
Modulation of Myotilin and Fylamin C in Various Muscle Diseases: A Microarray Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
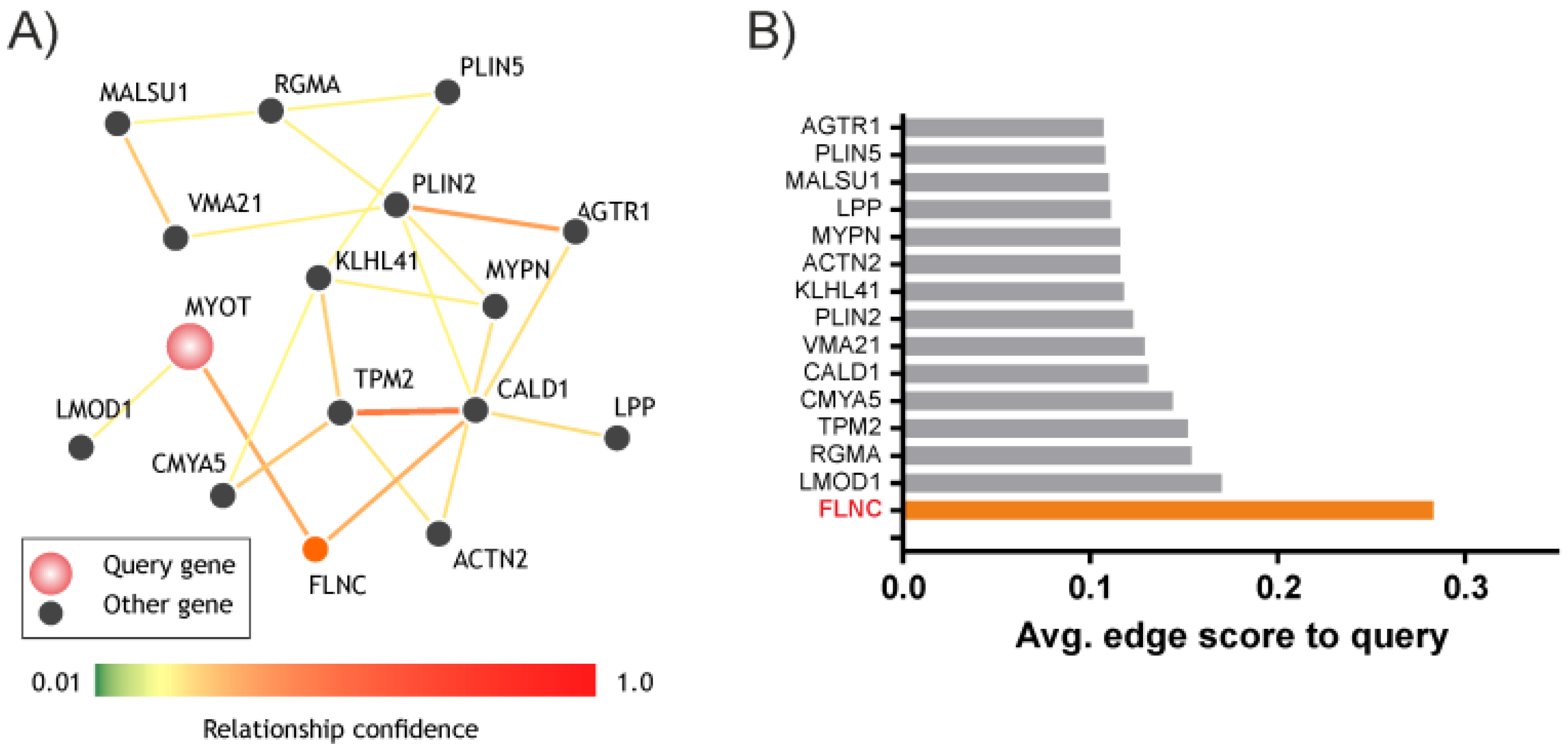
2.2. GIANT: Genome-Scale Integrated Analysis of Gene Networks in Tissues
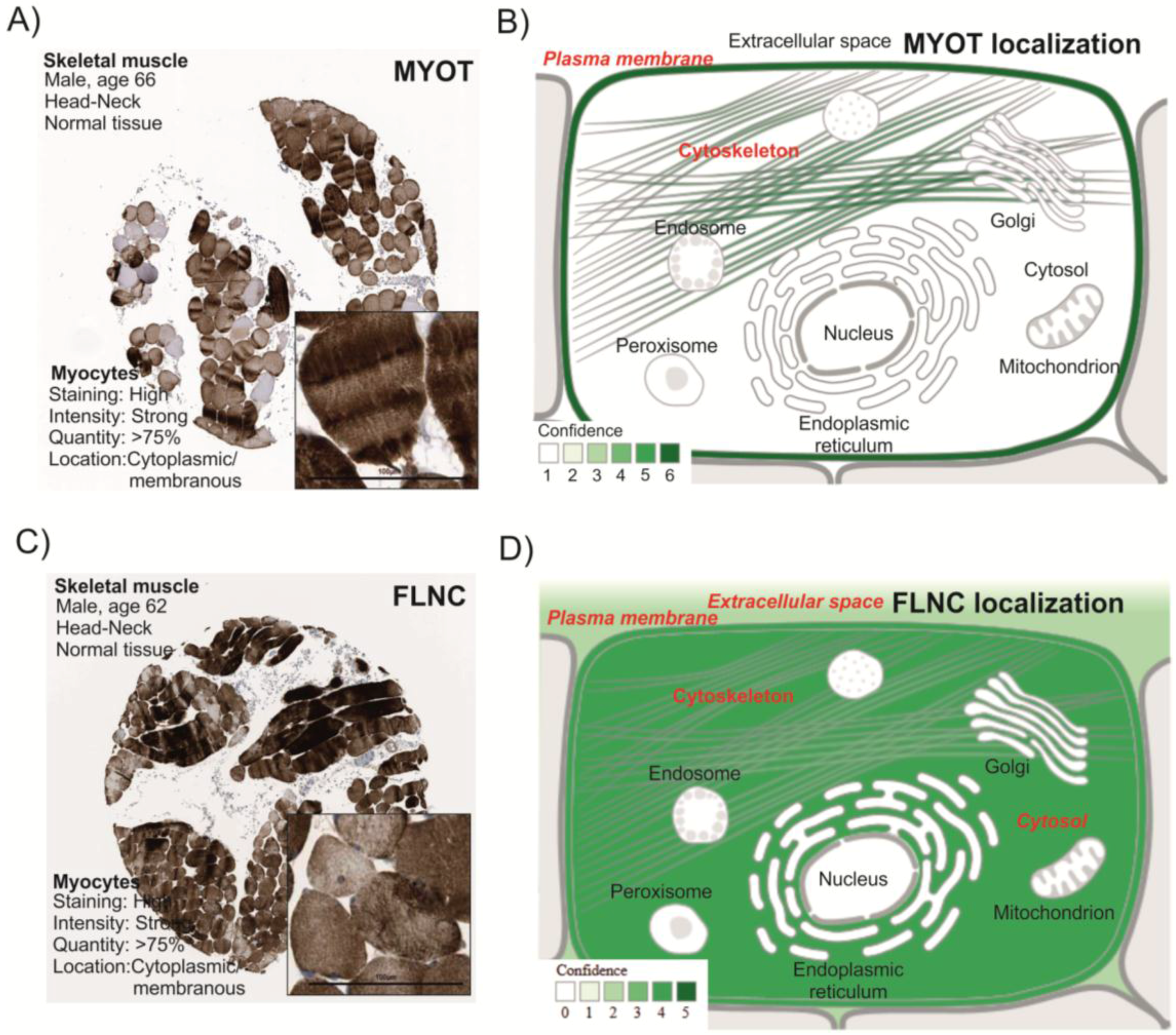
2.3. Immunohistochemestry (IH) by “the Human Protein Atlas”
- Consistent with extensive gene/protein characterization data
- Consistent with gene/protein characterization data
- Partly consistent with extensive gene/protein characterization data
- Partly consistent with gene/protein characterization data
- No available gene/protein characterization data
- Not consistent with gene/protein characterization data
- Not done
2.4. Analysis of Cellular Localization by "COMPARTMENTS"
2.5. Statistical Analysis
3. Results
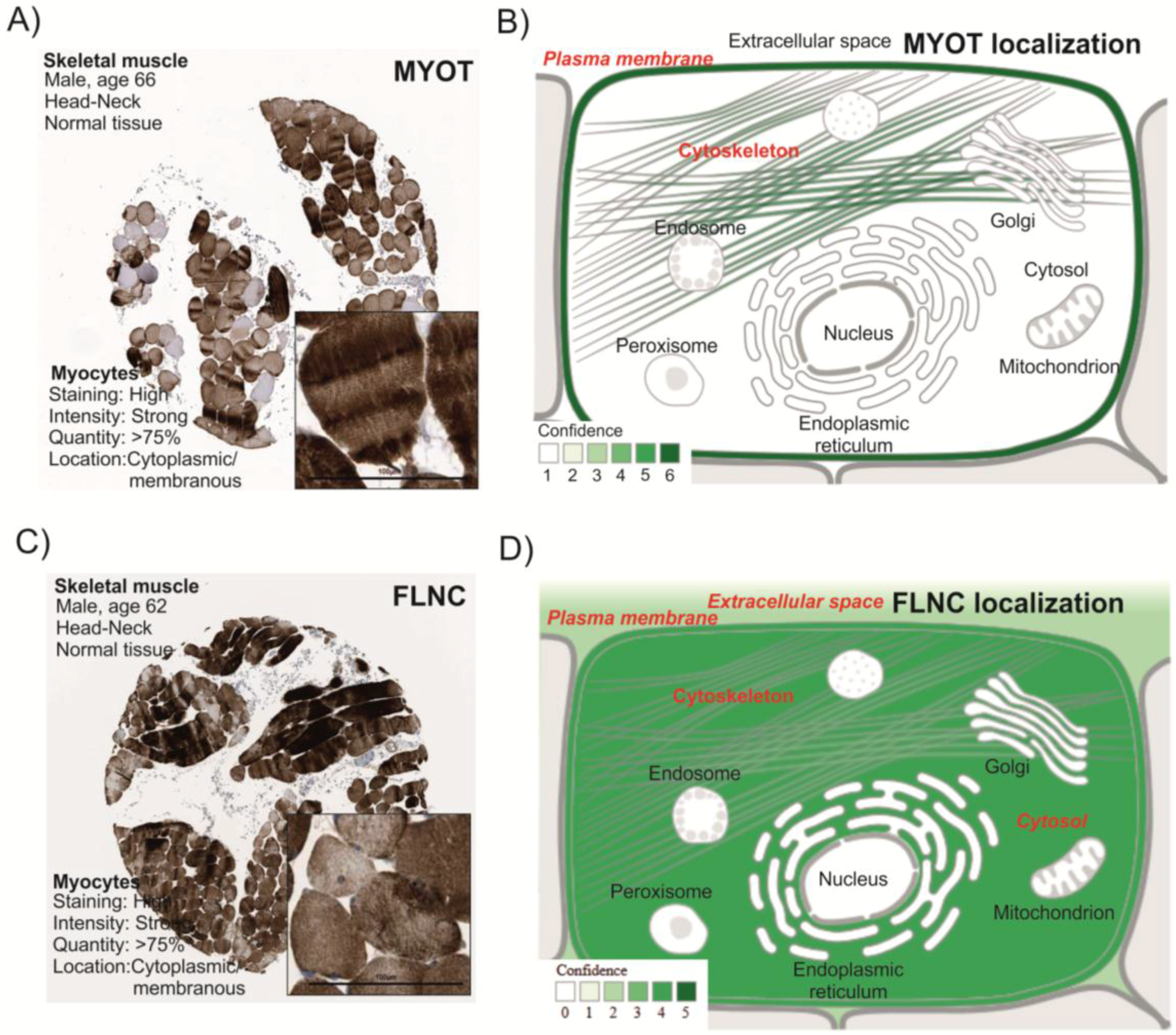
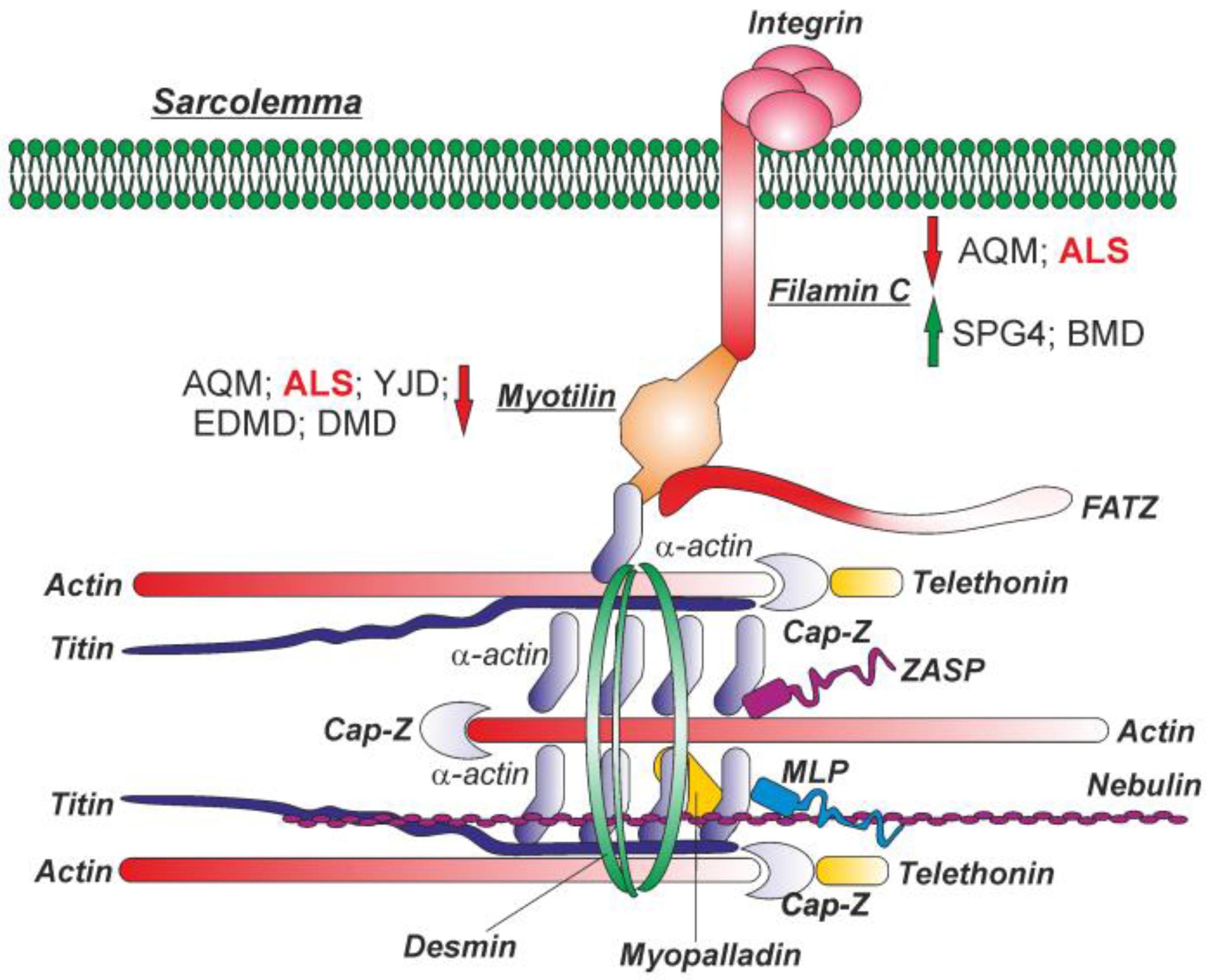
3.1. MYOT and FLNC Tissue and Cellular Localization
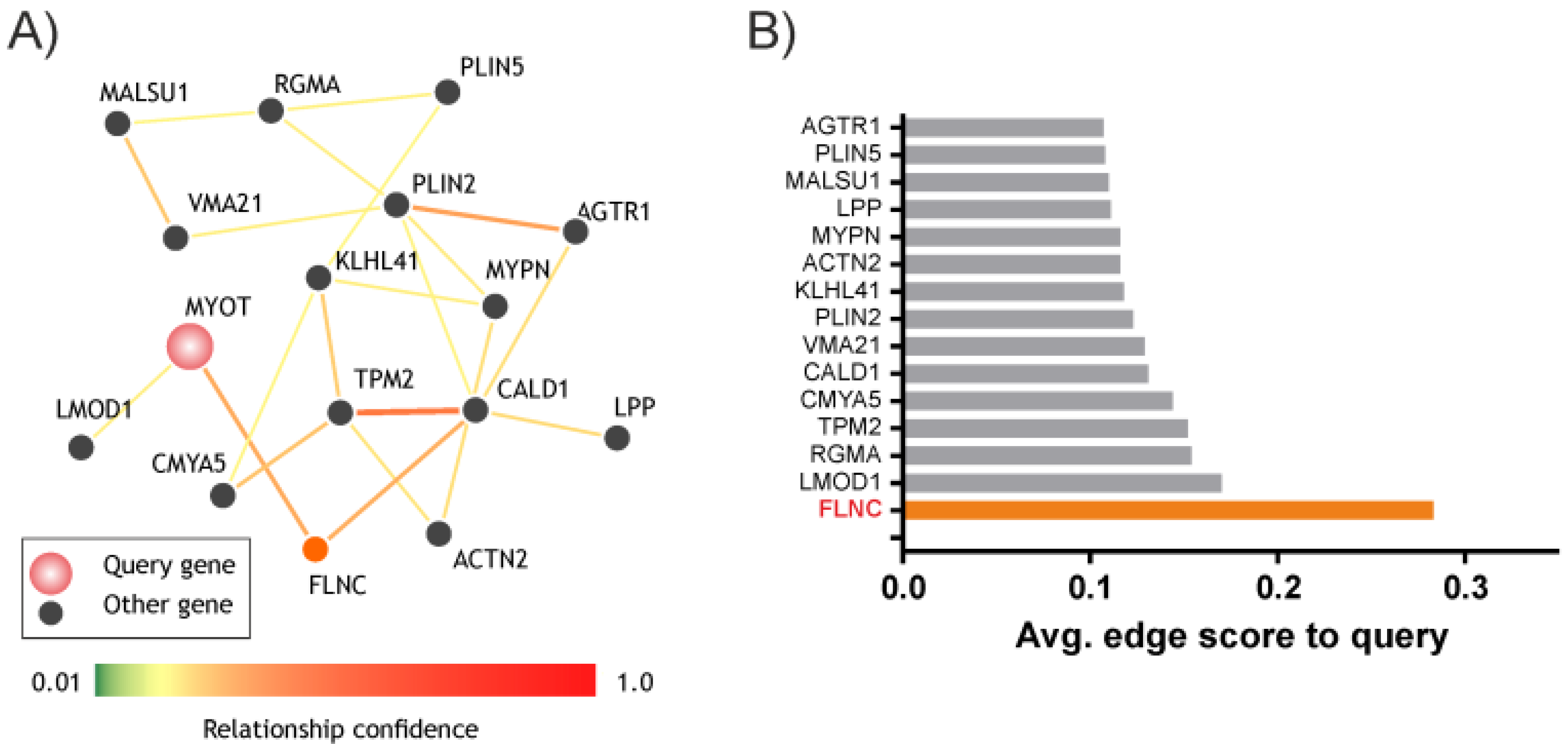
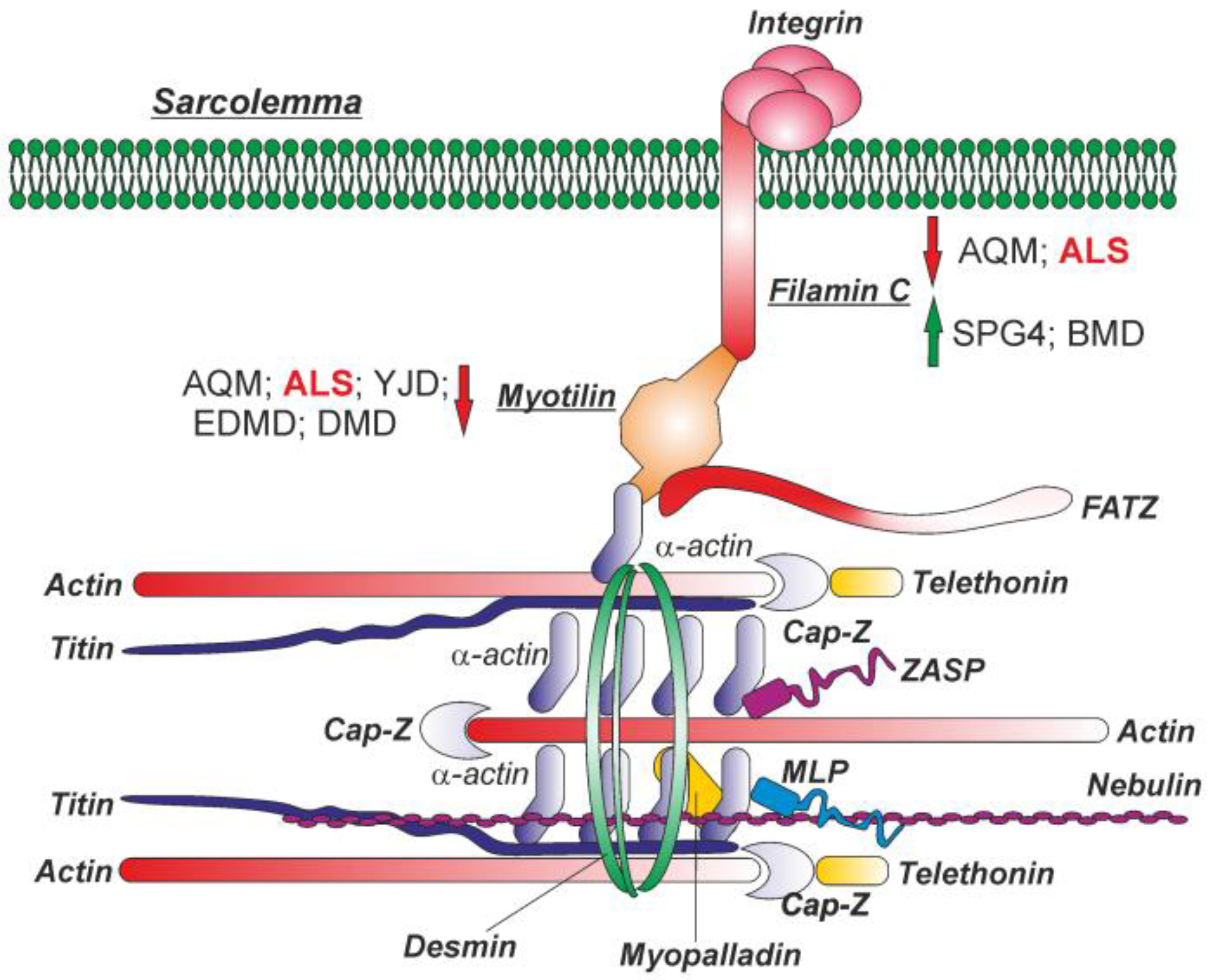
3.2. MYOT and FLNC Network
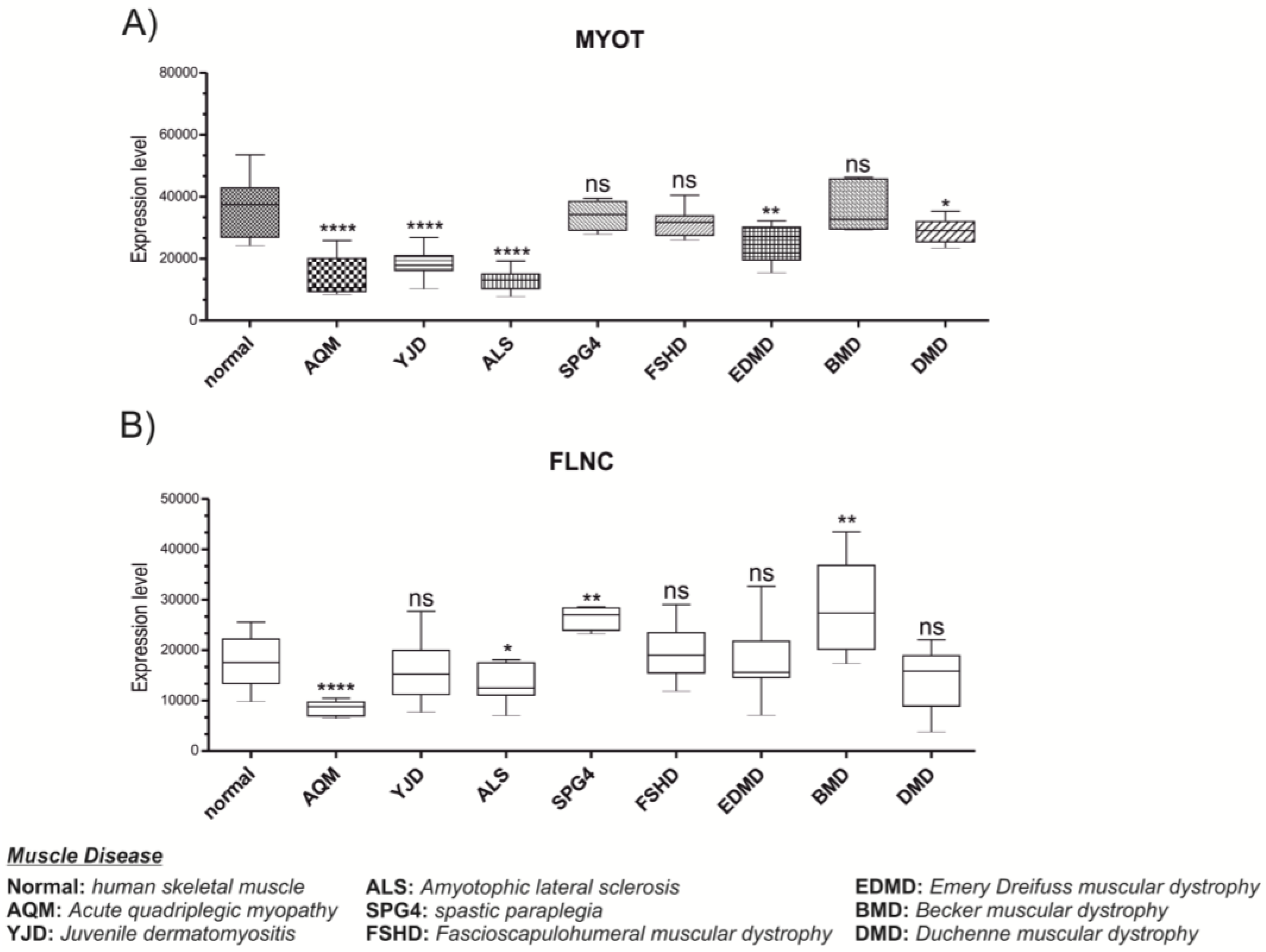
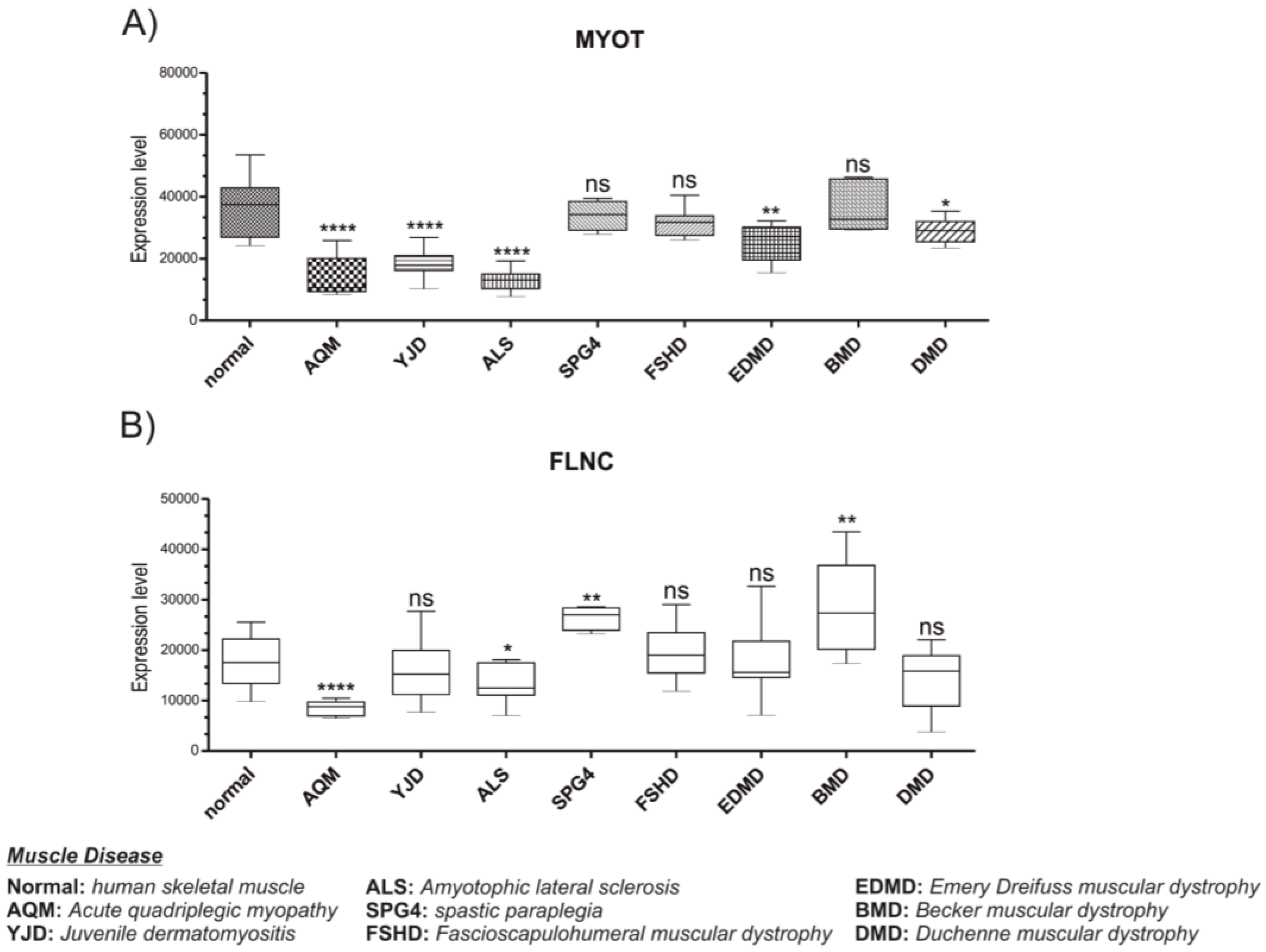
3.3. MYOT and FLNC Expression Levels in Various Muscle Diseases
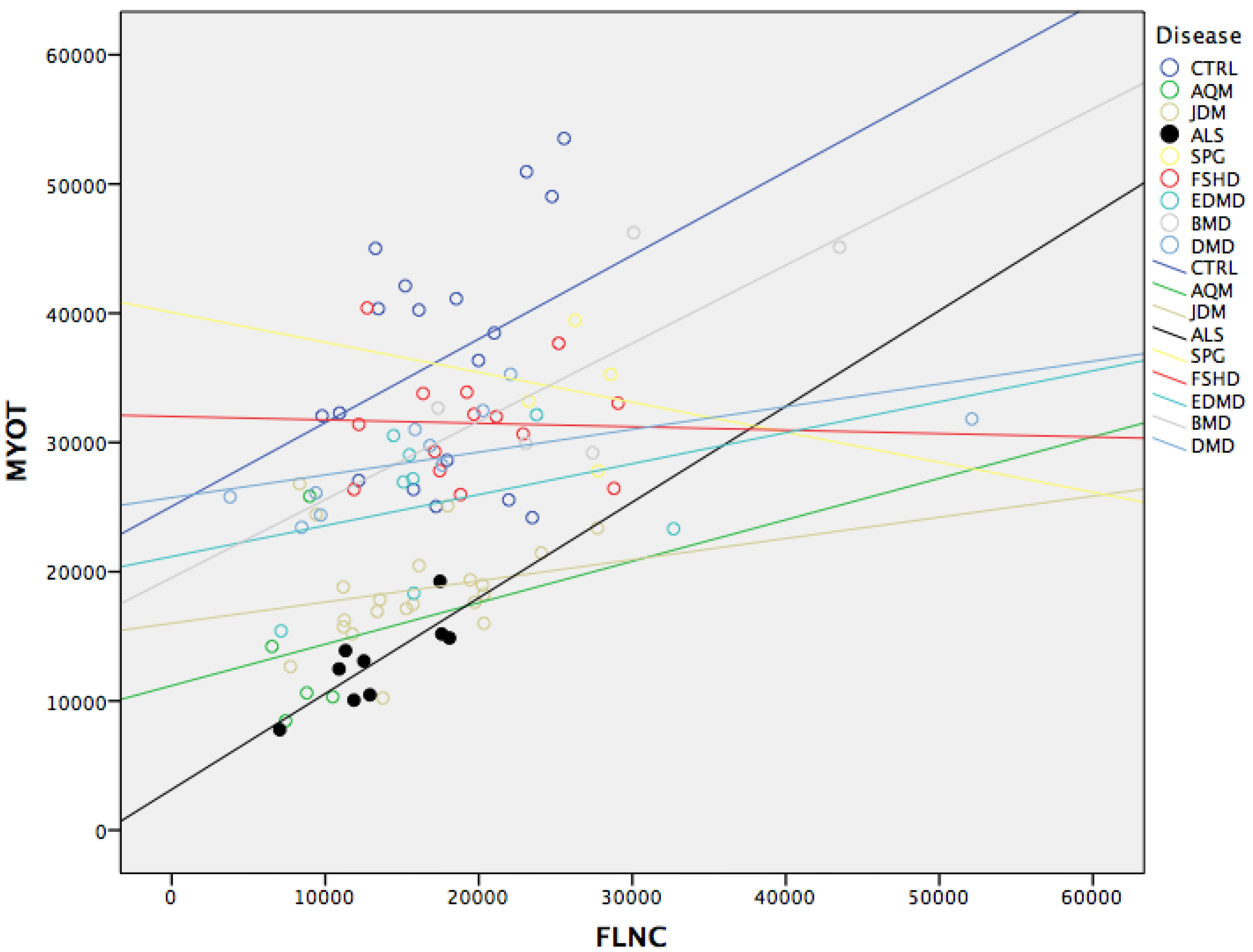
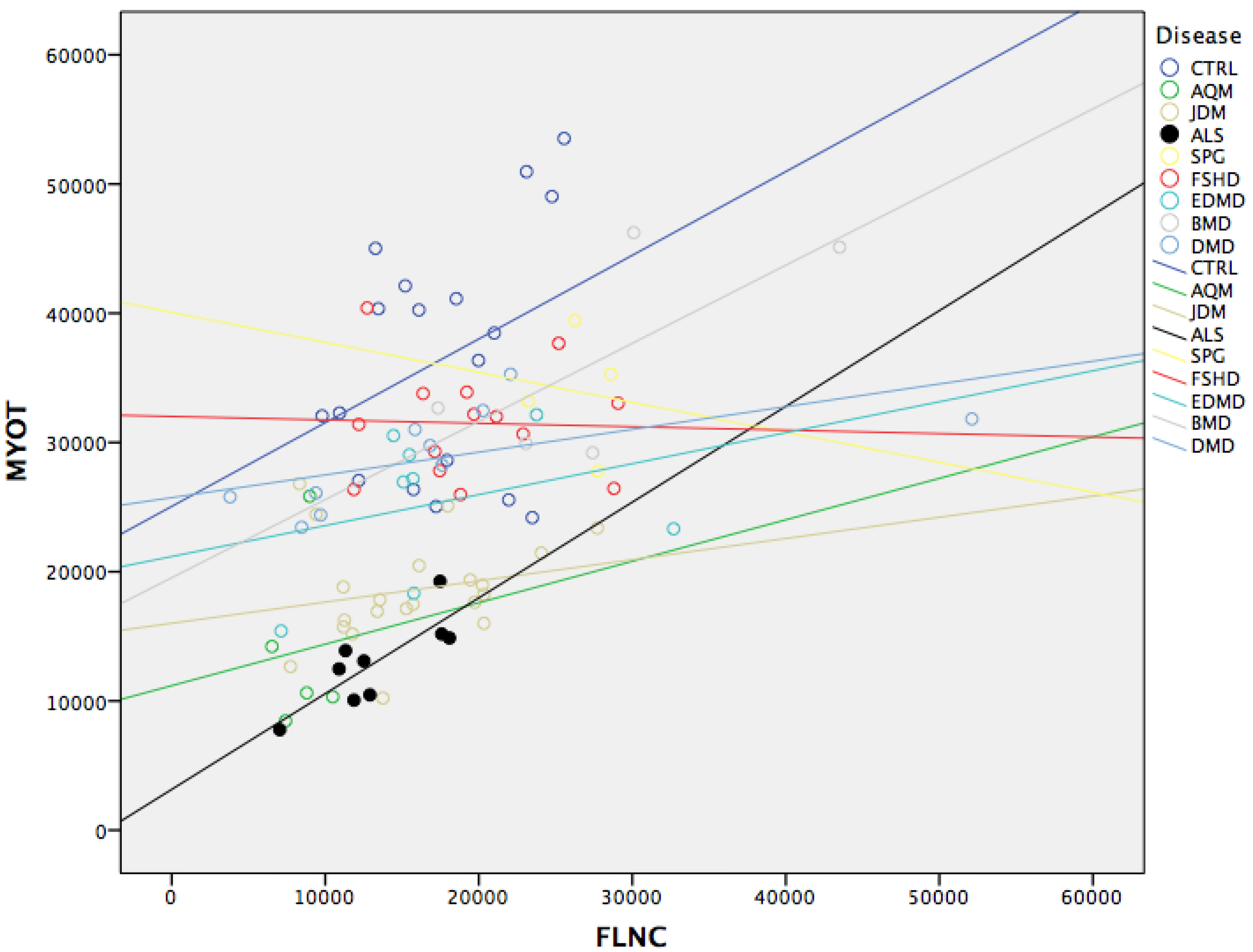
3.4. MYOT and FLNC Correlation
4. Discussion
Author Contributions
Conflicts of Interest
Abbreviations
| MYOT | myotilin |
| FLNC | filamin C, gamma |
| AQM | Acute Quadriplegic Myopathy |
| JDM | Juvenile Dermatomyositis |
| ALS | Amyotrophic Lateral Sclerosis |
| SPG4 | Spastic Paraplegia |
| FSHD | Fascioscapulohumeral Muscular Dystrophy |
| EDMD | Emery Dreifuss Muscular Dystrophy |
| BMD | Becker Muscular Dystrophy |
| DMD | Duchenne Muscular Dystrophy |
| GIANT | Genome-scale Integrated Analysis of gene Networks in Tissues |
| IH | Immunohistochemestry |
| FATZ | filamin-, actinin-, and telethonin-binding protein of the Z-disc |
| MLP | muscle LIM protein |
| ZASP | Z-band alternatively spliced PDZ-motif-containing protein |
References
- Grewal, P.K.; Hewitt, J.E. Glycosylation defects: A new mechanism for muscular dystrophy? Hum. Mol. Genet. 2003, 12, R259–R264. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.P.; Henriques, A. A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Antoine, J.C.; Camdessanche, J.P. Paraneoplastic disorders of the peripheral nervous system. Presse Med. 2013, 42, e235–e244. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.G.; Burns, A.S.; O’Ferrall, D.M.; Ditunno, J.F., Jr. Prevalence of upper motor neuron vs. lower motor neuron lesions in complete lower thoracic and lumbar spinal cord injuries. J. Spinal Cord Med. 2002, 25, 289–292. [Google Scholar] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Ann. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V. Current concepts in dystrophinopathies. Indian J. Pediatr. 2015, 82, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Lunt, P.W.; Compston, D.A.; Harper, P.S. Estimation of age dependent penetrance in facioscapulohumeral muscular dystrophy by minimising ascertainment bias. J. Med. Genet. 1989, 26, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; van der Maarel, S.; Padberg, G.W.; van Engelen, B.G. 171st ENMC international workshop: standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. NMD 2010, 20, 471–475. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M. Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 495–563. [Google Scholar] [CrossRef] [PubMed]
- De Smet, L. Emery-dreifuss muscular dystrophy. Genet. Counsel. 2004, 15, 91–94. [Google Scholar] [PubMed]
- Carstens, P.O.; Schmidt, J. Diagnosis, pathogenesis and treatment of myositis: Recent advances. Clin. Exp. Immunol. 2014, 175, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Rider, L.G.; Miller, F.W. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA 2011, 305, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Inflammatory muscle diseases. N. Engl. J. Med. 2015, 373, 393–394. [Google Scholar] [PubMed]
- Mira-Avendano, I.C.; Parambil, J.G.; Yadav, R.; Arrossi, V.; Xu, M.; Chapman, J.T.; Culver, D.A. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir. Med. 2013, 107, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Otey, C.A.; Rachlin, A.; Moza, M.; Arneman, D.; Carpen, O. The palladin/myotilin/myopalladin family of actin-associated scaffolds. Int. Rev. Cytol. 2005, 246, 31–58. [Google Scholar] [PubMed]
- Salmikangas, P.; Mykkanen, O.M.; Gronholm, M.; Heiska, L.; Kere, J.; Carpen, O. Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum. Mol. Genet. 1999, 8, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, P.F.; Wiesner, S.; Salmikangas, P.; Auerbach, D.; Himmel, M.; Kempa, S.; Hayess, K.; Pacholsky, D.; Taivainen, A.; Schroder, R.; et al. Indications for a novel muscular dystrophy pathway. Gamma-filamin, the muscle-specific filamin isoform, interacts with myotilin. J. Cell Biol. 2000, 151, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Gontier, Y.; Taivainen, A.; Fontao, L.; Sonnenberg, A.; van der Flier, A.; Carpen, O.; Faulkner, G.; Borradori, L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J. Cell Sci. 2005, 118, 3739–3749. [Google Scholar] [CrossRef] [PubMed]
- von Nandelstadh, P.; Gronholm, M.; Moza, M.; Lamberg, A.; Savilahti, H.; Carpen, O. Actin-organising properties of the muscular dystrophy protein myotilin. Exp. Cell Res. 2005, 310, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.; van der Ven, P.F.; Stocklein, W.; Furst, D.O. The limits of promiscuity: Isoform-specific dimerization of filamins. Biochemistry 2003, 42, 430–439. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, A.; Sonnenberg, A. Structural and functional aspects of filamins. Biochim. Biophys. 2001, 1538, 99–117. [Google Scholar] [CrossRef]
- Di Rosa, M.; Tibullo, D.; Saccone, S.; Distefano, G.; Basile, M.S.; Di Raimondo, F.; Malaguarnera, L. CHI3L1 nuclear localization in monocyte derived dendritic cells. Immunobiology 2016, 221, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.G.; Chan, Y.M.; Hack, A.A.; Brosius, M.; Rajala, M.; Lidov, H.G.; McNally, E.M.; Watkins, S.; Kunkel, L.M. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J. Cell Biol. 2000, 148, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, P.F.; Obermann, W.M.; Lemke, B.; Gautel, M.; Weber, K.; Furst, D.O. Characterization of muscle filamin isoforms suggests a possible role of γ-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil. Cytoskelet. 2000, 45, 149–162. [Google Scholar] [CrossRef]
- Takada, F.; Vander Woude, D.L.; Tong, H.Q.; Thompson, T.G.; Watkins, S.C.; Kunkel, L.M.; Beggs, A.H. Myozenin: An α-actinin- and γ-filamin-binding protein of skeletal muscle Z lines. Proc. Natl Acad Sci. USA 2001, 98, 1595–1600. [Google Scholar] [PubMed]
- Xie, Z.; Xu, W.; Davie, E.W.; Chung, D.W. Molecular cloning of human ABPl, an actin-binding protein homologue. Biochem. Biophys. Res. Commun. 1998, 251, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of filaminc (FLNC) results in severe defects in myogenesis and myotube structure. Mol. Cell. Biol. 2006, 26, 6522–6534. [Google Scholar] [CrossRef] [PubMed]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-myoD pathways in muscle regeneration. Brain A J. Neurol. 2006, 129, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Rockberg, J.; Uhlen, M. Prediction of antibody response using recombinant human protein fragments as antigen. Protein Sci. A Publ. Protein Soc. 2009, 18, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Ponten, F.; Gry, M.; Fagerberg, L.; Lundberg, E.; Asplund, A.; Berglund, L.; Oksvold, P.; Bjorling, E.; Hober, S.; Kampf, C.; et al. A global view of protein expression in human cells, tissues, and organs. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, C. The potential clinical impact of the tissue-based map of the human proteome. Expert Rev. Proteom. 2015, 12, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.X.; Pletscher-Frankild, S.; Tsafou, K.; Stolte, C.; O’Donoghue, S.I.; Schneider, R.; Jensen, L.J. COMPARTMENTS: Unification and visualization of protein subcellular localization evidence. Database 2014. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.A.; Horrigan, S.K.; Salmikangas, P.; Torian, U.M.; Viles, K.D.; Dancel, R.; Tim, R.W.; Taivainen, A.; Bartoloni, L.; Gilchrist, J.M.; et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. 2000, 9, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Clemen, C.S.; Olive, M.; Ferrer, I.; Goudeau, B.; Roth, U.; Badorf, P.; Wattjes, M.P.; Lutterbey, G.; Kral, T.; et al. Different early pathogenesis in myotilinopathy compared to primary desminopathy. Neuromuscul. Disord. NMD 2006, 16, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Penisson-Besnier, I.; Talvinen, K.; Dumez, C.; Vihola, A.; Dubas, F.; Fardeau, M.; Hackman, P.; Carpen, O.; Udd, B. Myotilinopathy in a family with late onset myopathy. Neuromuscul. Disord. NMD 2006, 16, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, E.; Hoffman, E.P. Limb-Girdle Muscular Dystrophy Overview. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Fong, C.T., Mefford, H.C., Smith, R.J.H., et al., Eds.; GeneReviews: Seattle, WA, USA, 1993. [Google Scholar]
- Polge, C.; Attaix, D.; Taillandier, D. Role of E2-Ub-conjugating enzymes during skeletal muscle atrophy. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Salmikangas, P.; van der Ven, P.F.; Lalowski, M.; Taivainen, A.; Zhao, F.; Suila, H.; Schroder, R.; Lappalainen, P.; Furst, D.O.; Carpen, O. Myotilin, the limb-girdle muscular dystrophy 1A (LGMD1A) protein, cross-links actin filaments and controls sarcomere assembly. Hum. Mol. Genet. 2003, 12, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.T.; Kanner, S.B.; Aruffo, A. Filamin binds to the cytoplasmic domain of the β1-integrin. Identification of amino acids responsible for this interaction. J. Biol. Chem. 1998, 273, 23304–23312. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.; Pallavicini, A.; Comelli, A.; Salamon, M.; Bortoletto, G.; Ievolella, C.; Trevisan, S.; Kojic, S.; Dalla Vecchia, F.; Laveder, P.; et al. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J. Biol. Chem. 2000, 275, 41234–41242. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.; van der Ven, P.F.; Vakeel, P.; Albinus, B.; Simonis, D.; Bendas, G.; Schenk, J.A.; Micheel, B.; Kley, R.A.; Furst, D.O. The sarcomeric Z-disc component myopodin is a multiadapter protein that interacts with filamin and α-actinin. Eur. J. Cell Biol. 2010, 89, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Kley, R.A.; Hellenbroich, Y.; van der Ven, P.F.; Furst, D.O.; Huebner, A.; Bruchertseifer, V.; Peters, S.A.; Heyer, C.M.; Kirschner, J.; Schroder, R.; et al. Clinical and morphological phenotype of the filamin myopathy: A Study of 31 German Patients. Brain A J. Neurol. 2007, 130, 3250–3264. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Hong, D.; Zhang, W.; Wang, Z.; Yuan, Y. A novel heterozygous deletion-insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large chinese family. Neuromuscul. Disord. NMD 2010, 20, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Shatunov, A.; Olive, M.; Odgerel, Z.; Stadelmann-Nessler, C.; Irlbacher, K.; van Landeghem, F.; Bayarsaikhan, M.; Lee, H.S.; Goudeau, B.; Chinnery, P.F.; et al. In-frame deletion in the seventh immunoglobulin-like repeat of filamin c in a family with myofibrillar myopathy. Eur. J. Hum. Genet. EJHG 2009, 17, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Vorgerd, M.; van der Ven, P.F.; Bruchertseifer, V.; Lowe, T.; Kley, R.A.; Schroder, R.; Lochmuller, H.; Himmel, M.; Koehler, K.; Furst, D.O.; et al. A mutation in the dimerization domain of filamin C causes a novel type of autosomal dominant myofibrillar myopathy. Am. J. Hum. Genet. 2005, 77, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of als-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanfilippo, C.; Di Rosa, M. Modulation of Myotilin and Fylamin C in Various Muscle Diseases: A Microarray Analysis. J. Funct. Morphol. Kinesiol. 2016, 1, 90-101. https://doi.org/10.3390/jfmk1010090
Sanfilippo C, Di Rosa M. Modulation of Myotilin and Fylamin C in Various Muscle Diseases: A Microarray Analysis. Journal of Functional Morphology and Kinesiology. 2016; 1(1):90-101. https://doi.org/10.3390/jfmk1010090
Chicago/Turabian StyleSanfilippo, Cristina, and Michelino Di Rosa. 2016. "Modulation of Myotilin and Fylamin C in Various Muscle Diseases: A Microarray Analysis" Journal of Functional Morphology and Kinesiology 1, no. 1: 90-101. https://doi.org/10.3390/jfmk1010090