Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update

1
Registrar (Otolaryngology, Head and Neck Surgery), Melbourne 3001, Australia
2
Department of Surgery, Monash University, Melbourne 3168, Australia
3
ENT—Otoneurology Unit, The Alfred, Melbourne 3004, Australia
*
Author to whom correspondence should be addressed.
J. Otorhinolaryngol. Hear. Balance Med. 2018, 1(1), 4; https://doi.org/10.3390/ohbm1010004
Submission received: 8 October 2017
/
Revised: 26 November 2017
/
Accepted: 3 December 2017
/
Published: 8 December 2017
Abstract
:Head and neck paraganglioma (HNPGL) are rare, highly vascular; typically slow growing and mostly benign neoplasms arising from paraganglia cells. HNPGL cause morbidity via mass effect on adjacent structures (particularly the cranial nerves), invasion of the skull base and, rarely, catecholamine secretion with associated systemic effects. The last decade has seen significant progress in the understanding of HNPGL genetics, with pertinent implications for diagnostic assessment and management of patients and their relatives. The implicated genes code for three of the five subunits of mitochondrial enzyme succinate dehydrogenase (SDH); recent literature reports that approximately one third of all HNPGL are associated with SDH mutations—a prevalence significantly greater than traditionally thought. There are distinct phenotypical syndromes associated with mutations in each individual SDH subunit (SDHD, SDHB, SDHC, and SDHAF2). This article focuses on the clinical features of HNPGL, the implications of HNPGL genetics, and the current evidence relating to optimal identification, investigation, and management options in HNPGL, which are supported by reference to a personal series of 60 cases. HNPGL require a systematic and thorough assessment to appropriately guide management decisions, and a suggested algorithm is presented in this article. Recent developments are particularly pertinent to surgeons of multiple disciplines, including otolaryngology, neurosurgery, vascular, and general surgery.
1. Introduction
Paraganglia are aggregations of neuroendocrine tissue with an embryonic migration pathway from skull base to pelvis. They are chemoreceptors influencing respiratory drive and catecholamine secretion [1,2,3,4,5,6].
Head and neck paraganglioma (HNPGL) or ‘glomus tumours’ are rare, highly vascular, typically slow growing, and mostly benign neoplasms arising from paraganglia cells; malignancy occurs in less than 5% of cases [2]. The majority of HNPGL are located and classified on the basis of their site of origin, namely: the jugular bulb (glomus jugulare), Jacobsen’s nerve plexus (glomus tympanicum), vagus nerve (glomus vagale), and carotid bifurcation (carotid body tumours), the last of which are the most common [4,6]. Other sites of HNPGL described include the paranasal sinuses, nasal cavity, parotid gland, orbit, larynx, thyroid and parathyroid glands, cervical sympathetic chain and oesophagus; but these are extremely uncommon [7,8]. HNPGL cause morbidity via mass effects on adjacent structures (cranial nerves, major blood vessels, invasion of the skull base and ear structures when within the temporal bone) [3,4,9,10,11]. HNPGL differ from paraganglioma of the abdomen, thorax, and adrenals as they very rarely secrete catecholamines, and are unique in their association with mutations in the mitochondrial enzyme succinate dehydrogenase [12]. Recent advances in the knowledge of HNPGL pathogenesis at a genetic and molecular level have greatly added to our understanding of biology of these lesions, with important implications for appropriate patient investigation and management—whether active treatment or observation. Genetic counselling must be offered to familial PGL patients and their families.
2. Aims
With reference to the HNPGL series of the senior author and review of the current literature, the aim of this paper is to highlight and discuss the pertinent features of HNPGL, with particular emphasis on the inherited forms, outlining potential implications of a genetically-acquired HNPGL.
We aim to outline an assessment framework for these tumours, which can be considered and followed by surgeons/clinicians of multiple subspecialties: including genetic analysis, investigation of excess catecholamine secretion, and imaging protocols in those at risk of having a heritable HNPGL.
3. Materials and Methods
All cases of confirmed HNPGL within the clinical practice over 26 years of a single surgeon (Vincent Cousins, Monash University, Melbourne, Australia) were analysed via direct access to patient medical records, with consent from identifiable patients. Alfred Health Human Research and Ethics Committee Approval (project number 412/11) was granted for this study.
De-identified data regarding epidemiological characteristics, SDH mutation genetics (where available), imaging, hormone secretion status, histology, management and on-going surveillance were entered into a Microsoft Excel© (Version 12.3.6, Santa Rosa, CA, USA) spreadsheet for analysis.
4. Results
A total of 60 cases of radiologically-diagnosed and mostly histologically-confirmed (91% histologically confirmed, five patients within the series were managed conservatively) HNPGL cases were identified and included in the study.
4.1. Epidemiological Factors
The average age of HNPGL presentation, including all anatomical locations was 50.5 years. The differences in average age of presentation and sex differences between anatomical locations of HNPGL are presented in Table 1.
4.2. Preoperative Catecholamine Screening
Ninety-five percent of patients in the series underwent urinary catecholamine screening at presentation. Two patients (3%) were found to have functional HNPGL with elevated levels of urinary catecholamines; one of these patients had elevated levels of dopamine, and one epinephrine. Additionally, 10 (17%) patients within the series had pre-operative serum metanephrine and normetanephine testing, with a further two patients were identified as having increased levels—neither of these patients had elevated urinary catecholamine levels detected on screening.
4.3. Genetic Testing
In this series, the SDH gene status was documented in 30 (50%) of cases. Genetic testing via immunohistochemistry techniques has been available at the interstate laboratory and utilised since 2000, 2001, and 2002 for SDHD, B, and C, respectively. Patients not having been tested within the series were assessed and treated prior to the routine availability of SDH mutation identification. Of the tested patients seven (23%) were found to have mutations in the SDH gene complex. The characteristics of these patients are summarised in Table 2.
The average age of HNPGL diagnosis in patients with SDH mutations was 34 years, in contrast to 50 years in patients whose SDH mutation screen was negative.
4.4. Management
The management undertaken in this series of patients (including recurrent tumours) is summarized in Table 3. Detailed analyses of treatment and outcomes are beyond the intention and scope of this article.
5. Discussion
HNPGL are rare and represent 0.03% of human tumours and 0.6% of head and neck tumours, having an estimated incidence of one in 30,000 [13,14]. Malignancy in HNPGL is extremely rare occurring in 1–3% of lesions [15]; there were no patients with malignant progression in our series.
It is reported that Carotid Body Tumours (CBT) are the most common HNPGL (60%). Glomus Tympanicum (GT) and Glomus Jugulare (GJ) are the next most common, each representing approximately 15% [16]. Within this cohort GT was the most common tumour. The over-representation of GT and GJ in the series is indicative of the senior author’s predominant area of practice in neuro-otology. The series is also limited in its sample size, and lack of SDH mutation testing for half of the patients (where the patients’ diagnoses and treatment pre-dates the availability of the test).
CBT reportedly show a male preponderance of 2:1, this trend was observed within this series. It is suggested that tympanicum, jugulare, and vagale lesions are more common in females, with incidences of up to six times higher [17,18]. Both GT and GJ were observed to be three times more common in females in this series. The mechanisms for sex distributions are not yet published.
Presenting symptoms of HNPGL are related to anatomical location and adjacent structures affected by the tumour. Pulsatile tinnitus was the most common presenting symptom for both GT and GJ here, being present in 80% of patients. Classically, GT lesions are suspected in the presence of pulsatile tinnitus and conductive hearing loss confirmed on audiometry. On physical examination these lesions are usually visualised through the tympanic membrane as a vascular tympanic mass on the promontory [10,19]. Glomus jugulare tumours are centred on the jugular bulb and may cause palsies of the ninth to eleventh cranial nerves. They may extend through the facial recess and mastoid air cells resulting in facial nerve palsy. In this series 27% of GJ had lower cranial nerve palsies at the time of diagnosis. These tumours may also invade the middle ear causing hearing loss and may be visualised as a vascular mass behind the lower segment of the tympanic membrane. The vascular nature of these tumours sees the masses blanch with air compression using a pneumatic otoscope; when this is noted clinically, it is referred to as Brown’s sign. The lesion can also present as a mass in the external auditory canal, occasionally with bleeding. Bruits are heard over the ear or mastoid in larger masses. Skull base tumours may also invade the posterior cranial fossa [2,10,17,19].
The GV and CBT tumours in this series presented as non-tender neck lumps. This is the normal presentation described for Glomus Vagale (GV) with occasional lower cranial nerve involvement [2,10,20]. CBT commonly present as painless, pulsatile swellings of the neck that are localised to the anterior border of sternocleidomastoid. The mass is classically freer to move in a horizontal rather than vertical axis (Fontaine’s Sign). Findings of a carotid bruit and/or pulsation on palpation strengthen the clinical suspicion of PGL as the diagnosis [2,19,21].
It is essential to note that 2–3% of HNPGL secrete clinically-significant levels of catecholamines, with signs such as labile hypertension, headache, malar flush, palpitations, and intractable tachycardia—these effects of catecholamine secretion can complicate anaesthesia (hypertensive crisis) and have end-organ morbidity (renal failure, cardiac arrhythmia). Paragangliomas (PGL) and phaeochromocytoma (PCC) have an abundance of enzyme catechol-O-methyltransferase (COMT), which is responsible for converting norepinephrine to normetanephrine and epinephrine to metanephrine. Screening for plasma metanephrines and normetanephrines will identify a secretory paraganglioma with 99% sensitivity [22]. In the catecholamine synthesis pathway, dopamine is converted to norepinephrine by enzyme dopamine beta-hydroxylase (DBH); elevated dopamine levels are specific for extra-adrenal paraganglioma, and may be secreted by some HNPGL [23]. These tests are not available at all centres and, alternatively, urinary catecholamine studies offer similar sensitivity [24]. Elevated levels of catecholamines should be optimally suppressed preoperatively to minimize anaesthetic risk, particularly from severe hypertension. Patients with evidence of catecholamine hypersecretion require radiological investigation for concurrent PGL and/or PCC that may be the source of hormone secretion prior to medical optimisation and any planned surgical management [10,17,25].
The last decade has seen significant progress in the understanding of HNPGL genetics, with pertinent implications for investigation and management of patients and their relatives [26]. Mutations of the mitochondrial enzyme succinate dehydrogenase (SDH): SDHD (11q23), SDHB (1p36), SDHC (1q23), SDHA (5p15), and SDHAF2 (11q12) and are associated with the development of familial paraganglioma and are reported to account for up to one third of HNPGL [27]. Other genetic syndromes are associated with HNPGL, but with lower frequency including von-Hippel-Lindau (VHL, 4–10%), RET proto-oncogene syndromes (MEN 2a and 2b, 1–5%), and NF1 (1–5%) [28,29] SDH mutations are inherited in an autosomal dominant fashion, with complex genomic imprinting differences and variable penetrance between SDH sub-types [8,30] SDHD mutations have high tumour penetrance (85% by age 50) and can be inherited via maternal or paternal genes. However, tumours will not develop if maternal in origin, where the allele is silenced in the epigenetic “parent-of-origin” type phenomenon (so-called maternal imprinting). Therefore, tumours will only develop in SDHD mutations inherited from the father [11,31], the rare SDHAF2 mutations also exhibit this pattern [29]. SDHB mutations by contrast have lower tumour penetrance, with approximately 45% developing tumours by age 50 and without genomic imprinting [32]. The inherited forms of HNPGL occur at a younger age [11,33,34], this trend was replicated within this series. In addition, 23% of HNPGL patients tested had an SDH complex mutation, in keeping with the rates reported in the recent literature.
Each of the SDH mutations described to date has specific phenotypic characteristics, which have been grouped and described as individual paraganglioma “syndromes” (PGL1–5). These syndromes are not only associated with HNPGL, but also the development of phaeochromocytoma, gastrointestinal tumours, renal cancers and, rarely, pituitary tumours [29].
5.1. PGL-1
5.2. PGL-2
Paraganglioma syndrome 2 (PGL-2) is linked to mutations in the SDHAF2 gene on 11q12. There are now four reported families with this mutation; these patients have a propensity for multiple head and neck lesions, however, no phaeochromocytoma or extra-adrenal paragangliomas have been described, and no malignancies have been reported [31,35].
5.3. PGL-3
5.4. PGL-4
SDHB mutations on 1p36 are associated with the PGL4 syndrome. These patients have a very high risk of concurrent phaeochromocytoma (approximately 20%) or paragangliomas arising outside the head and neck [11,31,38]. HNPGL in PGL4 are predisposed to malignant transformation (reported up to one-third), and associated with more aggressive lesions with higher incidence of recurrence post-resection [14,35,39]. SDHB mutations are also associated with higher rates of breast cancer, papillary thyroid carcinoma, and neuroblastoma [40]. No malignant lesions were found in our series.
5.5. PGL-5
Mutations in the SDHA gene are very rare (account for less than 5% of all paragangliomas) with very low penetrance. Mutations in SDHA may be associated with gastrointestinal tumours and pituitary adenomas [29,41].
Large datasets have reported the SDHD mutation as most common, followed by SDHB. The financial cost for the three gene tests is in excess of $2500 AUD. It is, therefore, recommended to selectively screen for SDH mutations commencing with SDHD and progressing to SDHB if the preceding screen is negative. HNPGL can be a rare finding in other syndromes including Multiple Endocrine Neoplasia Type 2A and 2B (mutations in RET gene), some sub types of von Hippel-Lindau disease and Neurofibromatosis Type I. In the absence of SDHD or SDHB gene mutations the genetic markers for these syndromes, along with SDHC may also be screened if clinical suspicion remains high [2,33,42].
The current consensus is that all HNPGL/pheochromocytoma patients with confirmed SDH mutations should have genetic counselling offered to all first-degree relatives to assess their risk of inheriting PGL and/or PCC tumours [33,43]. Early diagnosis of asymptomatic tumours provides the opportunity of reducing tumour and treatment morbidity leading to better patient outcomes. Large studies have justified this recommendation with 13% of asymptomatic relatives screened being found to have unidentified HNPGL or PCC [32].
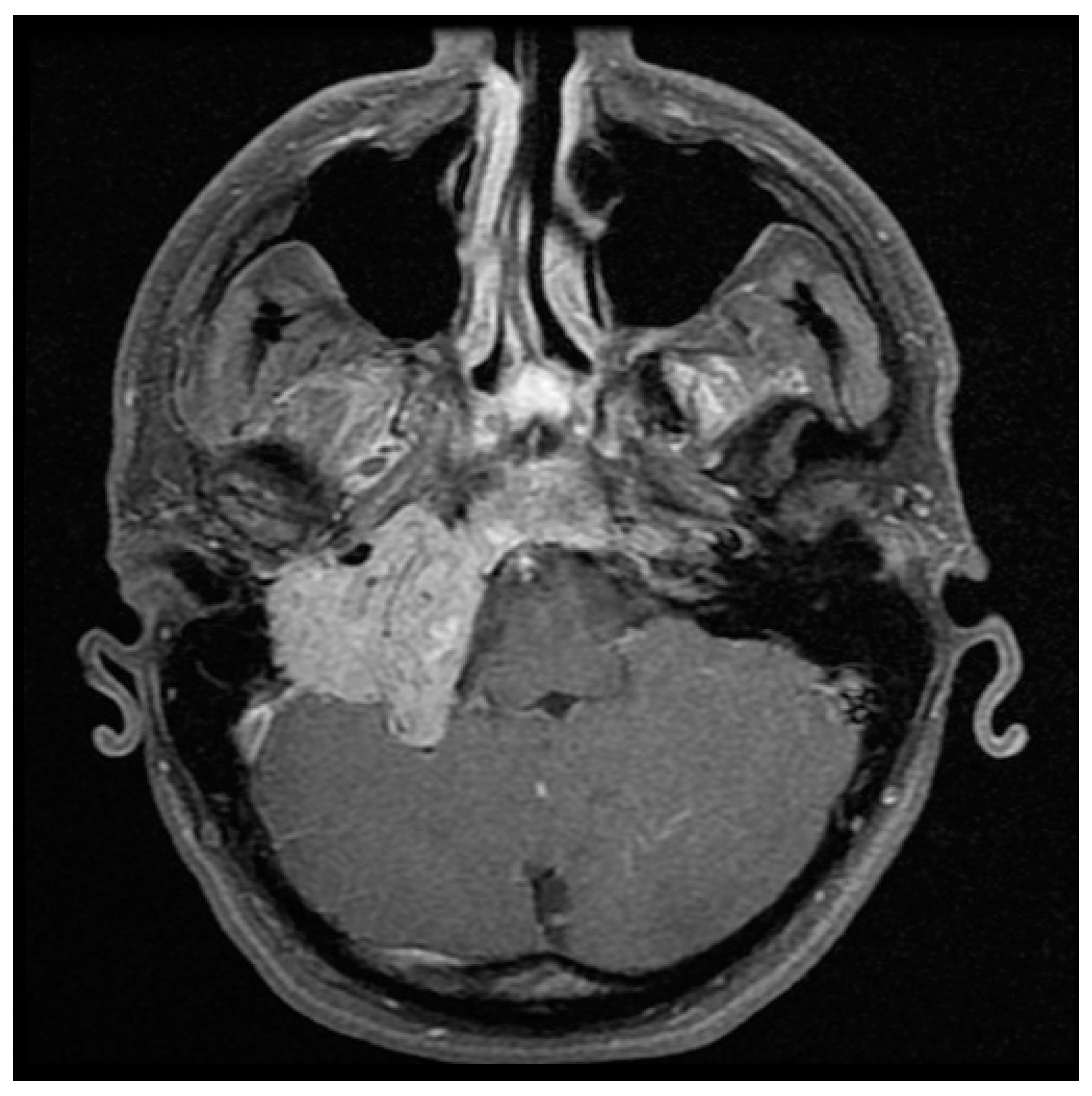
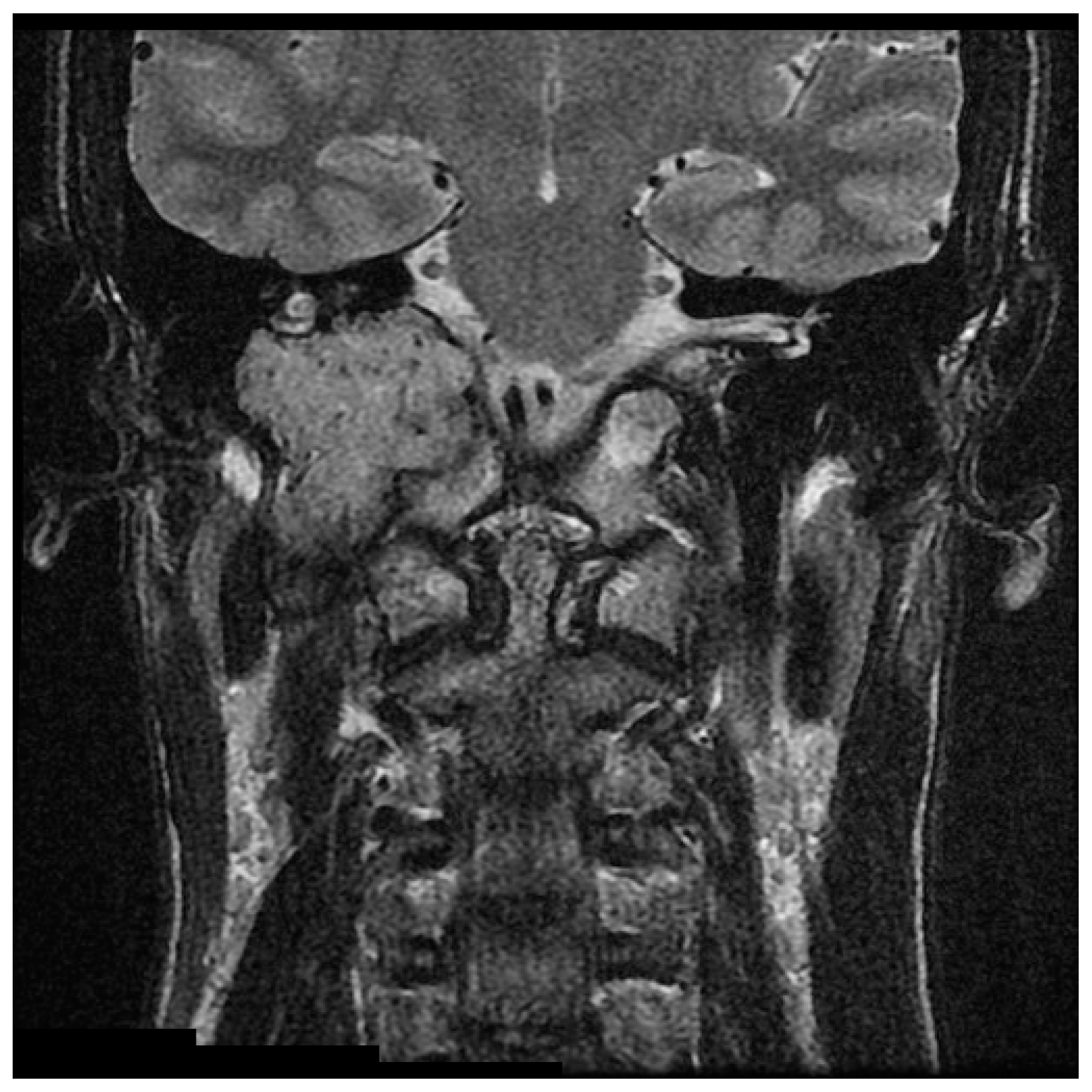
Detailed imaging is necessary in initial assessment and management planning for HNPGL. B-mode sonography and Doppler ultrasonography are non-invasive and cost-effective first line modalities of investigation for cervical masses, but lack specificity for definitive diagnosis and treatment [2,19,44]. MRI is highly sensitive in evaluating HNPGL (Figure 1 and Figure 2) [45]. On T1 weighted images HNPGL appear hypointense with a characteristic ‘speckled’ appearance. The T1 images with gadolinium enhancement show early uptake owing to the marked hypervascularity of the lesions, with serpentine flow-voids giving a characteristic ‘salt and pepper’ appearance [2,45,46,47].
Further information regarding flow velocities and collateral vascular supply is gained with MRA/MRV sequences [46]. High-resolution CT scans show small middle ear lesions very well and give important information regarding bony destruction in the temporal bone and skull base [45].
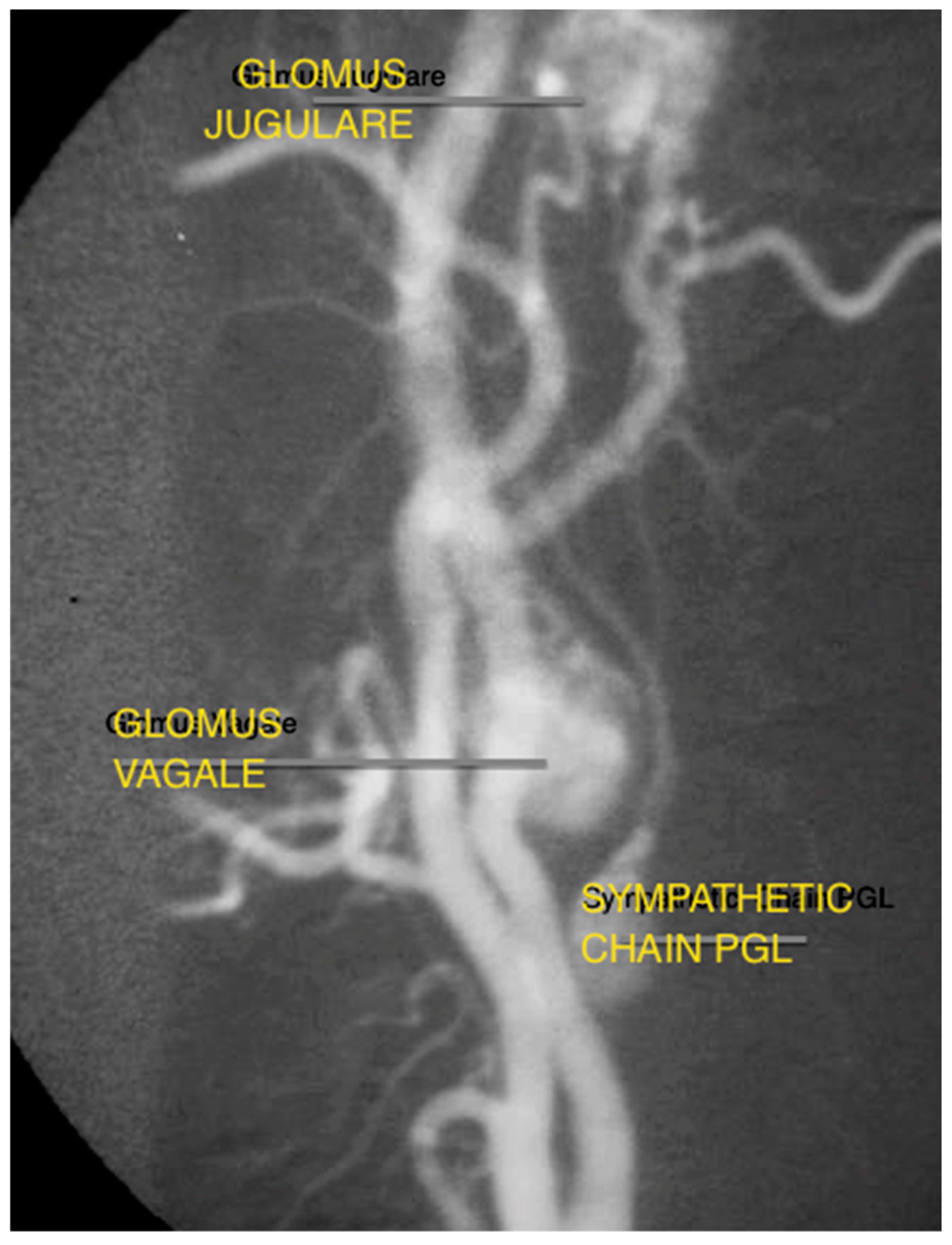
Despite the evolution of MR and CT technology, digital subtraction angiography (DSA) remains the gold standard in evaluating tumour vascularity, and is advocated close to the time of surgery to allow embolization, if required (Figure 3) [19]. It can also identify multiple vascular tumours in the head and neck.
Radioactive nucleotide agents in conjunction with SPECT have been recently acknowledged as useful adjuncts to traditional radiology for HNPGL; I123MIBG, F-DOPA, 18-F-flurodopa (F-FDA) have all been utilized although with modest sensitivity and specificity [48,49,50]. Recent reports have identified 68Ga-DOTATATE as a more sensitive and specific marker in extra-adrenal PGL [51].
Jugulo-tympanic tumours are generally classified by either the Fisch or Glasscock-Jackson staging systems [52,53,54,55]. The detail shown in the CT and MRI scans together enable accurate staging, greatly assisting selection of surgical approaches. The Shamblin classification of carotid body tumours is similarly useful [56].
Treatment must be individualised based on patient factors (age, co-morbidities) and tumour factors (size, location, involved local structures, intracranial extension, multiple tumours) [16].
The morbidity associated with treatment of HNPGL is significant, and given the slow growing and mostly benign nature of these lesions observation and supportive management may be appropriate, particularly in patients of advanced age, patients with significant co-morbidities, and those who have not developed cranial nerve deficits from their tumour(s) [34].
The goal of surgical resection is complete tumour removal, and is the optimal modality if this can be achieved with minimal surgical morbidity. Cranial nerve palsies represent the most common complication [2]. Surgical approaches range from tympanotomy or mastoidectomy to extensive infratemporal approaches depending on the site of origin and size of the tumour. Intracranial tumour is usually best treated surgically. The goal of radiotherapy is to reduce tumour size and to halt/control growth. Radiotherapy via external beam radiotherapy or stereotactic radiosurgery has shown excellent local control rates (>95%) for primary management in HNPGL [57,58,59]. Radiotherapy is not without complications; the major being failure to control the tumour [58]. Osteoradionecrosis of the mandible and tympanic bone can complicate radiation therapy to the ear, along with cranial nerve deficits depending on tumour location [10].
There is no consensus regarding the optimal treatment modality regarding HNPGL, and a combination may be appropriate. Familial HNPGL are more likely to be recurrent, or multiple in nature and close post-treatment follow-up is advised. Although there is no acknowledged protocol for long-term follow-up, surveillance examination and imaging every 1–2 years is advocated [59].
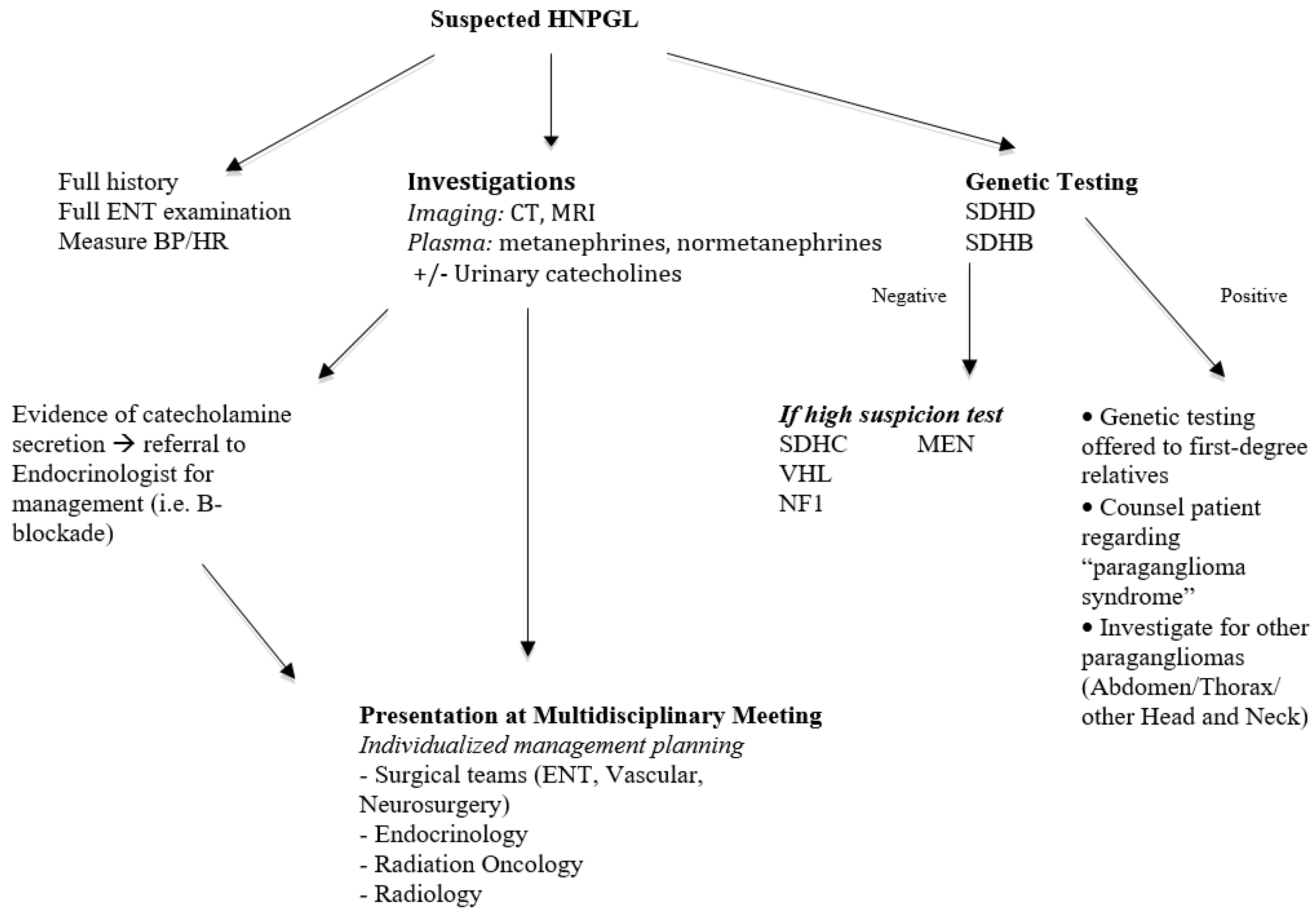
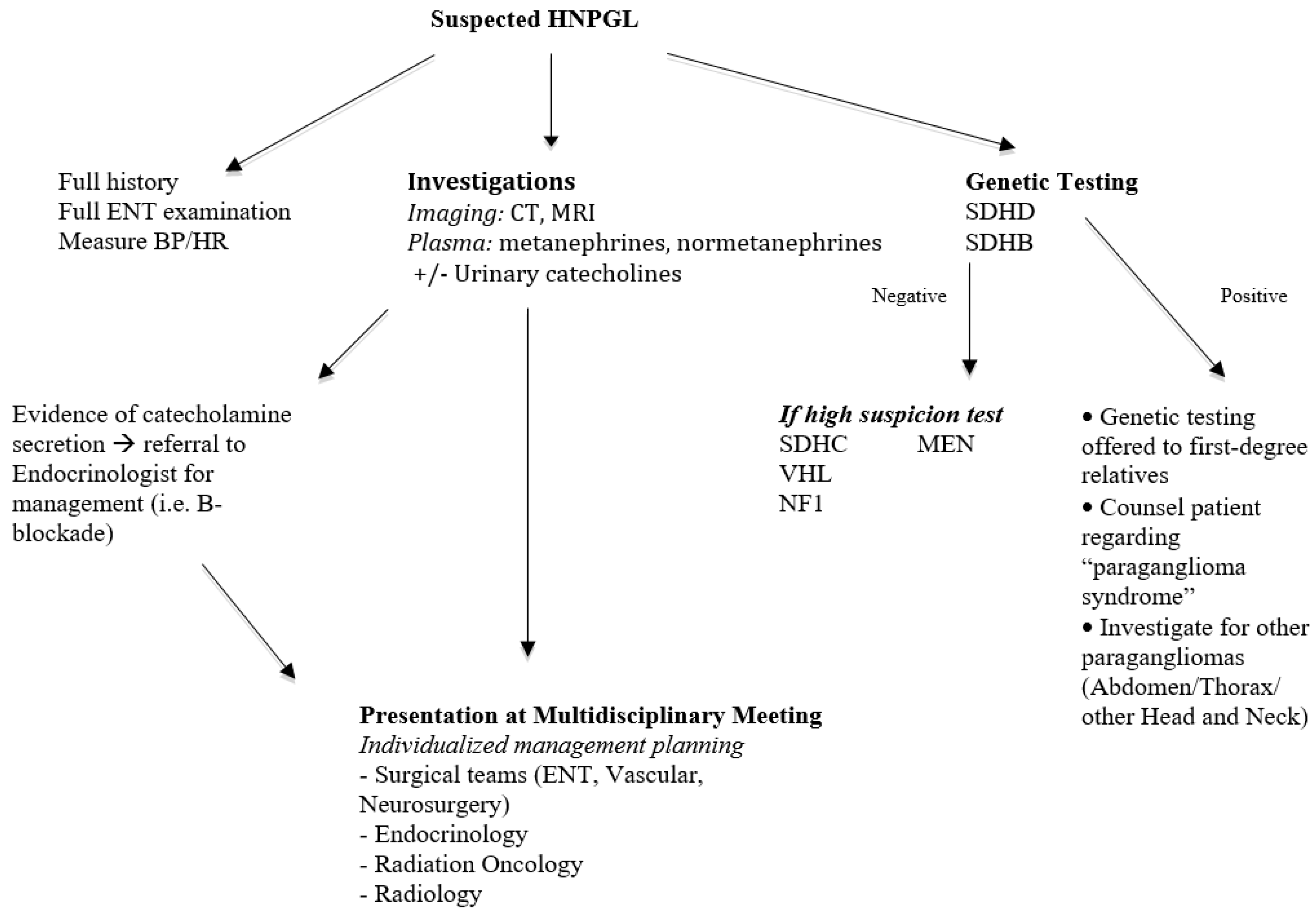
Based on the literature and our experience, a proposed algorithm for assessment of a patient with HNPGL suspicious for a hereditary form is presented in Figure 4.
6. Conclusions
HNPGL are rare tumours requiring a systematic approach to diagnosis, investigation, and management involving multimodal surgical, medical, genetics, and oncological input for optimal management. All HNPGL patients must be assessed for SDH mutations given the contemporary understanding of inheritance patterns related to this condition, and the implications for the patient and their relatives. Patients with familial PGL have a greater chance of multiple tumours, hormone secretion, malignancy, and greater rates of recurrent/residual tumour. The natural history of inherited HNPGL is different to sporadic cases, as observed in the literature and our dataset—identification of these patients has important implications in the monitoring and management of these patients and their families.
There is a small, but finite, number of HNPGL patients that secrete excess catecholamines. This secretion can greatly affect the safety of general anaesthesia for these patients and, therefore, all should be screened.
The optimal management of HNPGL depends on comprehensive assessment, including these medical measures and detailed imaging. Treatment must be individualized for each patient.
Author Contributions
Nathan Hayward and Vincent Cousins conceived and designed the article; Nathan Hayward collated and analysed the data; Vincent Cousins contributed patient files and access to notes/images; Nathan Hayward performed the literature review and wrote the article; and Vincent Cousins provided academic and editorial support and guidance throughout the development of the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baysal, B.E. Hereditary paraganglioma targets diverse paraganglia. J. Med. Genet. 2002, 39, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.C.; Ridder, G.J.; Schipper, J. Paragangliomas of the head and neck: Diagnosis and treatment. Fam. Cancer 2005, 4, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Dziegelewski, P.T.; Knox, A.; Liu, R.; Hung, R.; Harris, J. Familial Paraganglioma Syndrome: Applying Genetic Screening in Otolaryngology. J. Otolaryngol. Head Neck Surg. 2010, 39, 646–653. [Google Scholar]
- Martin, T.P.C.; Irving, R.; Maher, E.R. The genetics of paragangliomas: A review. Clin. Otolaryngol. 2007, 37, 7–11. [Google Scholar]
- Schiavi, F.; Savvoukidis, T.; Trabalzini, F.; Grego, F.; Piazza, M.; Amista, P.; Dematte, S.; Del Piano, A.; Cecchini, M.E.; Erlic, Z.; et al. Paraganglioma syndrome: SDHB, SDHC, and SDHD mutations in head and neck paragangliomas. Ann. N. Y. Acad. Sci. 2006, 1073, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Thabet, M.H.; Kotob, H. Cervical paragangliomas: Diagnosis, management and complications. J. Laryngol. Otol. 2001, 115, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Myssiorek, D. Head and neck paragangliomas: An overview. Otolaryngol. Clin. N. Am. 2001, 34, 829–836. [Google Scholar] [CrossRef]
- Boedeker, C.C. Paragangliomas and paraganglioma syndromes. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 2011, 10. [Google Scholar] [CrossRef]
- Badenhop, R.F.; Jansen, J.C.; Fagan, P.A.; Lord, R.S.; Wang, Z.G.; Foster, W.J.; Schofield, P.R. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J. Med. Genet. 2004, 41, e99. [Google Scholar] [CrossRef] [PubMed]
- Semaan, M.; Megerian, C. Current assessment and management of glomus tumours. Curr. Opin. Otolaryngol. Head Neck Surg. 2008, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Offergeld, C.; Brase, C.; Yaremchuk, S.; Mader, I.; Rischke, H.C.; Glasker, S.; Schmid, K.W.; Wiech, T.; Preuss, S.F.; Suarez, C.; et al. Head and neck paragangliomas: Clinical and molecular genetic classification. Clinics 2012, 67, 19–28. [Google Scholar] [CrossRef]
- Bikhazi, P.H.; Messina, L.; Mhatre, A.N.; Goldstein, J.A.; Lalwani, A.K. Molecular pathogenesis in sporadic head and neck paraganglioma. Laryngoscope 2000, 110, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Barich, F.; Karnell, L.H.; Robinson, R.A.; Zhen, W.K.; Gantz, B.J.; Hoffman, H.T. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer 2002, 94, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Manolidis, S.; Shohet, J.A.; Jackson, C.G.; Glasscock, M.E., 3rd. Malignant glomus tumors. Laryngoscope 1999, 109, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Mann, W.J.; Amedee, R.G. Management of head and neck paragangliomas: Review of 120 patients. Head Neck 2009, 31, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.; Kudva, Y.C.; Ebersold, M.J.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A.; Young, W.F., Jr. Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J. Clin. Endocrinol. Metab. 2001, 86, 5210–5216. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.B.; Koeller, K.K.; Adair, C.F. From the archives of the AFIP. Paragangliomas of the head and neck: Radiologic-pathologic correlation. Armed Forces Institute of Pathology. Radiographics 1999, 19, 1605–1632. [Google Scholar] [CrossRef] [PubMed]
- Pellitteri, P.K.; Rinaldo, A.; Myssiorek, D.; Gary Jackson, C.; Bradley, P.J.; Devaney, K.O.; Shaha, A.R.; Netterville, J.L.; Manni, J.J.; Ferlito, A. Paragangliomas of the head and neck. Oral Oncol. 2004, 40, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.B.; Boon, M.S.; Atkins, J.P.; Lowry, L.D. Vagal paraganglioma: The Jefferson experience. Otolaryngol. Head Neck Surg. 2000, 122, 482–487. [Google Scholar] [PubMed]
- Rodriguez-Cuevas, H.; Lau, I.; Rodriguez, H.P. High-altitude paragangliomas diagnostic and therapeutic considerations. Cancer 1986, 57, 672–676. [Google Scholar] [CrossRef]
- Colen, T.Y.; Mihm, F.G.; Mason, T.P.; Roberson, J.B. Catecholamine-secreting paragangliomas: Recent progress in diagnosis and perioperative management. Skull Base 2009, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Osinga, T.E.; Korpershoek, E.; de Krijger, R.R.; Kerstens, M.N.; Dullaart, R.P.; Kema, I.P.; van der Laan, B.F.; van der Horst-Schrivers, A.N.; Links, T.P. Catecholamine-Synthesizing Enzymes Are Expressed in Parasympathetic Head and Neck Paraganglioma Tissue. Neuroendocrinology 2015, 101, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G. Screening for pheochromocytomas and paragangliomas. Curr. Hypertens. Rep. 2012, 14, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, E.; Bresciani, S.; Panaia, R.; Delmastro, E.; Malinverni, G.; Gabriele, P. Hereditary paraganglioma syndrome associated with SDHD gene mutations: A patient with multicentric presentation treated with radiotherapy. Case report. Tumori 2011, 97, 214–220. [Google Scholar] [PubMed]
- Hensen, E.F.; Bayley, J.P. Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam. Cancer 2011, 10, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, C.M.; Solorzano, C.C. Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment. Surg. Oncol. Clin. N. Am. 2016, 25, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.C.; Hensen, E.F.; Neumann, H.P.; Maier, W.; van Nederveen, F.H.; Suarez, C.; Kunst, H.P.; Rodrigo, J.P.; Takes, R.P.; Pellitteri, P.K.; et al. Genetics of hereditary head and neck paragangliomas. Head Neck 2014, 36, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Erlic, Z.; Boedeker, C.C.; Rybicki, L.A.; Robledo, M.; Hermsen, M.; Schiavi, F.; Falcioni, M.; Kwok, P.; Bauters, C.; et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: Cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res. 2009, 69, 3650–3656. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, W.M.; Amdur, R.J.; Vaysberg, M.; Mendenhall, C.M.; Werning, J.W. Head and neck paragangliomas. Head Neck 2011, 33, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kunst, H.P.; Rutten, M.H.; de Monnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.; Marres, H.A.; Jansen, J.C.; Kremer, H.; Bayley, J.P.; Cremers, C.W. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [PubMed]
- Schiavi, F.; Boedeker, C.C.; Bausch, B.; Peczkowska, M.; Gomez, C.F.; Strassburg, T.; Pawlu, C.; Buchta, M.; Salzmann, M.; Hoffmann, M.M.; et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 2005, 294, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Mewes, T.; Rossmann, H.; Fottner, C.; Schneider-Raetzke, B.; Bartsch, O.; Schreckenberger, M.; Lackner, K.J.; Amedee, R.G.; Mann, W.J. Head and neck paragangliomas: Report of 175 patients (1989–2010). Head Neck 2012, 34, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dwight, T.; Benn, D.E.; Clarkson, A.; Vilain, R.; Lipton, L.; Robinson, B.G.; Clifton-Bligh, R.J.; Gill, A.J. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am. J. Surg. Pathol. 2013, 37, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Pheochromocytoma and extra-adrenal paraganglioma: Updates. Arch. Pathol. Lab. Med. 2008, 132, 1272–1284. [Google Scholar] [PubMed]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Stoeckli, S.J.; Schuknecht, B.; Alkadhi, H.; Fisch, U. Evaluation of paragangliomas presenting as a cervical mass on color-coded Doppler sonography. Laryngoscope 2002, 112, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R. Imaging and management of head and neck paragangliomas. Eur. Radiol. 2005, 15, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.; Schepers, A.; de Bruine, F.T.; Liauw, L.; Mertens, B.J.; van der Mey, A.G.; van Buchem, M.A. The value of MR angiography techniques in the detection of head and neck paragangliomas. Eur. J. Radiol. 2004, 52, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.; Verbist, B.M.; Mertens, B.J.; van der Mey, A.G.; van Buchem, M.A. Head and neck paragangliomas: Improved tumor detection using contrast-enhanced 3D time-of-flight MR angiography as compared with fat-suppressed MR imaging techniques. AJNR Am. J. Neuroradiol. 2004, 25, 863–870. [Google Scholar] [PubMed]
- Milardovic, R.; Corssmit, E.P.; Stokkel, M. Value of 123I-MIBG Scintigraphy in Paraganglioma. Neuroendocrinology 2010, 91, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hoegerle, S.; Ghanem, N.; Altehoefer, C.; Schipper, J.; Brink, I.; Moser, E.; Neumann, H.P. 18F-DOPA positron emission tomography for the detection of glomus tumours. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, A.; Shulkin, B.L.; Uprimny, C.; Frech, A.; Gasser, R.W.; Url, C.; Gautsch, K.; Madleitner, R.; Nilica, B.; Sprinzl, G.M.; et al. 68Ga-DOTATOC PET/CT provides accurate tumour extent in patients with extraadrenal paraganglioma compared to 123I-MIBG SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Oldring, D.; Fisch, U. Glomus tumors of the temporal region: Surgical therapy. Am. J. Otol. 1979, 1, 7–18. [Google Scholar] [PubMed]
- Jackson, C.G.; Glasscock, M.E., 3rd; Harris, P.F. Glomus Tumors. Diagnosis, classification, and management of large lesions. Arch. Otolaryngol. 1982, 108, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Fisch, U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann. Otol. Rhinol. Laryngol. 1982, 91, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Alaani, A.; Chavda, S.V.; Irving, R.M. The crucial role of imaging in determining the approach to glomus tympanicum tumours. Eur. Arch. Otorhinolaryngol. 2009, 266, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Shamblin, W.R.; ReMine, W.H.; Sheps, S.G.; Harrison, E.G., Jr. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am. J. Surg. 1971, 122, 732–739. [Google Scholar] [CrossRef]
- Lightowlers, S.; Benedict, S.; Jefferies, S.J.; Jena, R.; Harris, F.; Burton, K.E.; Burnet, N.G. Excellent local control of paraganglioma in the head and neck with fractionated radiotherapy. Clin. Oncol. 2010, 22, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Gilbo, P.; Morris, C.G.; Amdur, R.J.; Werning, J.W.; Dziegielewski, P.T.; Kirwan, J.; Mendenhall, W.M. Radiotherapy for benign head and neck paragangliomas: A 45-year experience. Cancer 2014, 120, 3738–3743. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, W.M.; Amdur, R.J. Radiotherapy for Head and Neck Paraganglioma. Oper. Tech. Otolaryngol. 2016, 27, 55–57. [Google Scholar] [CrossRef]
- Capatina, C.; Ntali, G.; Karavitaki, N.; Grossman, A.B. The management of head-and-neck paragangliomas. Endocr. Relat. Cancer 2013, 20, R291–R305. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Axial T1 with gadolinium MRI of large right glomus jugulare.
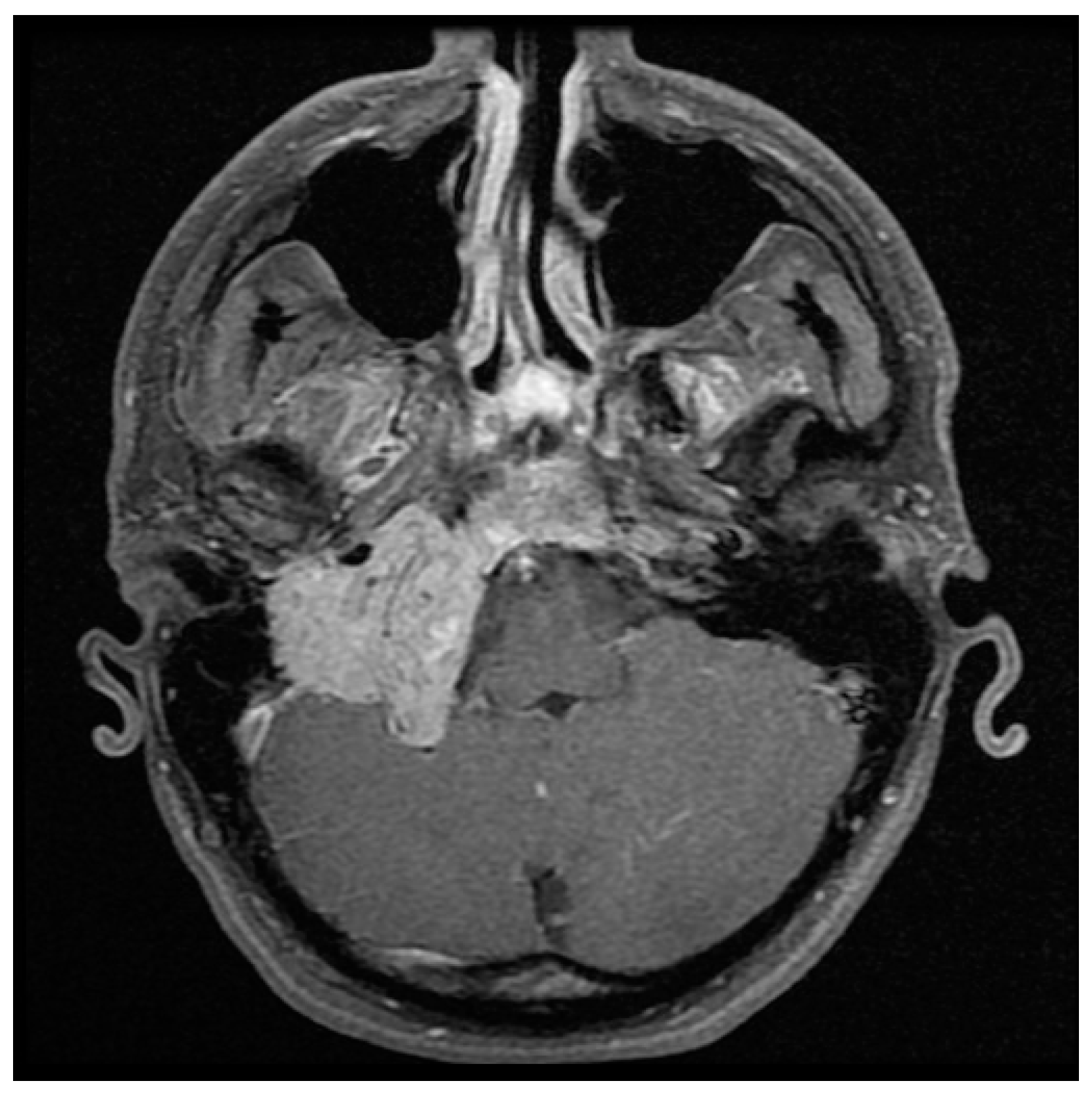
Figure 2.
Coronal T2 with gadolinium MRI of the large right glomus jugulare showing numerous vascular flow voids.
Figure 2.
Coronal T2 with gadolinium MRI of the large right glomus jugulare showing numerous vascular flow voids.

Figure 3.
Angiography capture of concurrent glomus tympanicum, glomus vagale, and cervical sympathetic chain paraganglioma.
Figure 3.
Angiography capture of concurrent glomus tympanicum, glomus vagale, and cervical sympathetic chain paraganglioma.
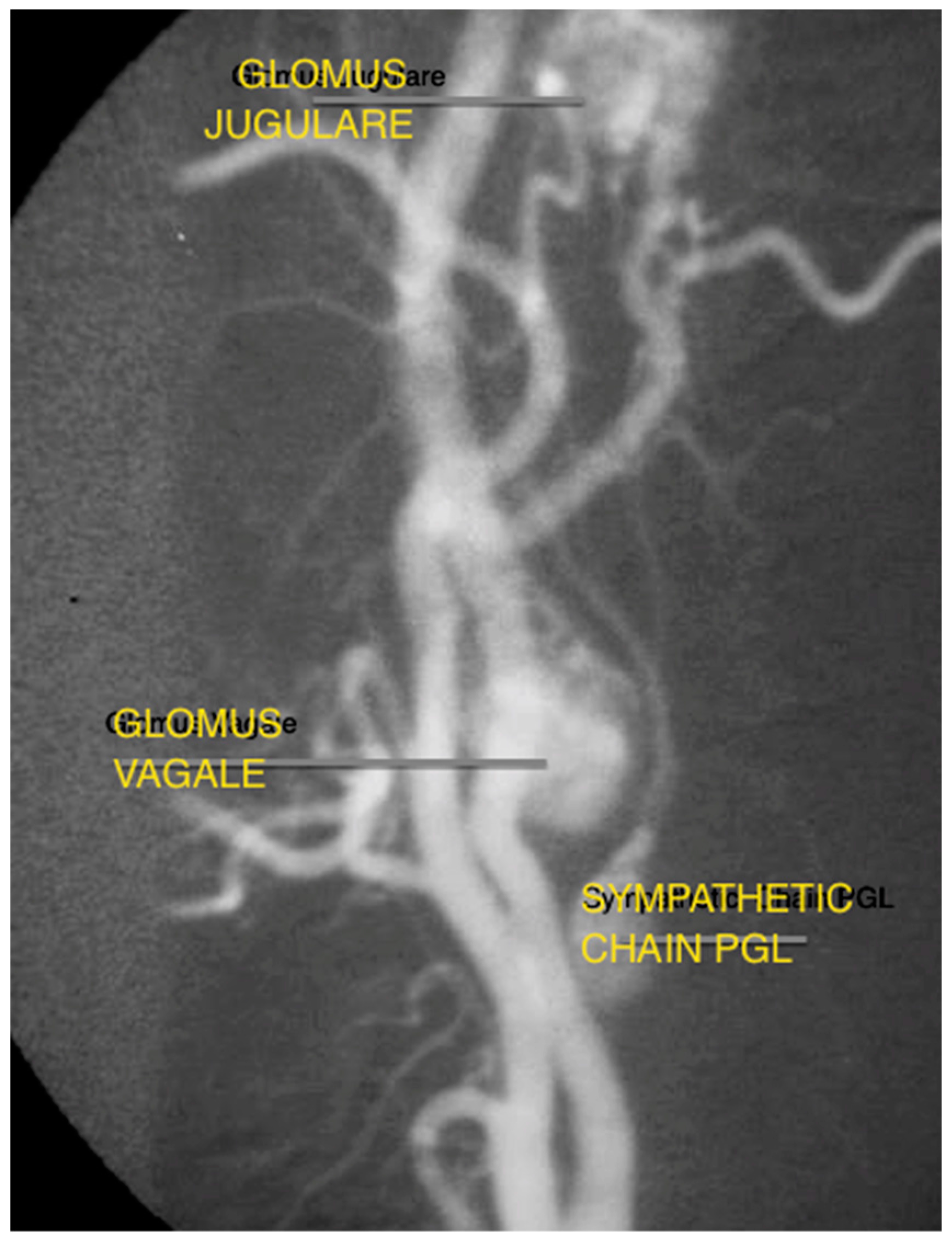
Figure 4.
Proposed assessment algorithm for HNPGL.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sex and average age of presentation for HNPGL.
| Glomus Tympanicum | Glomus Jugulare | Glomus Vagale | Carotid Body Tumours | |
|---|---|---|---|---|
| Total Patients (n = 60) | 32 | 20 | 3 | 5 |
| Mean age and Range at presentation (Years) | 52.6 years (30–82 years) | 50.8 years (21–80 years) | 47.0 years (27–57 years) | 38.6 years (30–56 years) |
| Sex differences (Female:Male) | 3:1 | 3:1 | 2:1 | 2:3 |
Table 2.
Characteristics of HNPGL patients with SDH gene mutations.
| Age/Sex (Diagnosis) | Tumour | SDH Mutation | Specific Detail |
|---|---|---|---|
| 34F | Glomus tympanicum | SDHB | Developed extensive recurrence requiring further excision six years post initial surgery |
| 21M | Glomus jugulare | SDHB | Catecholamine secreting tumour. Mother past history phaeochromocytoma. Ongoing elevated catecholamines post planned subtotal excision. |
| 47F | Glomus tympanicum | SDHD | Multiple lesions: contralateral Carotid Body Tumour diagnosed one year post glomus tympanicum excision |
| 31F | Glomus jugulare | SDHD | Monitoring |
| 57F 27F | Glomus vagale Glomus jugulare | SDHB (Presumed SDHB) | Relative (aunt) within pedigree with carotid body tissue |
| 20M | Glomus jugulare | SDHB | Multiple recurrence (four excisions over 20 years, multiple surgeons) |
| 32F | Carotid Body Tumour | SDHD | Multiple lesions: bilateral glomus jugulare concurrently (non-operative management) |
Table 3.
Summary of the management undertaken in this series of HNPGL patients.
| Glomus Tympanicum (n = 32) | Glomus Jugulare (n = 20) | Glomus Vagale (n = 3) | Carotid Body Tumours (n = 4) |
|---|---|---|---|
| Tympanotomy: 23 | Surgery only: 14 (Infratemporal fossa) | Excision: 2 | Excision: 4 |
| Mastoidectomy: 14 | Five cases combined with Neurosurgery | Observation:1 | |
| Observation: 4 | Radiation only: 4 | ||
| Surgery + Radiotherapy: 5 | |||
| Totals: 41 | Totals: 23 | Totals: 3 | Totals: 4 |
This table includes operations for subsequent/recurrent lesions (four multifocal).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hayward, N.; Cousins, V. Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update. J. Otorhinolaryngol. Hear. Balance Med. 2018, 1, 4. https://doi.org/10.3390/ohbm1010004
AMA Style
Hayward N, Cousins V. Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update. Journal of Otorhinolaryngology, Hearing and Balance Medicine. 2018; 1(1):4. https://doi.org/10.3390/ohbm1010004
Chicago/Turabian StyleHayward, Nathan, and Vincent Cousins. 2018. "Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update" Journal of Otorhinolaryngology, Hearing and Balance Medicine 1, no. 1: 4. https://doi.org/10.3390/ohbm1010004