
Alcohol Inhibits Organic Dust-Induced ICAM-1 Expression on Bronchial Epithelial Cells

 and
and 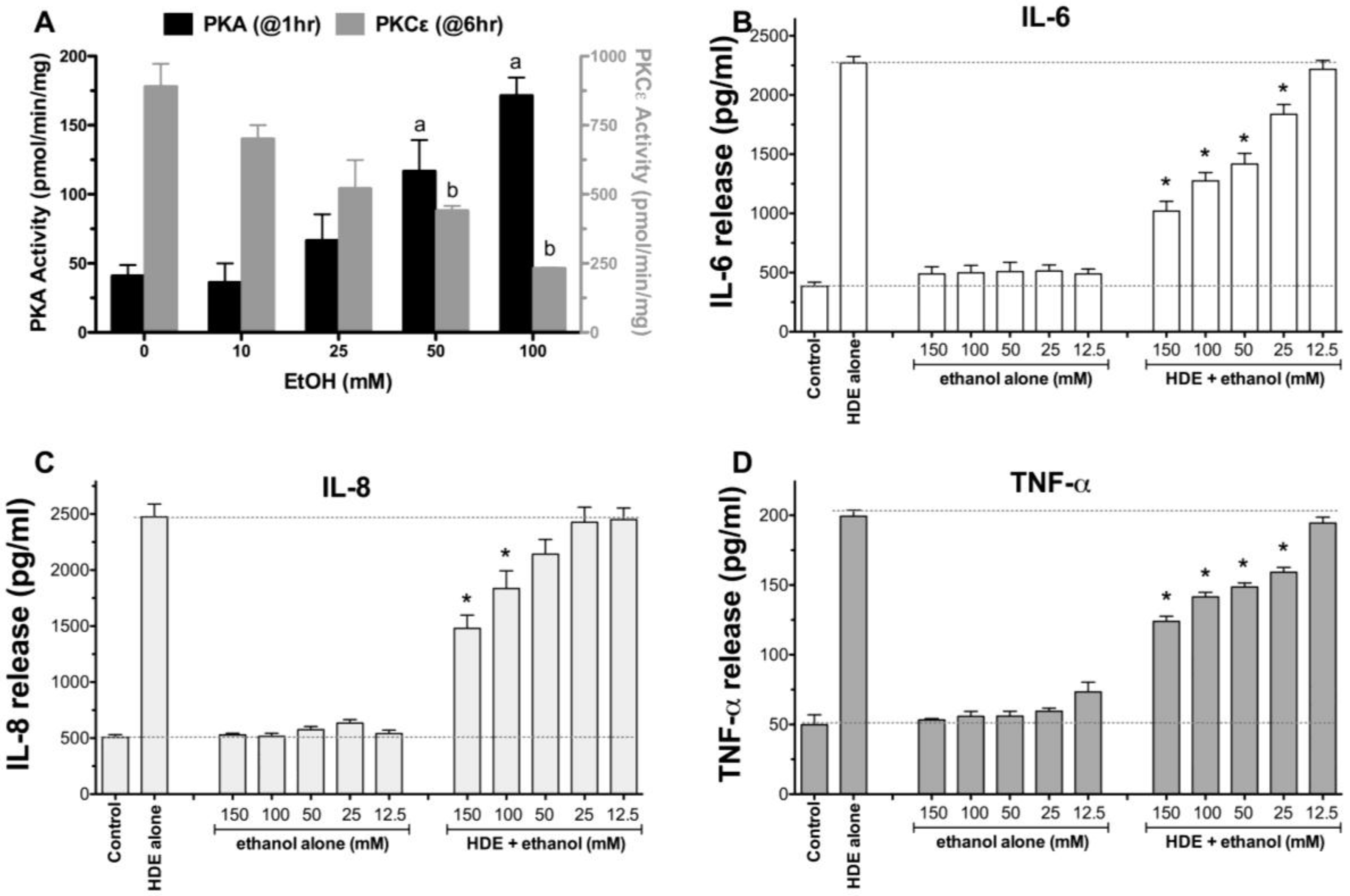{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
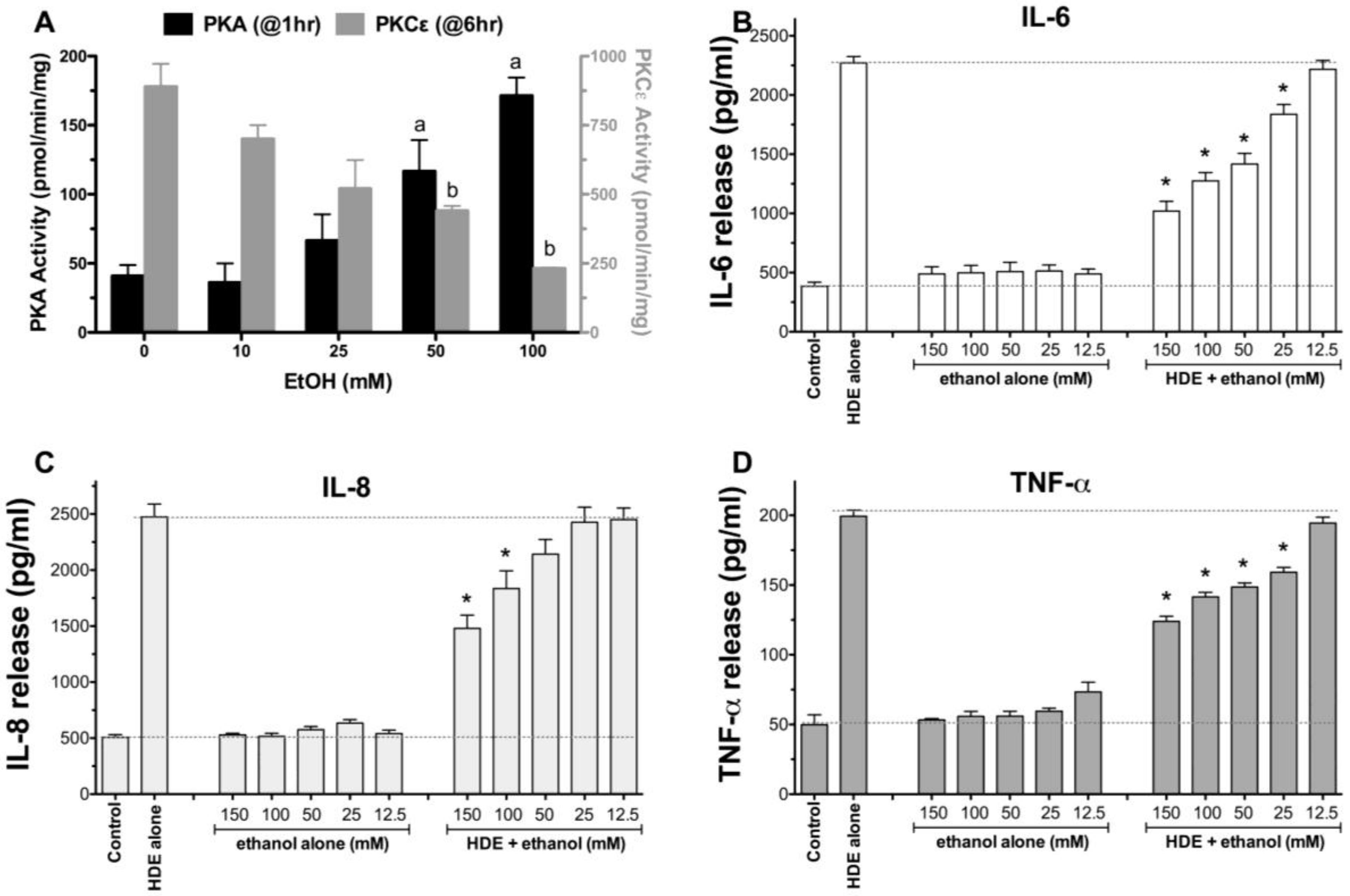
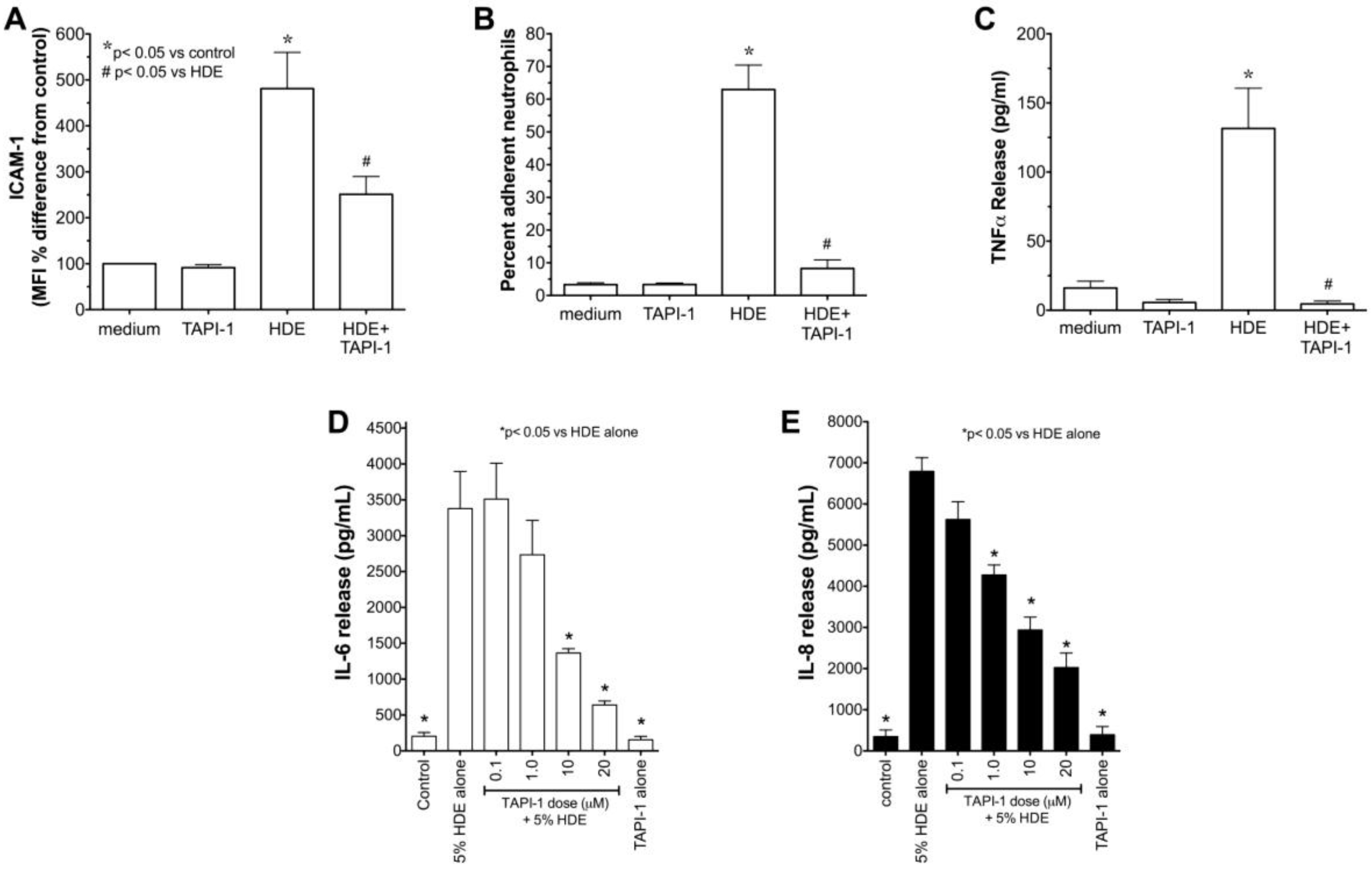
2.1. Alcohol Dose-Dependently Alters HDE-Stimulated PKC Activity and Cytokine Release
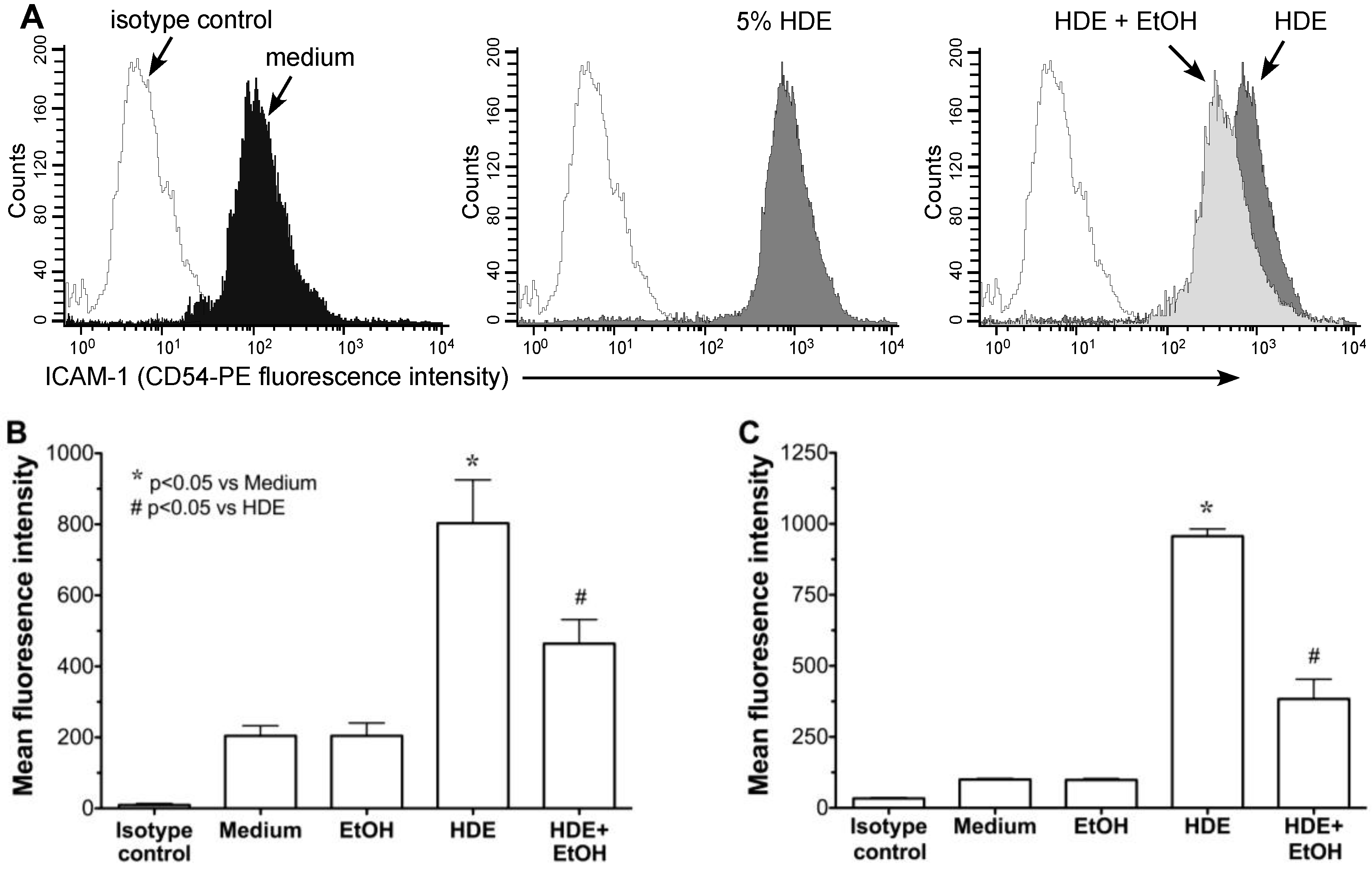
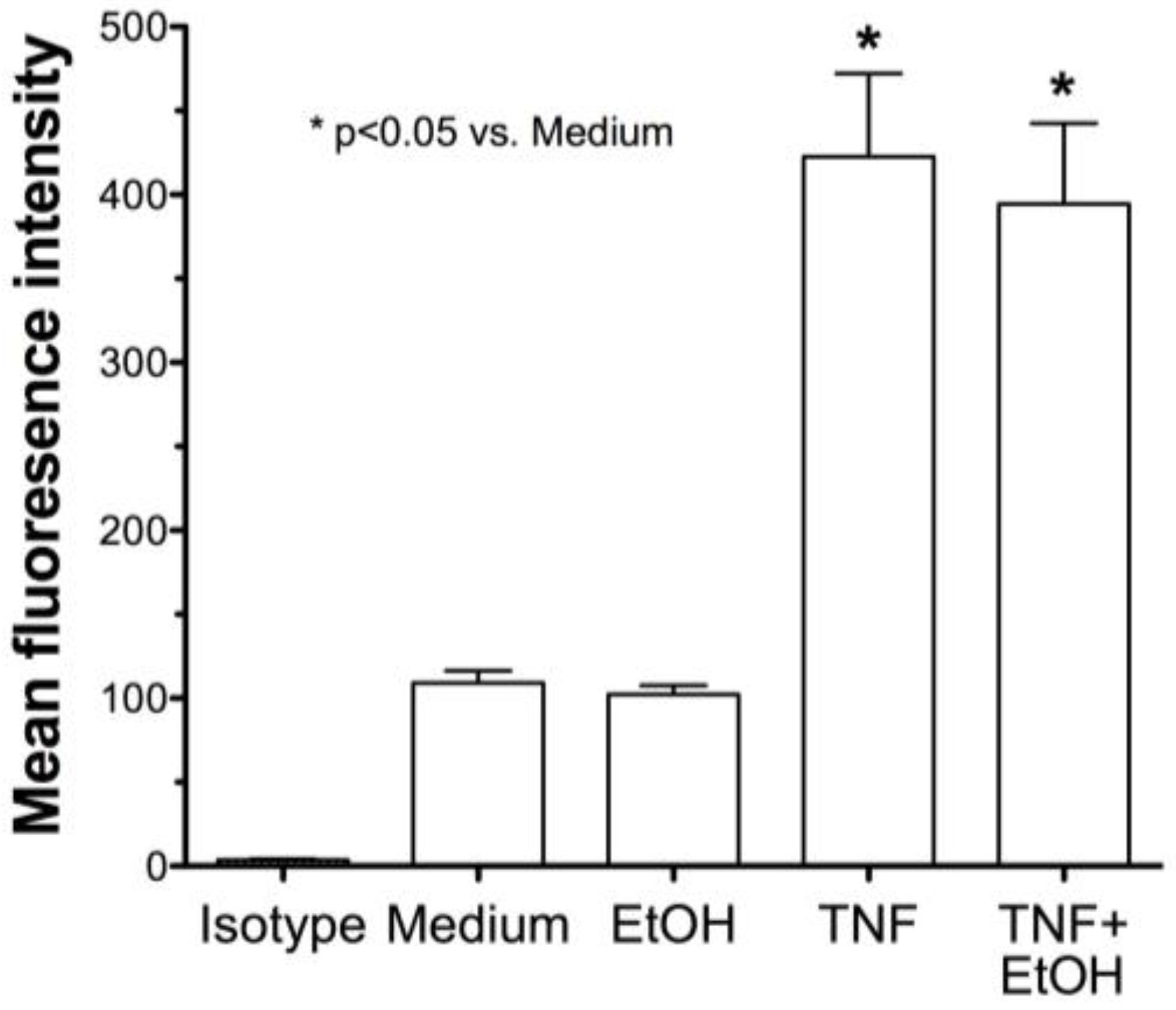
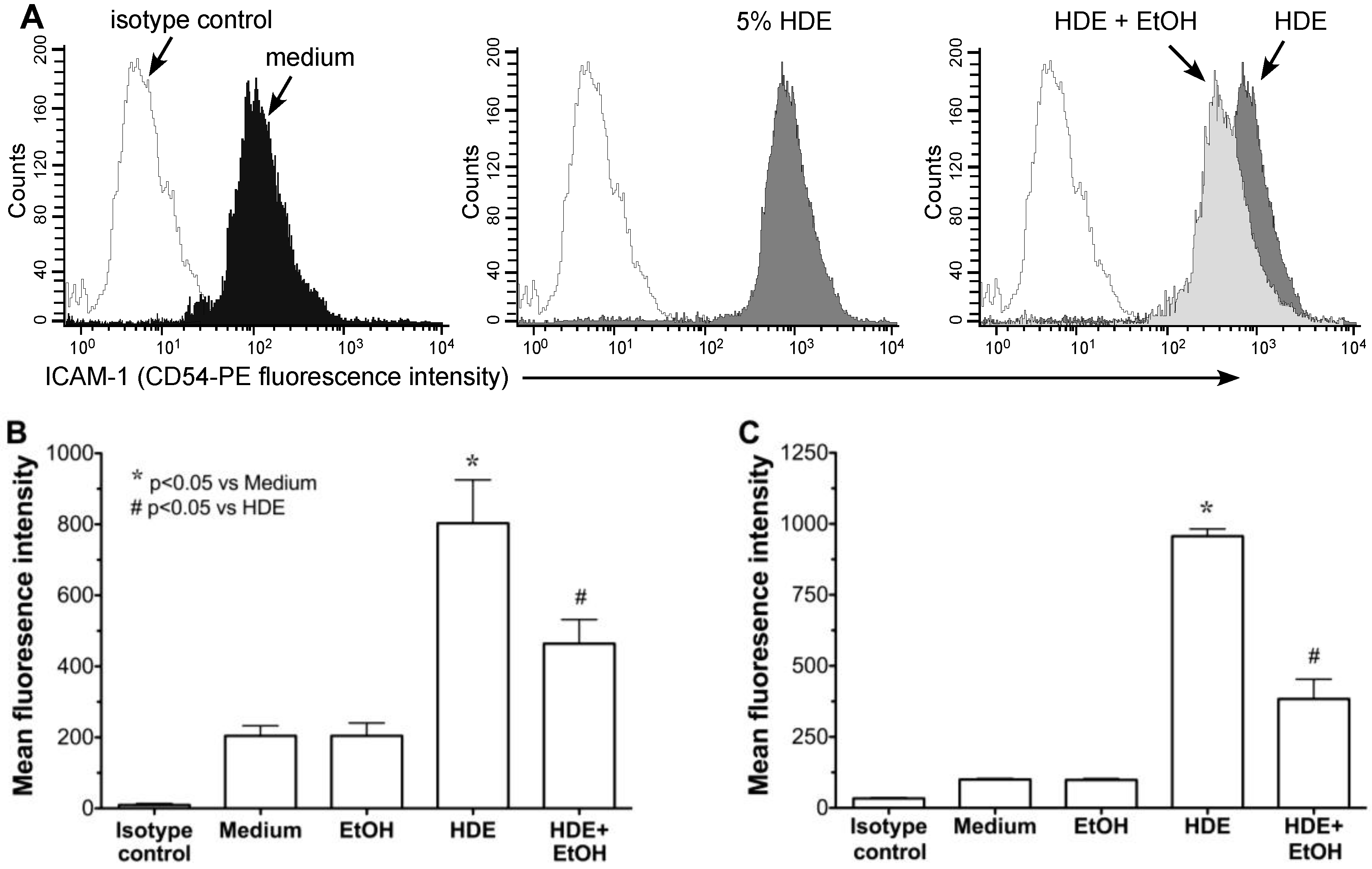
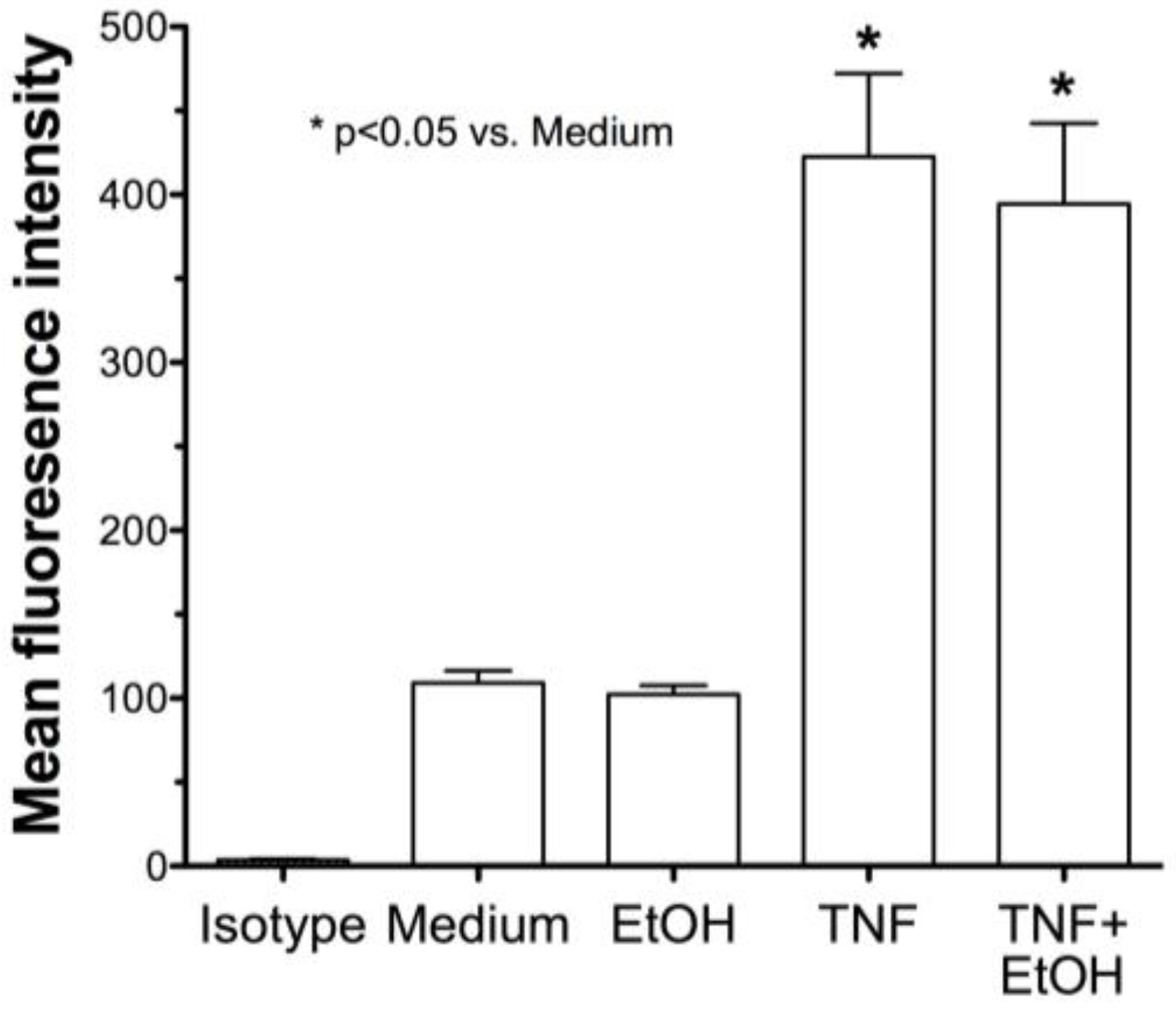
2.2. Alcohol Exposure Decreases HDE-Mediated ICAM-1 Expression
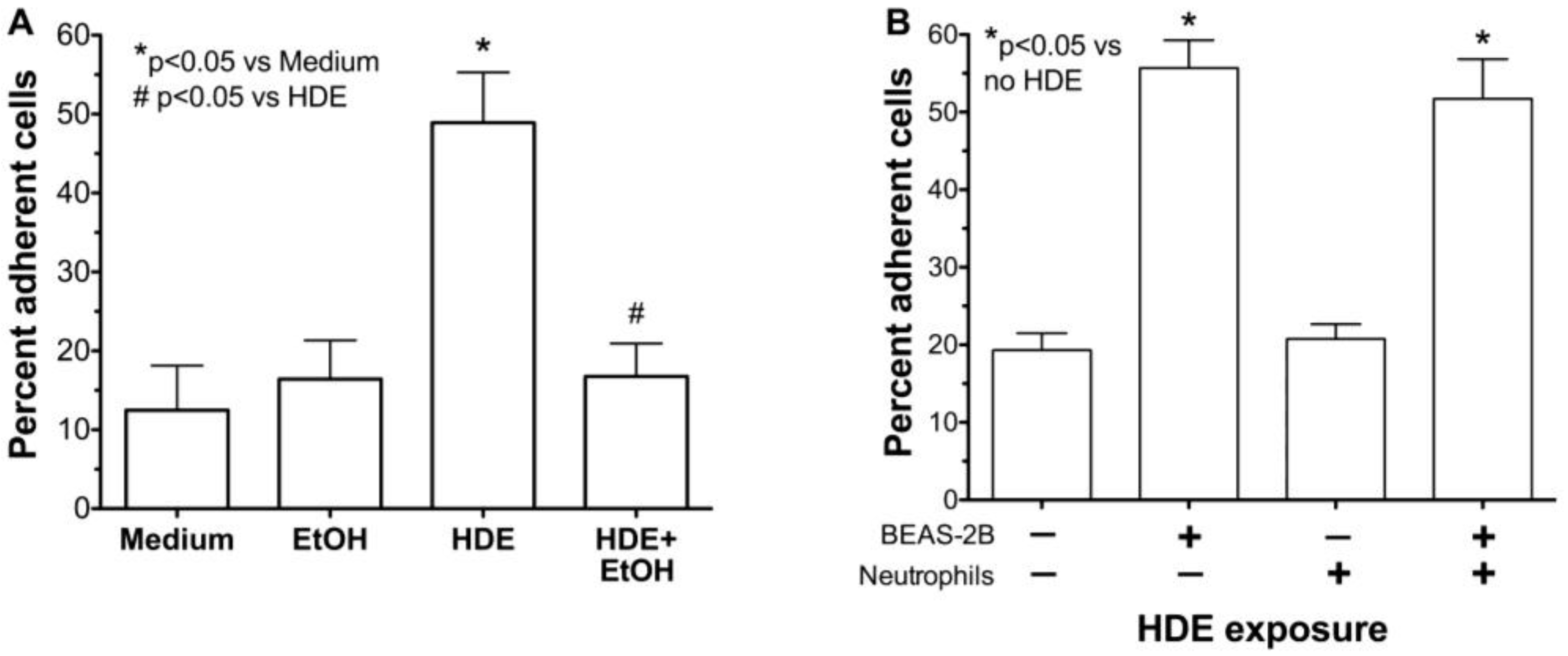
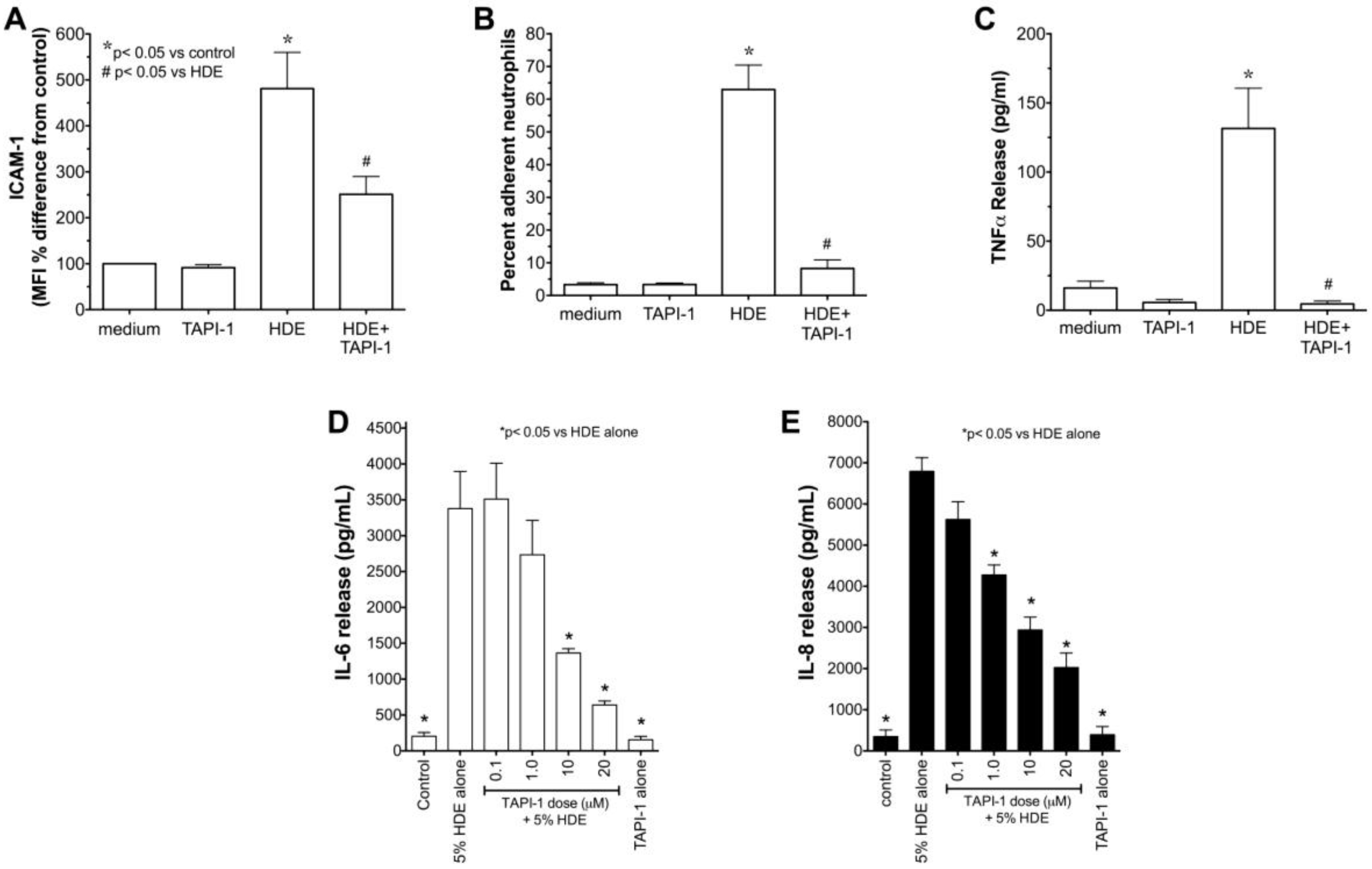
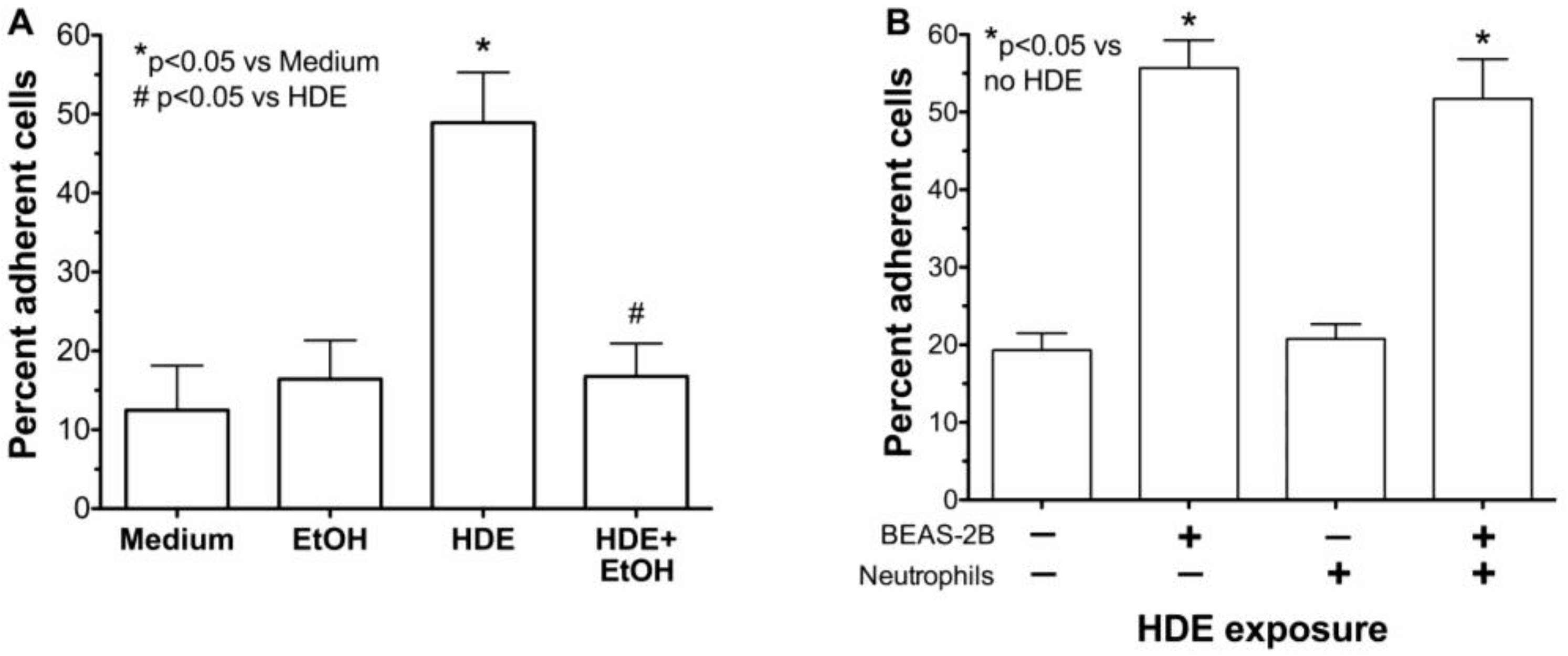
2.3. Alcohol Decreases HDE-Mediated Neutrophil Adhesion
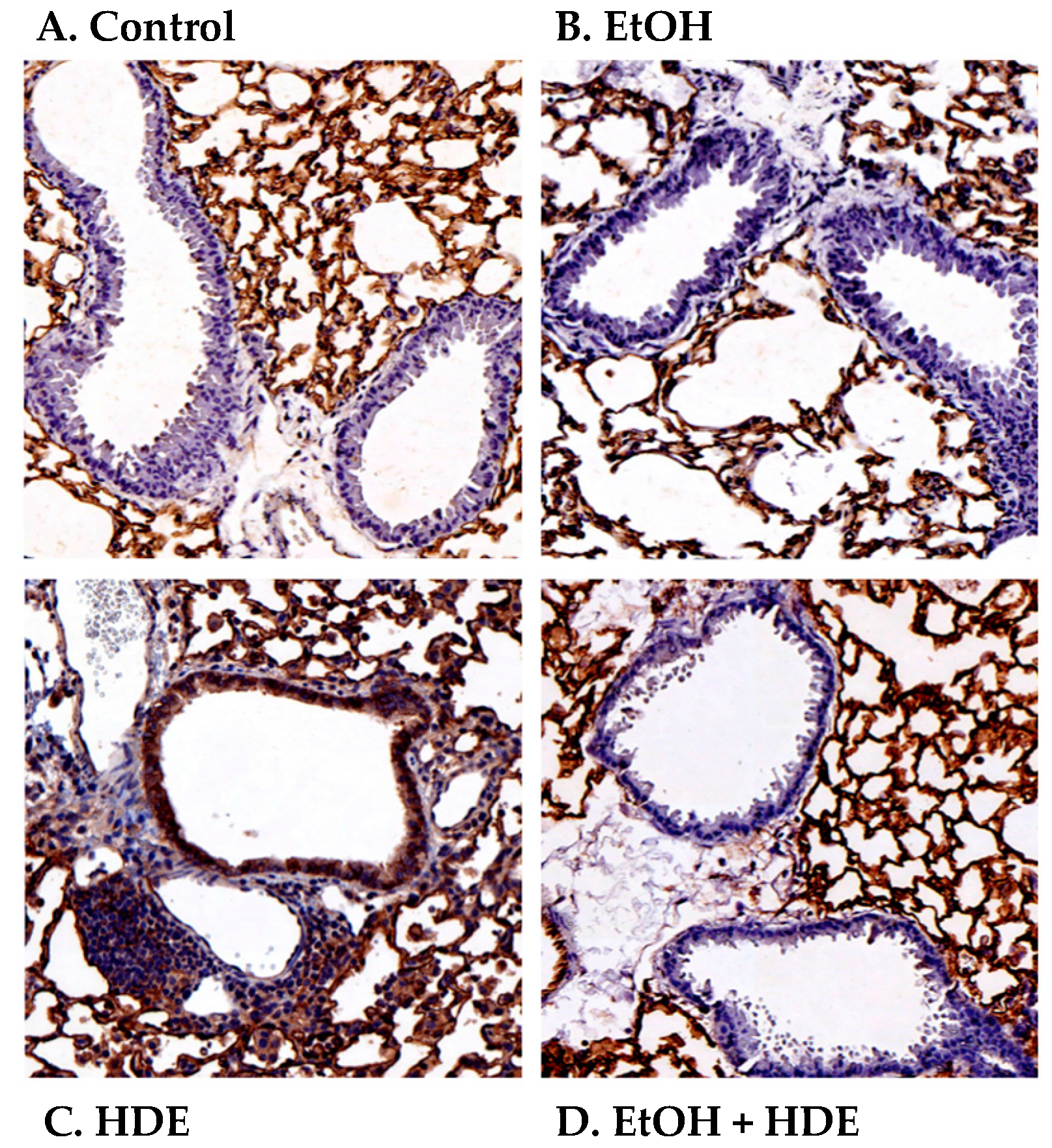
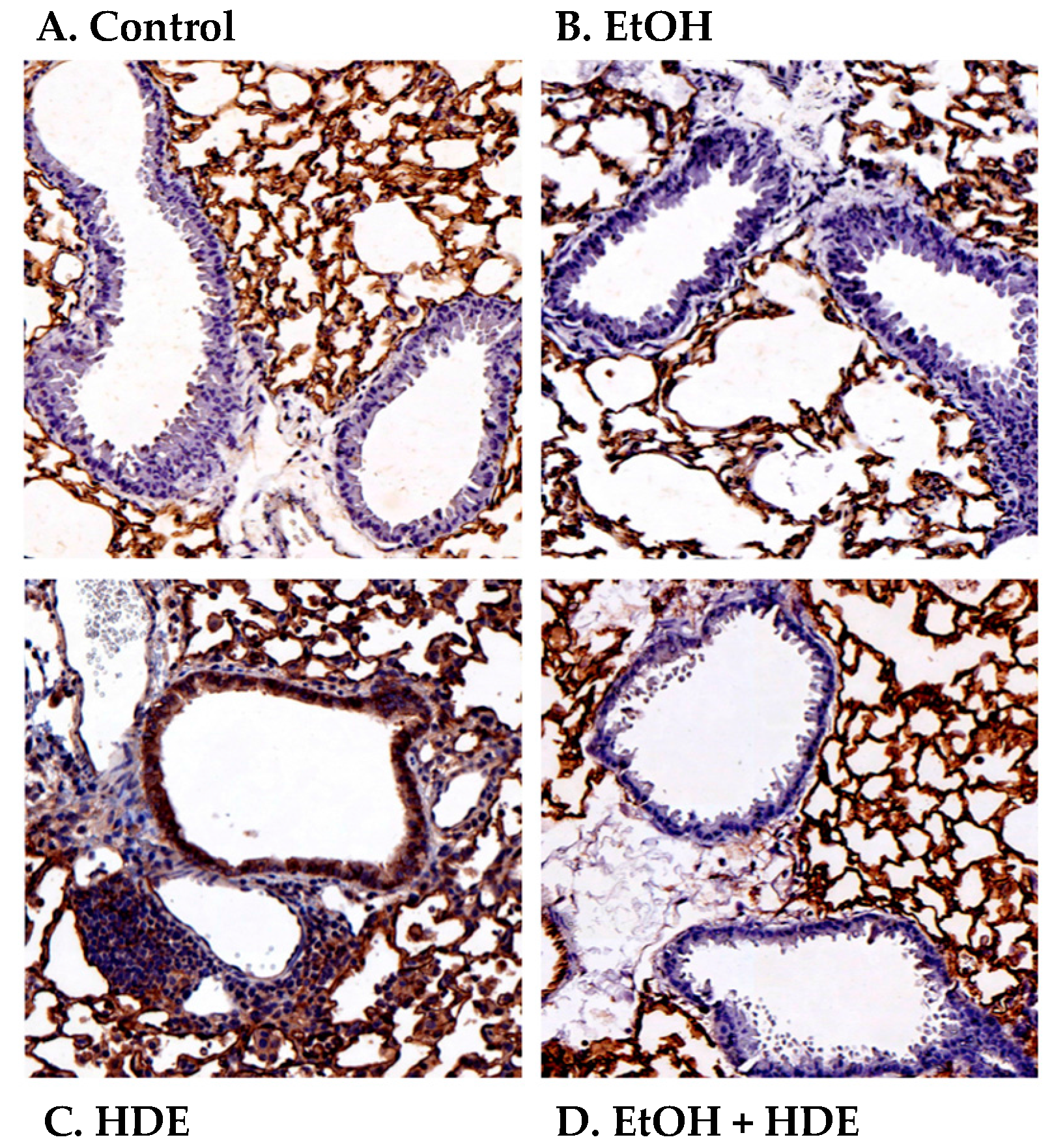
2.4. Alcohol Decreases In Vivo HDE-Mediated ICAM-1 Expression in Murine Lung Tissue
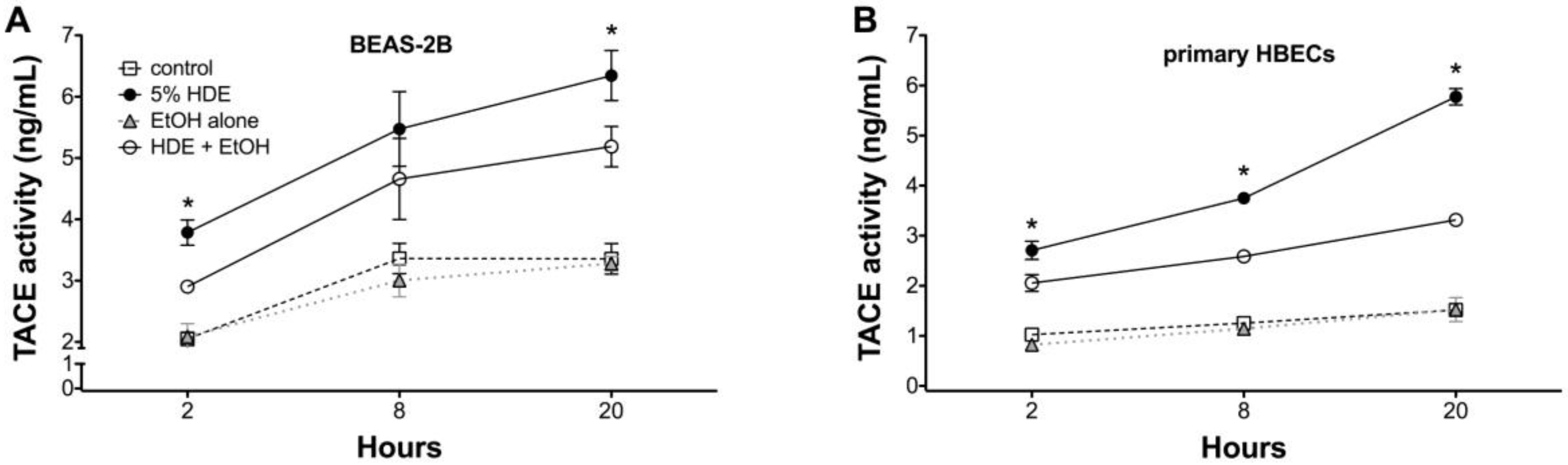
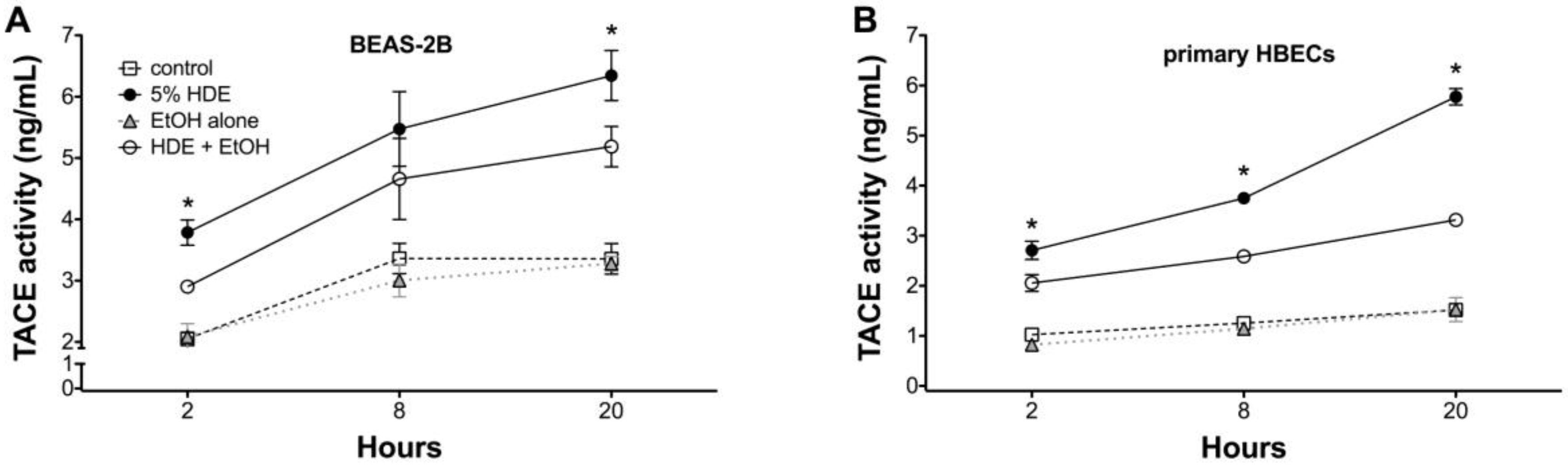
2.5. Alcohol Inhibits TACE Activity in Primary HBEC
3. Discussion
4. Materials and Methods
4.1. Cell Preparation
4.2. Preparation of HDE
4.3. Flow Cytometry Analysis for Epithelial Cells
4.4. Neutrophil Preparation
4.5. Neutrophil Adhesion Assay
4.6. Protein Kinase Activity Assay
4.7. Cytokine Release Assay
4.8. Animal Model and Exposure
4.9. Immunohistochemistry
4.10. TACE Activity Assay
4.11. Cell Viability Assay
4.12. Materials
4.13 Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- May, S.; Romberger, D.J.; Poole, J.A. Respiratory health effects of large animal farming environments. J. Toxicol. Environ. Health Part B 2012, 15, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Stemler, F.W.; Craig, F.N. Effects of respiratory equipment on endurance in hard work. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1977, 42, 28–32. [Google Scholar] [PubMed]
- Donham, K.J.; Cumro, D.; Reynolds, S.J.; Merchant, J.A. Dose-response relationships between occupational aerosol exposures and cross-shift declines of lung function in poultry workers: Recommendations for exposure limits. J. Occup. Environ. Med. 2000, 42, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Kaphalia, L.; Calhoun, W.J. Alcoholic lung injury: Metabolic, biochemical and immunological aspects. Toxicol. Lett. 2013, 222, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wheeler, K.; Bai, L.; Stallones, L.; Dong, Y.; Ge, J.; Xiang, H. Alcohol consumption and work-related injuries among farmers in Heilongjiang province, people’s republic of China. Am. J. Ind. Med. 2010, 53, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Stallones, L.; Xiang, H. Alcohol consumption patterns and work-related injuries among Colorado farm residents. Am. J. Prev. Med. 2003, 25, 25–30. [Google Scholar] [CrossRef]
- Brumby, S.; Kennedy, A.; Chandrasekara, A. Alcohol consumption, obesity, and psychological distress in farming communities-an australian study. J. Rural Health 2013, 29, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Pratt, D.S. Occupational health and the rural worker: Agriculture, mining, and logging. J. Rural Health 1990, 6, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Anthenelli, R.; Grandison, L. Effects of stress on alcohol consumption. Alcohol Res. 2012, 34, 381–382. [Google Scholar] [PubMed]
- Anthenelli, R.M. Overview: Stress and alcohol use disorders revisited. Alcohol Res. 2012, 34, 386–390. [Google Scholar] [PubMed]
- Wyatt, T.A.; Sisson, J.H.; Von Essen, S.G.; Poole, J.A.; Romberger, D.J. Exposure to hog barn dust alters airway epithelial ciliary beating. Eur. Respir. J. 2008, 31, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Slager, R.E.; Allen-Gipson, D.S.; Sammut, A.; Heires, A.; Devasure, J.; Von Essen, S.G.; Romberger, D.J.; Wyatt, T.A. Hog barn dust slows airway epithelial cell migration in vitro through a PKCα-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1469–L1474. [Google Scholar] [CrossRef] [PubMed]
- Romberger, D.J.; Bodlak, V.; Von Essen, S.G.; Mathisen, T.; Wyatt, T.A. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J. Appl. Physiol. 2002, 93, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Slager, R.E.; Heires, A.J.; Devasure, J.M.; Vonessen, S.G.; Poole, J.A.; Romberger, D.J. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am. J. Respir. Cell Mol. Biol. 2010, 42, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Poole, J.A.; Nordgren, T.M.; DeVasure, J.M.; Heires, A.J.; Bailey, K.L.; Romberger, D.J. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L643–L651. [Google Scholar] [CrossRef] [PubMed]
- Gerald, C.L.; Romberger, D.J.; DeVasure, J.M.; Khazanchi, R.; Nordgren, T.M.; Heires, A.J.; Sisson, J.H.; Wyatt, T.A. Alcohol decreases organic dust-stimulated airway epithelial TNF alpha through a nitric oxide and PKA-mediated inhibition of TACE. Alcoholism Clin. Exp. Res. 2016, 40, 273–283. [Google Scholar] [CrossRef] [PubMed]
- McCaskill, M.L.; Romberger, D.J.; Devasure, J.; Boten, J.; Sisson, J.H.; Bailey, K.L.; Poole, J.A.; Wyatt, T.A. Alcohol exposure alters mouse lung inflammation in response to inhaled dust. Nutrients 2012, 4, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Sisson, J.H.; May, K.; Wyatt, T.A. Nitric oxide-dependent ethanol stimulation of ciliary motility is linked to cAMP-dependent protein kinase (PKA) activation in bovine bronchial epithelium. Alcoholism Clin. Exp. Res. 1999, 23, 1528–1533. [Google Scholar] [CrossRef]
- Happel, K.I.; Nelson, S. Alcohol, immunosuppression, and the lung. Proc. Am. Thorac. Soc. 2005, 2, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Donham, K.J.; Rubino, M.; Thedell, T.D.; Kammermeyer, J. Potential health hazards to agricultural workers in swine confinement buildings. J. Occup. Med. 1977, 19, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M.; Kirychuk, S.; Drost, H.; Jacobson, L. Human health effects of dust exposure in animal confinement buildings. J. Agric. Saf. Health 2000, 6, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M. Lung involvement in organic dust exposure. Monaldi Arch. Chest Dis. 2000, 55, 62–65. [Google Scholar] [PubMed]
- Von Essen, S.G.; Auvermann, B.W. Health effects from breathing air near CAFOs for feeder cattle or hogs. J. Agromed. 2005, 10, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Goral, J.; Karavitis, J.; Kovacs, E.J. Exposure-dependent effects of ethanol on the innate immune system. Alcohol 2008, 42, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.; Kolls, J.K. Alcohol, host defence and society. Nat. Rev. Immunol. 2002, 2, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Sisson, J.H. Alcohol and airways function in health and disease. Alcohol 2007, 41, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Burnham, E.L. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit. Care Med. 2003, 31, S207–S212. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Harris, F.L.; Ping, X.D.; Gauthier, T.W. Chronic ethanol ingestion and the risk of acute lung injury: A role for glutathione availability? Alcohol 2004, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.E.; Happel, K.I.; Zhang, P.; Kolls, J.K.; Nelson, S. Focus on: Alcohol and the immune system. Alcohol Res. Health 2010, 33, 97–108. [Google Scholar] [PubMed]
- Messingham, K.A.; Faunce, D.E.; Kovacs, E.J. Alcohol, injury, and cellular immunity. Alcohol 2002, 28, 137–149. [Google Scholar] [CrossRef]
- Joshi, P.C.; Guidot, D.M. The alcoholic lung: Epidemiology, pathophysiology, and potential therapies. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L813–L823. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.K.; Sisson, J.H.; Wyatt, T.A. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am. J. Respir. Cell Mol. Biol. 2007, 36, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.D.; Kovacs, E.J. Organ-specific inflammation following acute ethanol and burn injury. J. Leukoc. Biol. 2008, 84, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Ostrovsky, Y. Endogenous ethanol—its metabolic, behavioral and biomedical significance. Alcohol 1986, 3, 239–247. [Google Scholar] [CrossRef]
- Johnson, R.A.; Noll, E.C.; Rodney, W.M. Survival after a serum ethanol concentration of 1 1/2%. Lancet 1982, 2, 1394. [Google Scholar] [CrossRef]
- Mathisen, T.; Von Essen, S.G.; Wyatt, T.A.; Romberger, D.J. Hog barn dust extract augments lymphocyte adhesion to human airway epithelial cells. J. Appl. Physiol. 2004, 96, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Tosi, M.F.; Stark, J.M.; Smith, C.W.; Hamedani, A.; Gruenert, D.C.; Infeld, M.D. Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: Effects on neutrophil-epithelial cell adhesion. Am. J. Respir. Cell Mol. Biol. 1992, 7, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Bloemen, P.G.; van den Tweel, M.C.; Henricks, P.A.; Engels, F.; Wagenaar, S.S.; Rutten, A.A.; Nijkamp, F.P. Expression and modulation of adhesion molecules on human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1993, 9, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Poole, J.A.; Mathisen, T.L.; Wyatt, T.A.; Von Essen, S.G.; Romberger, D.J. Toll-like receptor 2 is upregulated by hog confinement dust in an IL-6-dependent manner in the airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1049–L1054. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; Dooley, G.P.; Saito, R.; Burrell, A.M.; Bailey, K.L.; Romberger, D.J.; Mehaffy, J.; Reynolds, S.J. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J. Toxicol. Envion. Health Part A 2010, 73, 684–700. [Google Scholar] [CrossRef] [PubMed]
- Stout, S.L.; Wyatt, T.A.; Adams, J.J.; Sisson, J.H. Nitric oxide-dependent cilia regulatory enzyme localization in bovine bronchial epithelial cells. J. Histochem. Cytochem. 2007, 55, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Forget, M.A.; Sisson, J.H. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am. J. Pathol. 2003, 163, 1157–1166. [Google Scholar] [CrossRef]
- Zhang, Z.; Cork, J.; Ye, P.; Lei, D.; Schwarzenberger, P.O.; Summer, W.R.; Shellito, J.E.; Nelson, S.; Kolls, J.K. Inhibition of TNF-alpha processing and TACE-mediated ectodomain shedding by ethanol. J. Leukoc. Biol. 2000, 67, 856–862. [Google Scholar] [PubMed]
- Song, K.; Zhao, X.; Marrero, L.; Oliver, P.; Nelson, S.; Kolls, J.K. Alcohol reversibly disrupts TNF-α/TACE interactions in the cell membrane. Respir Res. 2005, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Reddel, R.R.; Gerwin, B.I.; Miyashita, M.; McMenamin, M.; Lechner, J.F.; Harris, C.C. Human bronchial epithelial cells with integrated SV40 virus T antigen genes retain the ability to undergo squamous differentiation. Differentiation 1988, 38, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Sisson, J.H.; Allen-Gipson, D.S.; McCaskill, M.K.; Boten, J.A.; DeVasure, J.M.; Bailey, K.L.; Poole, J.A. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase C epsilon-dependent manner. Am. J. Pathol. 2012, 181, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Coleman, R.A.; Zhu, X.; Alber, C.; Ballas, Z.K.; Waldschmidt, T.J.; Cook, R.T. Chronic ethanol consumption by mice results in activated splenic T cells. J. Leukoc. Biol. 2002, 72, 1109–1116. [Google Scholar] [PubMed]
- Spitzer, J.H.; Meadows, G.G. Modulation of perforin, granzyme, A.; and granzyme B in murine natural killer (NK), IL2 stimulated, N.K.; and lymphokine-activated killer cells by alcohol consumption. Cell. Immunol. 1999, 194, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; Wyatt, T.A.; Oldenburg, P.J.; Elliott, M.K.; West, W.W.; Sisson, J.H.; Von Essen, S.G.; Romberger, D.J. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1085–L1095. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyatt, T.A.; Canady, K.; Heires, A.J.; Poole, J.A.; Bailey, K.L.; Nordgren, T.M.; Romberger, D.J. Alcohol Inhibits Organic Dust-Induced ICAM-1 Expression on Bronchial Epithelial Cells. Safety 2017, 3, 5. https://doi.org/10.3390/safety3010005
Wyatt TA, Canady K, Heires AJ, Poole JA, Bailey KL, Nordgren TM, Romberger DJ. Alcohol Inhibits Organic Dust-Induced ICAM-1 Expression on Bronchial Epithelial Cells. Safety. 2017; 3(1):5. https://doi.org/10.3390/safety3010005
Chicago/Turabian StyleWyatt, Todd A., Kerry Canady, Art J. Heires, Jill A. Poole, Kristina L. Bailey, Tara M. Nordgren, and Debra J. Romberger. 2017. "Alcohol Inhibits Organic Dust-Induced ICAM-1 Expression on Bronchial Epithelial Cells" Safety 3, no. 1: 5. https://doi.org/10.3390/safety3010005