest breast cancer dataset to date (2000 tumors; referred to as METABRIC) and discovered that they have distinct clinical outcomes [47]. These results indicate that as our molecular understanding of diseases improves, we are likely to refine the disease classification with increasing numbers of molecular subtypes.
Overall, this means, in order to biologically validate 1400 different GRNs, each with 50,000 interactions, 1000 postdocs would need over 4000 years. A heroic endeavor of this magnitude could well be named the HUMAN GENE REGULATORY NETWORK PROJECT in analogy to its (little) brother, the Human Genome Project [51].
3.1. Enhancing Experimental Assays by Perturbing the System
In the above discussion, we focused entirely on the experimental techniques, neglecting the experimental design for their application. However, to infer “causal interactions”, it is known that observational data are problematic, and for this reason, experimental or interventional data need to be generated. In the following, we briefly describe a recently developed validation framework that is based on gene perturbations (gene knockdown [KD] or overexpression) in human cancer cell lines [52].
Our framework is based on the generation of independent, single-gene KD experiments that target a collection of genes in a network or pathway. For these, gene expression data are measured before and after the knockdowns. Based on this data, the performance of a network inference methods was assessed as follows:
Given a GRN inferred from data,
- A single gene knockdown experiment was selected from the collection that included all replicates as a validation set.
- The genes whose expressions are significantly affected by the perturbation were identified.
- The capacity of the network model to predict which genes are affected by the perturbation by focusing on connections local to the gene being perturbed was assessed.
- Steps 1–3 were repeated to assess the predictive power of the network model until all perturbations had been tested.
By using this quantitative validation framework, we showed that the integration of priors can significantly improve the quality of the inferred GRN [52]. However, there were three limitations. The first is that we only targeted a small set of eight key genes from the RAS signaling pathway, but we assessed their effects on other genes from the entire genome. Second, the performed KDs were for single genes which prevented us from assessing the effects of multiple KDs performed simultaneously. The third limitation was that we performed the KD experiments on two colorectal cancer cell lines. However, by extending our validation framework to a larger number of cell lines and by using additional single and multiple gene KDs, we should be able to further improve the robustness of the inferred networks. There is, therefore, a direct need to scale this validation framework to genome-scale networks by using single- and multi-gene perturbations.
3.2. Existing Data Repositories
We do not want to miss mentioning that there are existing data repositories from which information about interaction levels is available. For instance, the STRING database [53] provides information about protein–protein interactions. Another good recourse is IntAct [39], which also provides mainly information about protein interactions from over 275 species. An example of a database that provides information about transcription regulation is TRRUST [54].
4. General Considerations about Validation
Before we discuss the clinical validation of GRNs in the next section, we insert a general discussion about validation. This will clarify the need for different validation settings.
There are two important points to emphasize, which we will discuss in the following text in more detail.
- A validation is necessary because the entities to be validated are generated by a statistical model corresponding to its predictions.
- The entities to be predicted are scientifically meaningful for particular research fields.
Both of the above points hold for any validation and are, for this reason, generic rather than specific to the topic of our paper. Furthermore, both points are related to each other, and point (2) is a corollary of (1). However, for reasons of clarity, we separated them. The first point emphasizes the existence of a statistical model that predicts the entities to be validated. In the above biological context, the statistical model is an inference algorithm that infers GRNs, and the predicted entities are the interactions between gene products. In the clinical context below, the statistical model could be a classifier that predicts the disease stages of patients related to their survival times. Both predictions, the interactions between gene products and the disease stage of patients, are scientifically meaningful in their respective research fields, biology and medicine, but not necessarily beyond. For instance, the knowledge about the presence of a specific interaction between two proteins is not necessarily useful for a medical doctor when making a decision regarding the treatment of a patient depending on her/his disease stage if no additional information about the effect of this interaction on the patient/phenotype is available. Hence, in the clinical context, the specific validation of ‘interactions between proteins’ does not directly provide meaningful validation for the ‘disease stage’. This implies that validations are always context specific making it necessary to distinguish between biological and clinical validations.
5. Clinical Validation of GRNs
The clinical validation of GRNs assesses the utility of such networks in a clinical or biomedical context. For instance, GRNs have been used as network biomarkers for prognostic and diagnostic purposes [55,56]. This means that the underlying GRN is quantitatively assessed via its statistical classification or, in general, prediction abilities that can be brought into contact with clinical outcome variables, e.g., disease grade, survival time, or therapeutic response. Of course, this is not limited to GRNs, but also holds for other network types that could serve as structural biomarkers, e.g., [10,57,58,59,60,61].
Abstractly, one can consider this on a different semantic level of which GRNs provide us with information. Whereas in molecular biology, the unit of interest and biological meaning is the interaction between gene products, in a clinical context, it is the prediction ability of a network biomarker, e.g., for diagnostic purposes. Overall, this means that for biological validation, a GRN serves as a biological interaction map, whereas for a clinical validation, it is a black box statistical model whose composing components are not directly under scrutiny. Although, it is plausible to assume that a high quality GRN gives rise to high quality predictions, it is not conclusive to claim that an incomplete GRN is not capable of high quality clinical predictions. This could mean that certain types of association networks without a direct causal meaning of its components can also lead to fruitful results in such applications.
For the latter point, it is important to emphasize that the biological quality of a GRN in a clinical context can have a severe impact on the further usage of such a GRN, e.g., in pharmacogenomics. Whereas for networks with good predictive properties but limited causal explanation capabilities, a downstream application for drug design seems unmotivated, a causal molecular GRN with good clinical predictive abilities could very well serve as a fruitful starting point for this. Hence, despite the fact that in clinical validation of a GRN, its biological validation is of secondary interest, for the subsequent use in pharmacogenomic problems for drug design, it is crucial.
Despite this apparent simplicity, GRNs have been scarcely validated in a clinical context compared to non GRN-based approaches. For instance, recently, 45 cancer genomic tests were reviewed (Table 1 in [62]). Importantly, despite the fact that nearly half of them were recommended for clinical use, not one was based on a GRN. This mere fact indicates that, first, the structural information of GRNs might be too overwhelming to be used ’as is’ in the clinical practices and second, there is certainly a lack of algorithms that could simplify the application of GRNs for diagnostic and prognostic purposes. Interestingly, Chang et al. [62] also showed that a genomic test takes almost 16 years to be accepted in clinical practice. In part, this provides further explanation as to why current clinical tests exclude GRNs, because accurate computational approaches for GRN inference appeared years after the first gene signatures for diagnostic and prognostic purposes were suggested, e.g., ref. [63].
As a measure to promote the usage of GRNs for clinical problems in translational research, we suggest the establishment of a public database to provide direct access to inferred GRNs. In addition, this database needs to be complemented with appropriate open access analysis tools in order to study relevant clinical questions. Overall, such an infrastructure would allow us to focus on the underlying clinical problems without the need to worry about the vast number of intricate details of every analysis step for inferring and analyzing GRNs before the actual clinical problem can be studied. This would free a tremendous amount of resources that could be directly dedicated to the investigation of the clinical validation of GRNs. Here, it is important to emphasize that the database as well as the analysis tools need to be open access, because this ensures the reproducibility of the obtained results by other groups. In a biomedical context, the involvement of humans and patients in achieving and demonstrating reproducibility is highly non trivial in general, and probably even more challenging for studies involving GRNs due to the additional layers of analysis complexity.
Higher-Level Networks
We want to finish by mentioning that there can be also higher-level networks beyond GRN that are useful for clinical applications. By higher-level networks, we mean networks where nodes correspond to a higher organizational unit than a gene. For instance, in [64], pathway networks (PN) were studied in which nodes correspond to pathways. It has been shown that these pathway networks can be useful for the in-silico identification of drug targets when utilizing the concept of pathway cross-talk inhibition (PCI), introduced by ref. [65]. However, the PNs are more abstract than GRNs because they represent summarized information of groups of genes and their interactions and hence, the question of their biological validation is different to GRNs.
6. Conclusions
In this paper, we discussed the question of whether it is possible to biologically validate inferred gene regulatory networks experimentally. As a result we found, in principle, a positive answer; however, practically, this appears to only be feasible in an iterative manner involving a heroic number of follow-up experiments rather than a single experiment and hence, requires a significant amount of time. Furthermore, this presupposes that GRNs need to be stored in public databases that allow easy access and storage of such follow-up results so that they can be seemingly integrated with each other.
We also discussed the utility of GRNs in clinical applications and argued that the clinical validation of a GRN is different from its biological validation, demonstrating by this that any validation step needs to be integrated into a practical context that defines the semantic meaning of a GRN. Whereas in molecular biology the semantic meaning is already on the level of molecular interactions, e.g., transcription regulations or protein bindings, in medicine and the clinical practice, it is beyond this level, at the level of clinical outcome, e.g., survival time. Furthermore, the current lack of applications of GRNs in the clinical practice similar to genomic tests for gene signatures points to an urgent developmental need in this direction. This would result in a shifting away from black box statistical models towards model-based statistical methods which are arguably more problem specific and additionally, have a meaningful biological interpretation.
We are of the opinion that due to the complexity of gene regulatory networks, and thus the many perspectives on their applications and interpretations, it is of the utmost importance to maintain a constant discussion about these aspects to further enhance our understanding. Due to the extend of this problem, we think this can only be tackled by establishing the HUMAN GENE REGULATORY NETWORK PROJECT with a meaningful organizational structure for the problem specific categories required for biological and clinical validation.
Author Contributions
F.E.-S. conceived the study. F.E.-S. and M.D. contributed to the writing of the manuscript and approved the final version.
Funding
M.D. thanks the Austrian Science Funds for supporting this work (project P30031).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ghahramani, Z. Probabilistic machine learning and artificial intelligence. Nature 2015, 521, 452. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436. [Google Scholar] [CrossRef] [PubMed]
- Witten, D.M.; Friedman, J.H.; Simon, N. New Insights and Faster Computations for the Graphical Lasso. J. Comput. Graph. Stat. 2011, 20, 892–900. [Google Scholar] [CrossRef]
- De Matos Simoes, R.; Emmert-Streib, F. Bagging statistical network inference from large-scale gene expression data. PLoS ONE 2012, 7, e33624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opgen-Rhein, R.; Strimmer, K. Learning causal networks from systems biology time course data: An effective model selection procedure for the vector autoregressive process. BMC Bioinform. 2007, 8, S3. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R. Next-Generation DNA Sequencing Methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, J.; Wyrwicz, L.S. Integrating genomics, proteomics and bioinformatics in translational studies of molecular medicine. Expert Rev. Mol. Diagn. 2009, 9, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Oltvai, Z.N. Network Biology: Understanding the Cell’s Functional Organization. Nat. Rev. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Lee, E.; Liu, Y.T.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Dehmer, M.; Jurisica, I. Knowledge discovery and interactive data mining in bioinformatics-state-of-the-art, future challenges and research directions. BMC Bioinform. 2014, 15, I1. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Hidalgo, D.; Domínguez-Cejudo, M.A.; Amore, G.; Brockmann, A.; Lemos, M.C.; Córdoba, A.; Casares, F. A Hh-driven gene network controls specification, pattern and size of the Drosophila simple eyes. Development 2012, 082172. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Hidalgo, D.; Zurita, A.C.; Fernández, M.C.L. Complex networks evolutionary dynamics using genetic algorithms. Int. J. Bifurc. Chaos 2012, 22, 1250156. [Google Scholar] [CrossRef]
- Aguilar-Hidalgo, D.; Lemos, M.C.; Córdoba, A. Evolutionary dynamics in gene networks and inference algorithms. Computation 2015, 3, 99–113. [Google Scholar] [CrossRef]
- Ideker, T.; Krogan, N.J. Differential network biology. Mol. Syst. Biol. 2012, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulatory network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H. Genome network medicine: Innovation to overcome huge challenges in cancer therapy. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Yachie-Kinoshita, A.; Onishi, K.; Ostblom, J.; Langley, M.A.; Posfai, E.; Rossant, J.; Zandstra, P.W. Modeling signaling-dependent pluripotency with Boolean logic to predict cell fate transitions. Mol. Syst. Biol. 2018, 14, e7952. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, E.R. Validation of inference procedures for gene regulatory networks. Curr. Genom. 2007, 8, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, E.R. Validation of gene regulatory networks: Scientific and inferential. Brief. Bioinform. 2011, 12, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A. What does biologically meaningful mean? A perspective on gene regulatory network validation. Genome Biol. 2011, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Schaffter, T.; Marbach, D.; Floreano, D. GeneNetWeaver: In silico benchmark generation and performance profiling of network inference methods. Bioinformatics 2011, 27, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Van den Bulcke, T.; Van Leemput, K.; Naudts, B.; van Remortel, P.; Ma, H.; Verschoren, A.; De Moor, B.; Marchal, K. SynTReN: A generator of synthetic gene expression data for design and analysis of structure learning algorithms. BMC Bioinform. 2006, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmert-Streib, F.; Glazko, G.; Altay, G.; de Matos Simoes, R. Statistical inference and reverse engineering of gene regulatory networks from observational expression data. Front. Genet. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, J. Causality: Models, Reasoning, and Inference; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2000. [Google Scholar]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction, and Search; Springer: New York, NY, USA, 1993. [Google Scholar]
- Emmert-Streib, F. The Chronic Fatigue Syndrome: A Comparative Pathway Analysis. J. Comput. Biol. 2007, 14, 961–972. [Google Scholar] [CrossRef] [PubMed]
- De Matos Simoes, R.; Dehmer, M.; Emmert-Streib, F. Interfacing cellular networks of S. cerevisiae and E. coli: Connecting dynamic and genetic information. BMC Genom. 2013, 14, 324. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse Engineering of Regulatory Networks in Human B Cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef] [PubMed]
- De Matos Simoes, R.; Dehmer, M.; Emmert-Streib, F. B-cell lymphoma gene regulatory networks: Biological consistency among inference methods. Front. Genet. 2013, 4, 281. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; de Matos Simoes, R.; Mullan, P.; Haibe-Kains, B.; Dehmer, M. The gene regulatory network for breast cancer: Integrated regulatory landscape of cancer hallmarks. Front. Genet. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; de Matos Simoes, R.; Glazko, G.; McDade, S.; Haibe-Kains, B.; Holzinger, A.; Dehmer, M.; Campbell, F. Functional and genetic analysis of the colon cancer network. BMC Bioinform. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey-Smith, P. Theory and Reality: An Introduction to the Philosophy of Science; Science and Its Conceptual Foundations Series; University of Chicago Press: Chicago, IL, USA, 2003. [Google Scholar]
- Kell, D.B.; Oliver, S.G. Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. BioEssays 2004, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Bruce, J. Chemical Cross-Linking for Protein? Protein Interaction Studies. In Mass Spectrometry of Proteins and Peptides; Methods in Molecular Biology; Lipton, M., Pasa-Tolic, L., Eds.; Humana Press: New York, NY, USA, 2009; Volume 492, pp. 283–293. [Google Scholar]
- Liu, Q.; Dinu, I.; Adewale, A.; Potter, J.; Yasui, Y. Comparative evaluation of gene-set analysis methods. BMC Bioinform. 2007, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Roth, S.Y. Current Protocols in Molecular Biology; Chapter Identification of Protein Interactions by Far Western Analysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2009, 38, D525–D531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.; Lieb, J. ChIP-chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 2004, 83, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B.L.; Hu, G.; Zhao, K. ChIP-Seq: Technical considerations for obtaining high-quality data. Nat. Immunol. 2011, 12, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Seeholzer, S.; Ohh, M. Identification of associated proteins by coimmunoprecipitation. In Molecular Cloning—A Laboratory Manual; Sambrook, J., Russell, D.W., Eds.; CSHL Press: New York, NY, USA, 2002; Volume 3, pp. 18–60. [Google Scholar]
- Joung, J.K.; Ramm, E.I.; Pabo, C.O. A bacterial two-hybrid selection system for studying protein–DNA and protein–protein interactions. Proc. Natl. Acad. Sci. USA 2000, 97, 7382–7387. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J. Vis. Exp. 2011, 50, 2638. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; McKusick, V.A. Online Mendelian inheritance in man (OMIM). Hum. Mutat. 2000, 15, 57–61. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.; He, X.; Perou, C. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Haibe-Kains, B.; Desmedt, C.; Loi, S.; Culhane, A.C.; Bontempi, G.; Quackenbush, J.; Sotiriou, C. A Three-Gene Model to Robustly Identify Breast Cancer Molecular Subtypes. J. Natl. Cancer Inst. 2012, 104, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Olsen, C.; Fleming, K.; Prendergast, N.; Rubio, R.; Emmert-Streib, F.; Bontempi, G.; Haibe-Kains, B.; Quackenbush, J. Inference and validation of predictive gene networks from biomedical literature and gene expression data. Genomics 2014, 103, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2017, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, R.; Efroni, S. Gene expression and network-based analysis reveals a novel role for hsa-miR-9 and drug control over the p38 network in glioblastoma multiforme progression. Genome Med. 2011, 3, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehmer, M.; Mueller, L.; Emmert-Streib, F. Quantitative Network Measures as Biomarkers for Classifying Prostate Cancer Disease States: A Systems Approach to Diagnostic Biomarkers. PLoS ONE 2013, 8, e77602. [Google Scholar] [CrossRef] [PubMed]
- Cun, Y.; Frohlich, H. Network and Data Integration for Biomarker Signature Discovery via Network Smoothed T-Statistics. PLoS ONE 2013, 8, e73074. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, H. Including network knowledge into Cox regression models for biomarker signature discovery. Biom. J. 2014, 56, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, G.; Chen, L. Prediction and early diagnosis of complex diseases by edge-network. Bioinformatics 2014, 30, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, W.; Yu, X.; Liu, X.; Li, M.; Liu, R.; Chen, L. Edge biomarkers for classification and prediction of phenotypes. Sci. China Life Sci. 2014, 57, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, T.; Sun, S.y.; Wang, Y.; Zhu, H.; Chen, L. Network biomarkers reveal dysfunctional gene regulations during disease progression. FEBS J. 2013, 280, 5682–5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Q.; Tingle, S.R.; Filipski, K.K.; Khoury, M.J.; Lam, T.K.; Schully, S.D.; Ioannidis, J.P. An overview of recommendations and translational milestones for genomic tests in cancer. Genet. Med. 2014, 17, 431. [Google Scholar] [CrossRef] [PubMed]
- Van De Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. In silico identification of drug target pathways in breast cancer subtypes using pathway cross-talk inhibition. J. Transl. Med. 2018, 16, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, S.; Igea, A.; Arroyo, R.; Alcalde, V.; Canovas, B.; Orozco, M.; Nebreda, A.R.; Aloy, P. Quantification of pathway cross-talk reveals novel synergistic drug combinations for breast cancer. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
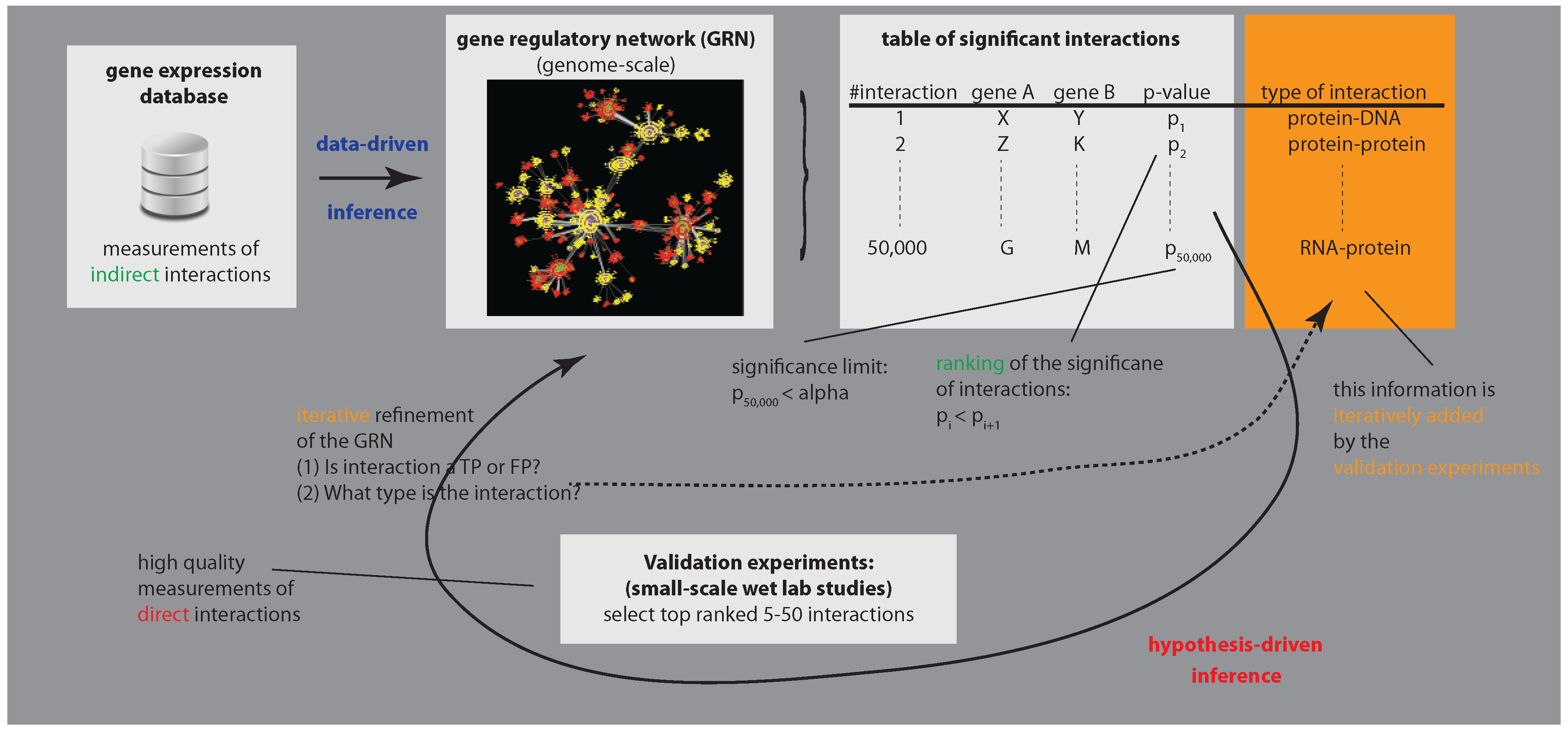
Figure 1.
The figure illustrates the process of biological validation of an inferred gene regulatory network. Due to the size of a GRN, here, arbitrarily but realistically chosen to contain 50,000 interactions, single follow-up wet lab experiments tested only a tiny fraction of all statistically significant interactions. For this reason, the whole network could only iteratively be validated.
Figure 1.
The figure illustrates the process of biological validation of an inferred gene regulatory network. Due to the size of a GRN, here, arbitrarily but realistically chosen to contain 50,000 interactions, single follow-up wet lab experiments tested only a tiny fraction of all statistically significant interactions. For this reason, the whole network could only iteratively be validated.


{kind=link}
Table 1.
Overview of gene regulatory networks (GRN), transcriptional regulatory networks (TRN), and protein interaction networks (PIN) for various organisms and phenotype states.
Table 1.
Overview of gene regulatory networks (GRN), transcriptional regulatory networks (TRN), and protein interaction networks (PIN) for various organisms and phenotype states.
| Organism (Network Type) | # of Interactions | References |
|---|---|---|
| E. coli (GRN) | 21,820 | [28] |
| S. cerevisiae (GRN) | 27,493 | [4] |
| B-cell lymphoma (GRN) | 129,000; 57,905 | [29,30] |
| Breast cancer (GRN) | 180,171 | [31] |
| Colon cancer (GRN) | 135,194 | [32] |
| S. cerevisiae (TRN) | 12,873 | [28] |
| S. cerevisiae (PIN) | 112,562 | [28] |
| Human (TRN) | 51,871 | [30] |
| Human (PIN) | 185,433 | [30] |
Table 2.
Overview of experimental techniques to biologically validate molecular interactions by measuring direct interactions.
Table 2.
Overview of experimental techniques to biologically validate molecular interactions by measuring direct interactions.
| Experimental Technique | Type of Interaction | Reference |
|---|---|---|
| ChIP-chip/ ChIP-seq | protein–DNA interaction | [40,41] |
| Co-immunoprecipitation | protein–protein interaction | [42] |
| Yeast two-hybrid | protein–protein interaction | [43] |
| Crosslinking Protein Interaction Analysis | protein–protein interaction | [35] |
| Label Transfer Chemistry | protein–protein interaction | [36] |
| Far-Western Blot Analysis | protein–protein interaction | [37] |
| BiFCassays | protein–protein interaction | [38] |
| iCLIP | protein–RNA interaction | [44] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Emmert-Streib, F.; Dehmer, M. Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations? Mach. Learn. Knowl. Extr. 2019, 1, 138-148. https://doi.org/10.3390/make1010008
AMA Style
Emmert-Streib F, Dehmer M. Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations? Machine Learning and Knowledge Extraction. 2019; 1(1):138-148. https://doi.org/10.3390/make1010008
Chicago/Turabian StyleEmmert-Streib, Frank, and Matthias Dehmer. 2019. "Inference of Genome-Scale Gene Regulatory Networks: Are There Differences in Biological and Clinical Validations?" Machine Learning and Knowledge Extraction 1, no. 1: 138-148. https://doi.org/10.3390/make1010008