Accelerating NMR-Based Structural Studies of Proteins by Combining Amino Acid Selective Unlabeling and Fast NMR Methods


Abstract
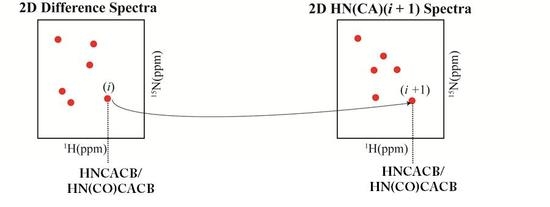
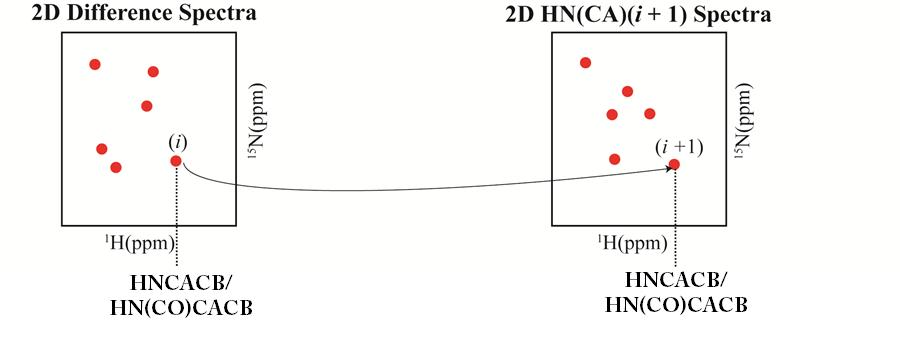
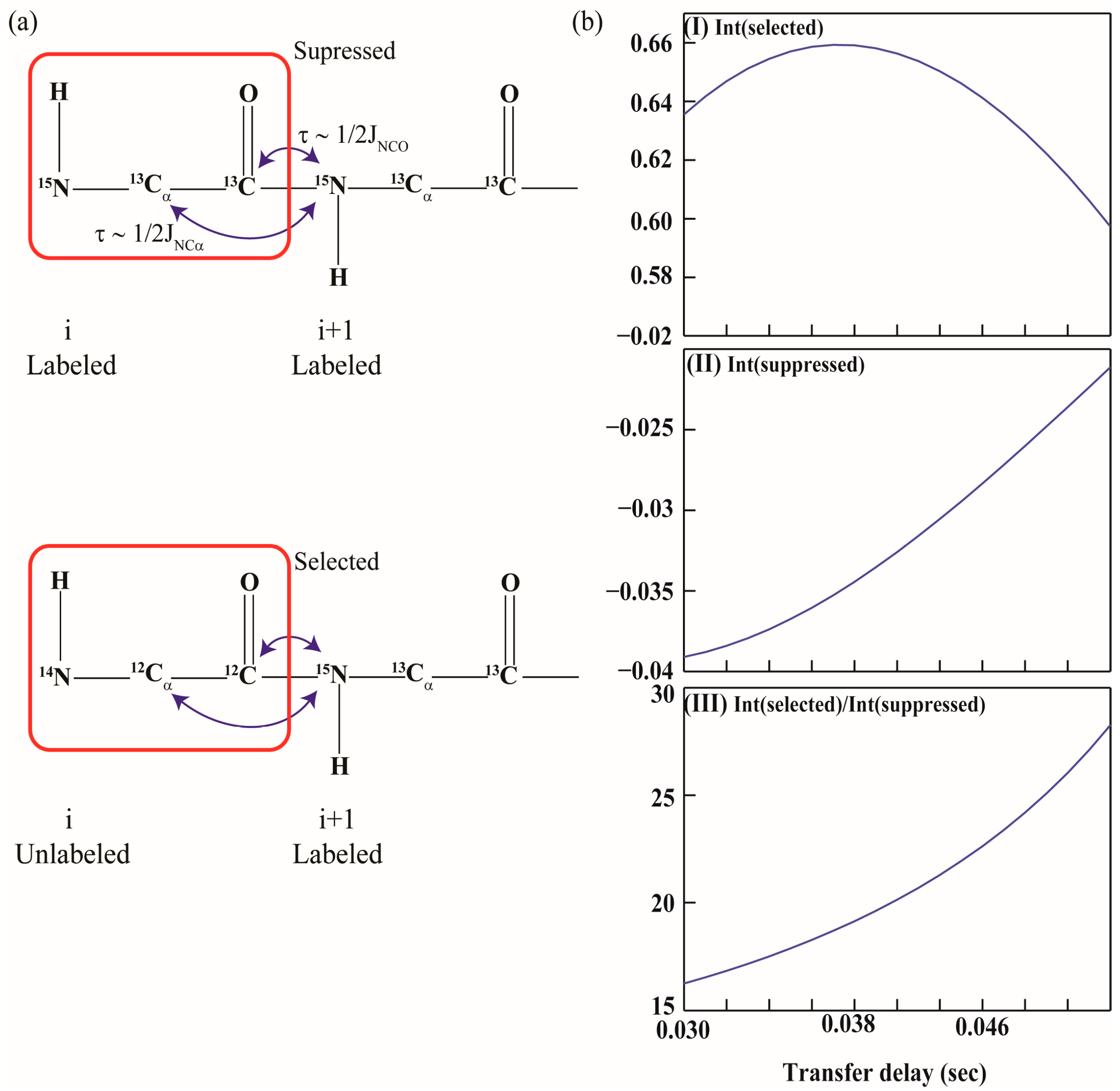
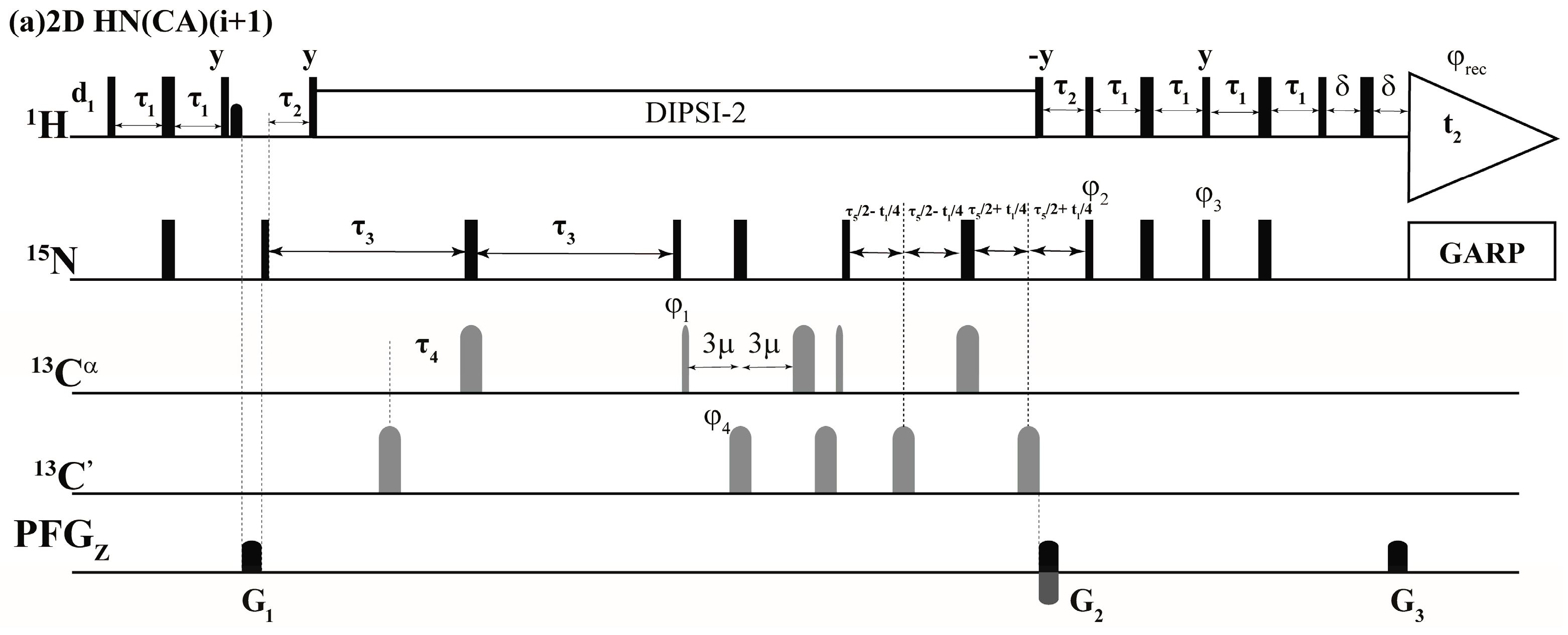
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
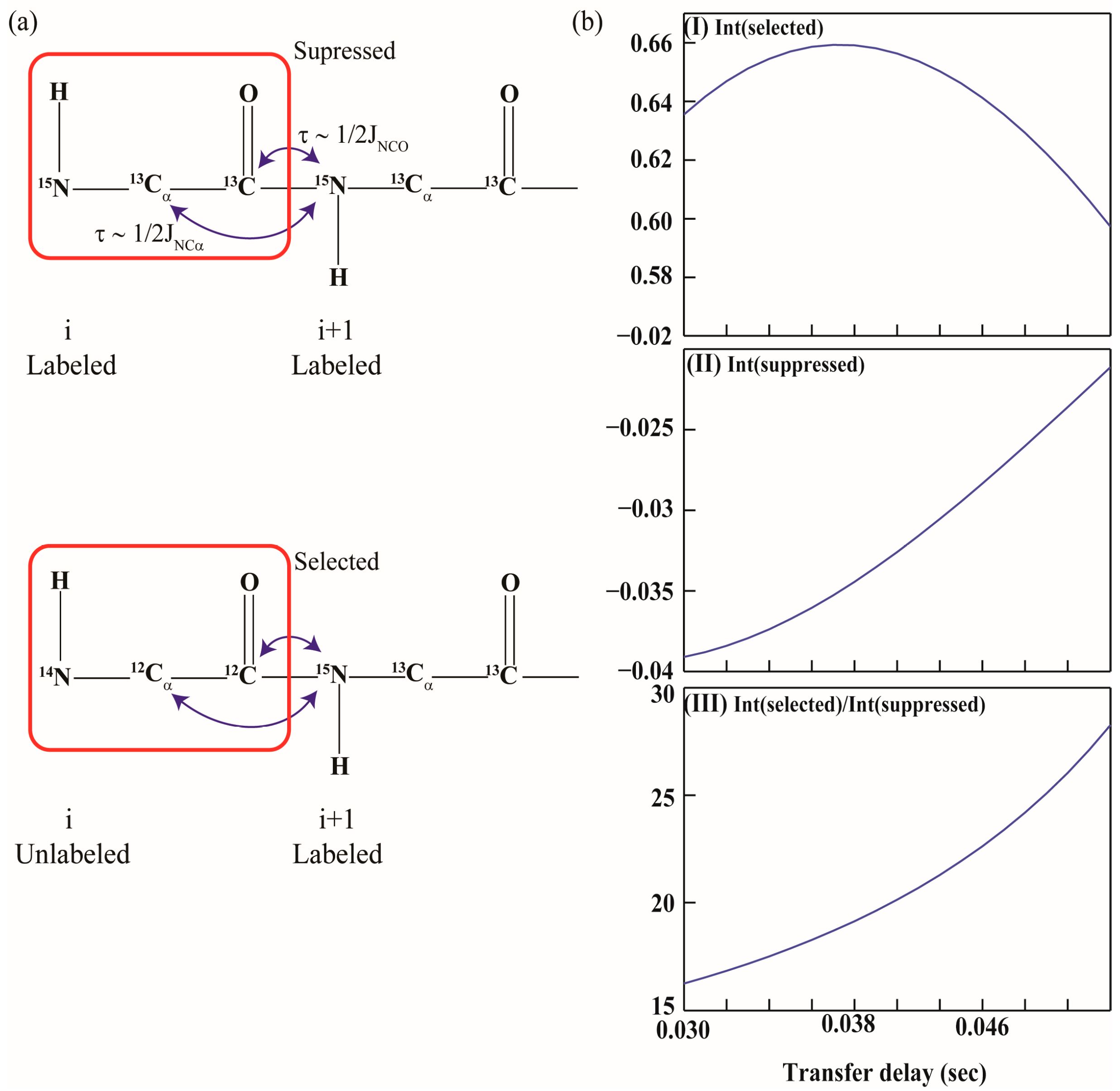
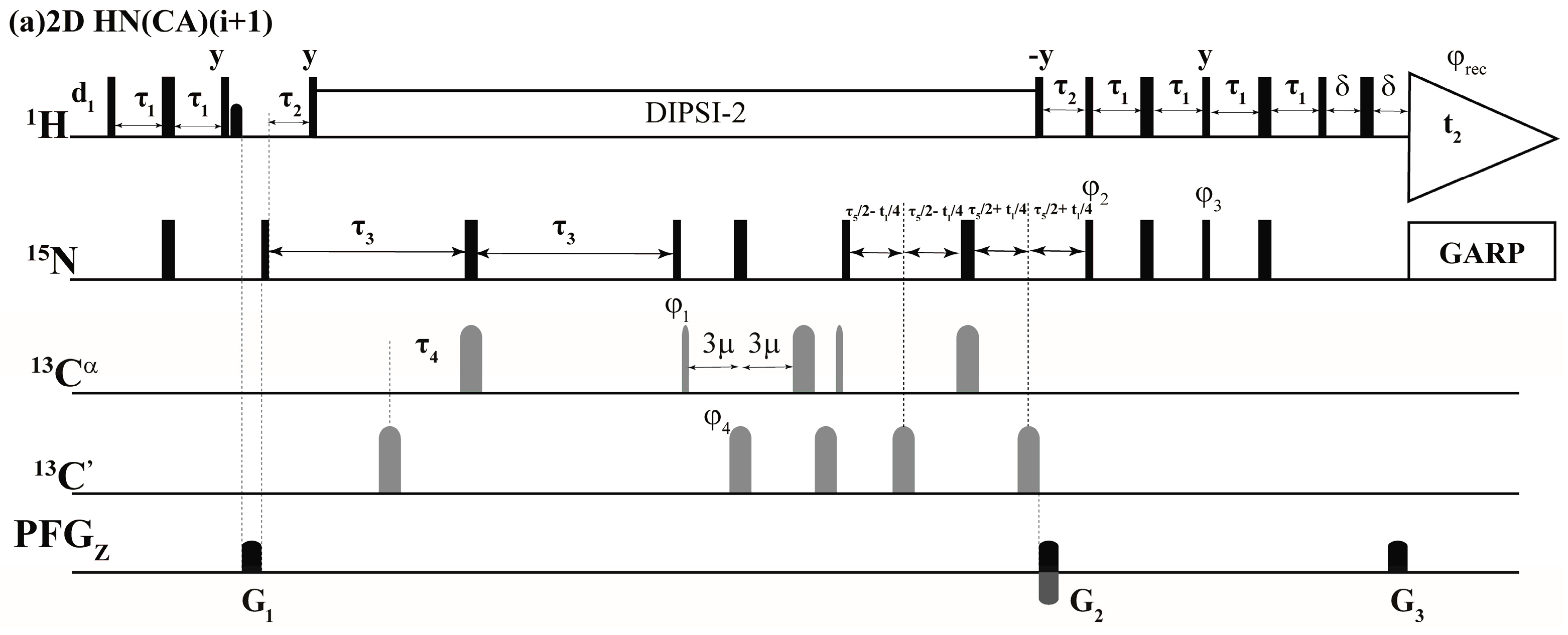
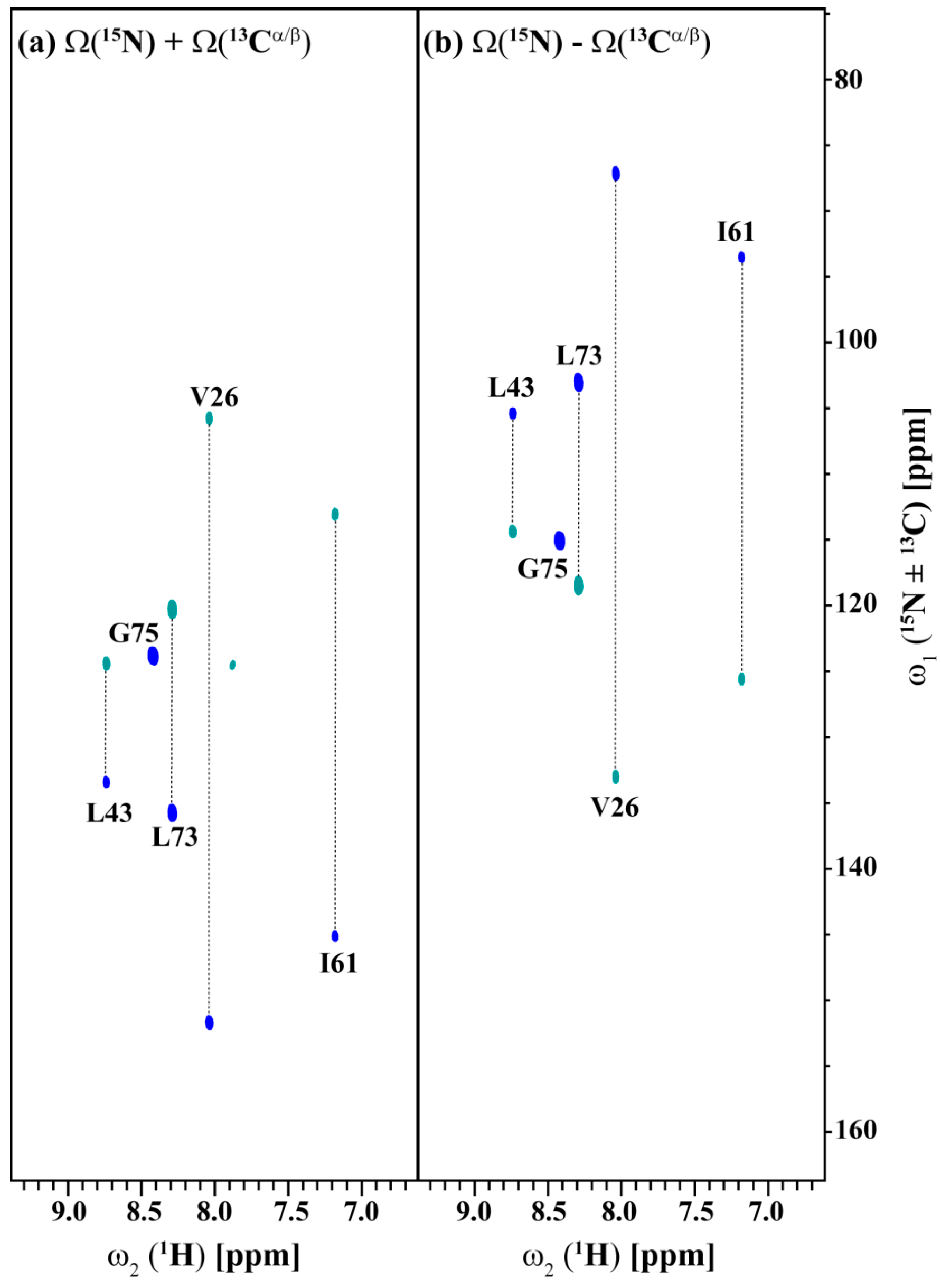
2.1. Implementation of NMR Experiments
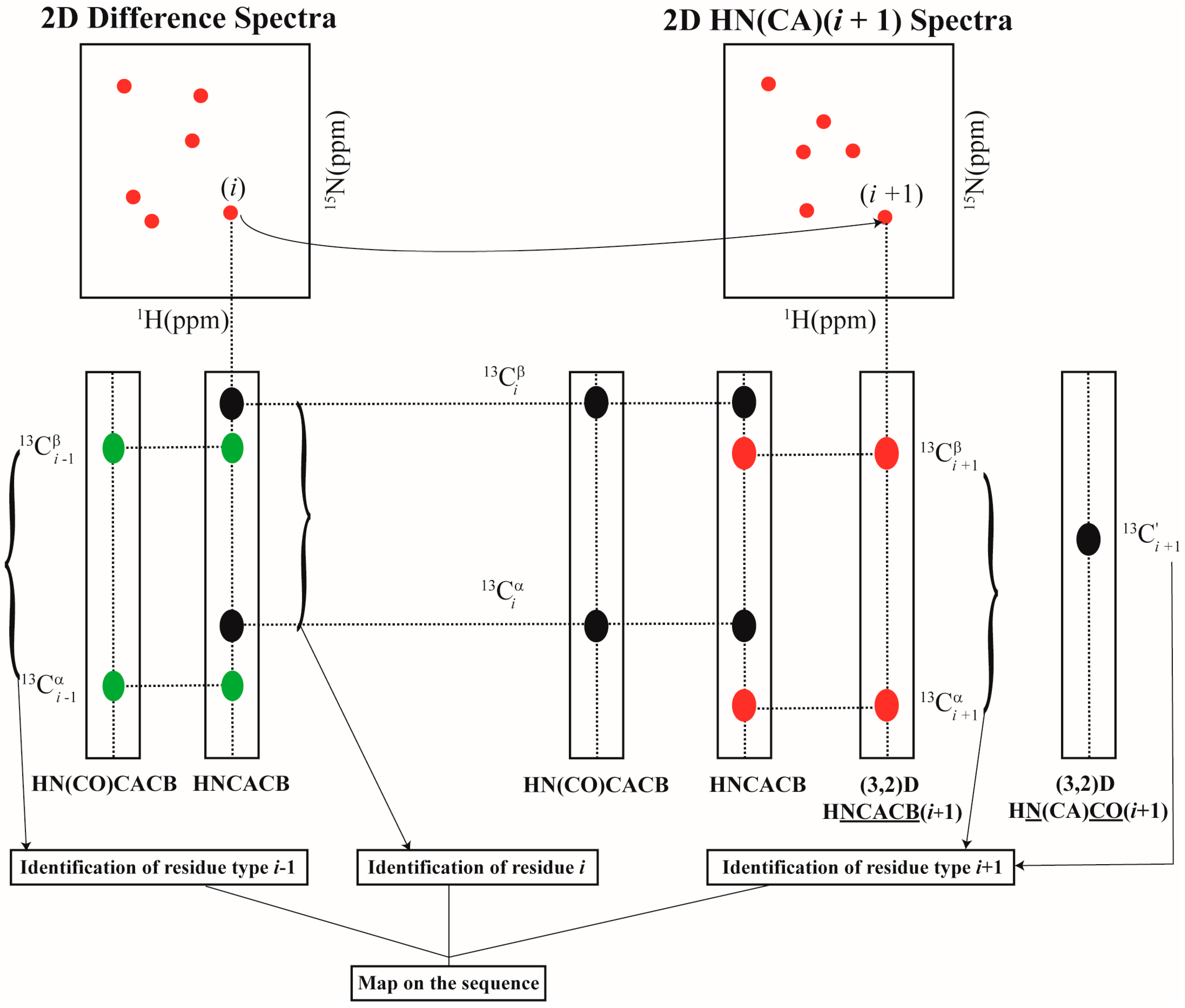
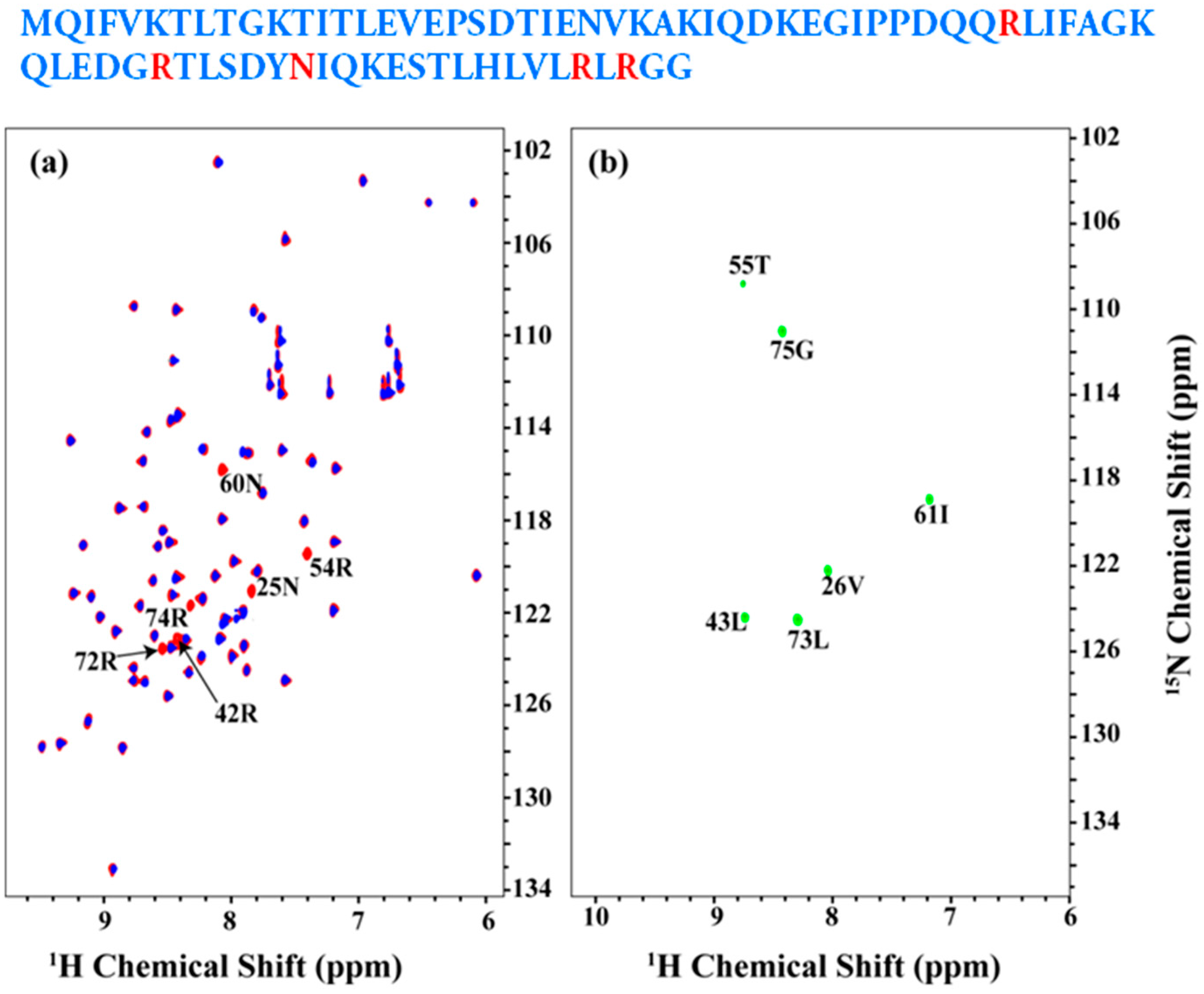
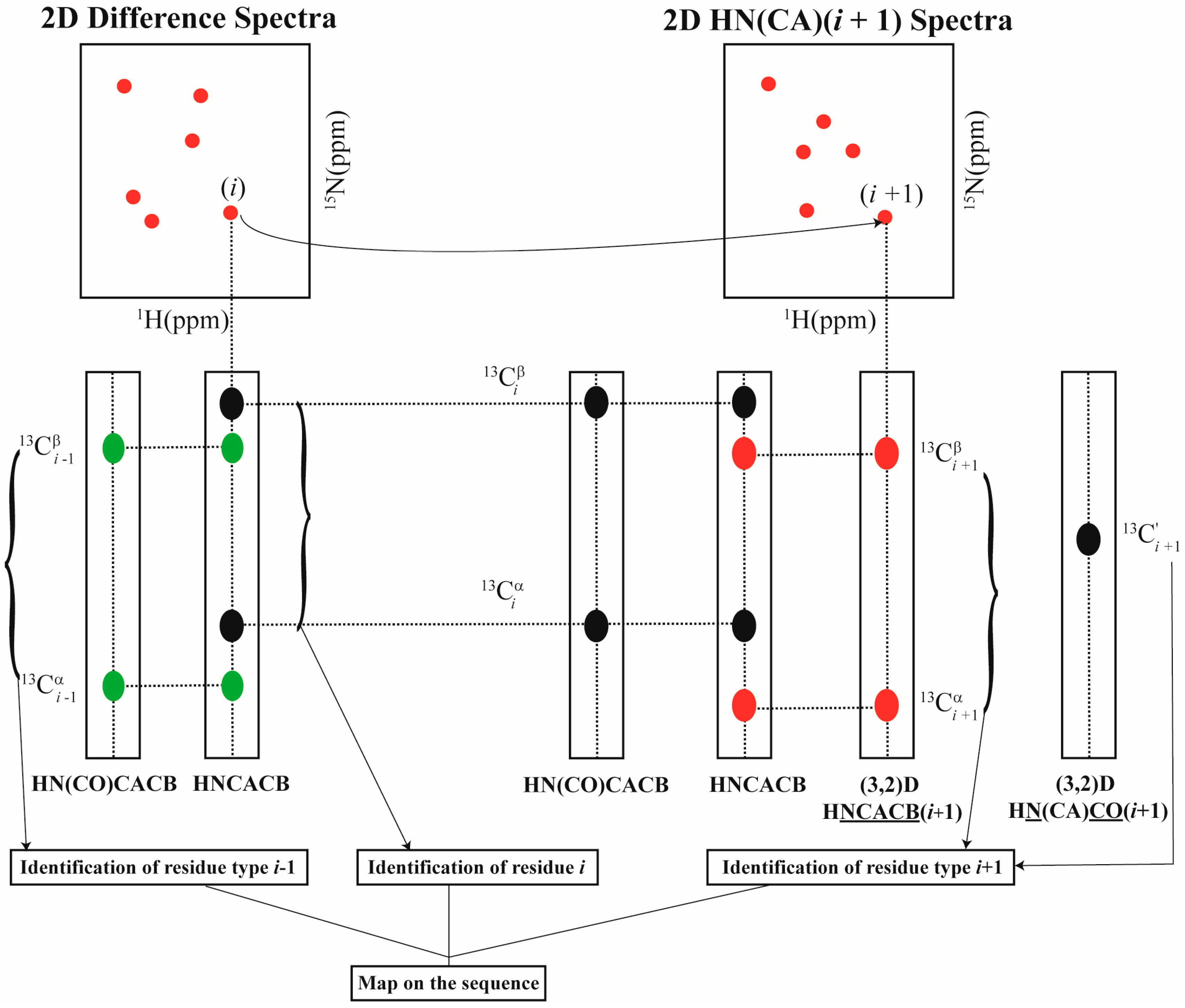
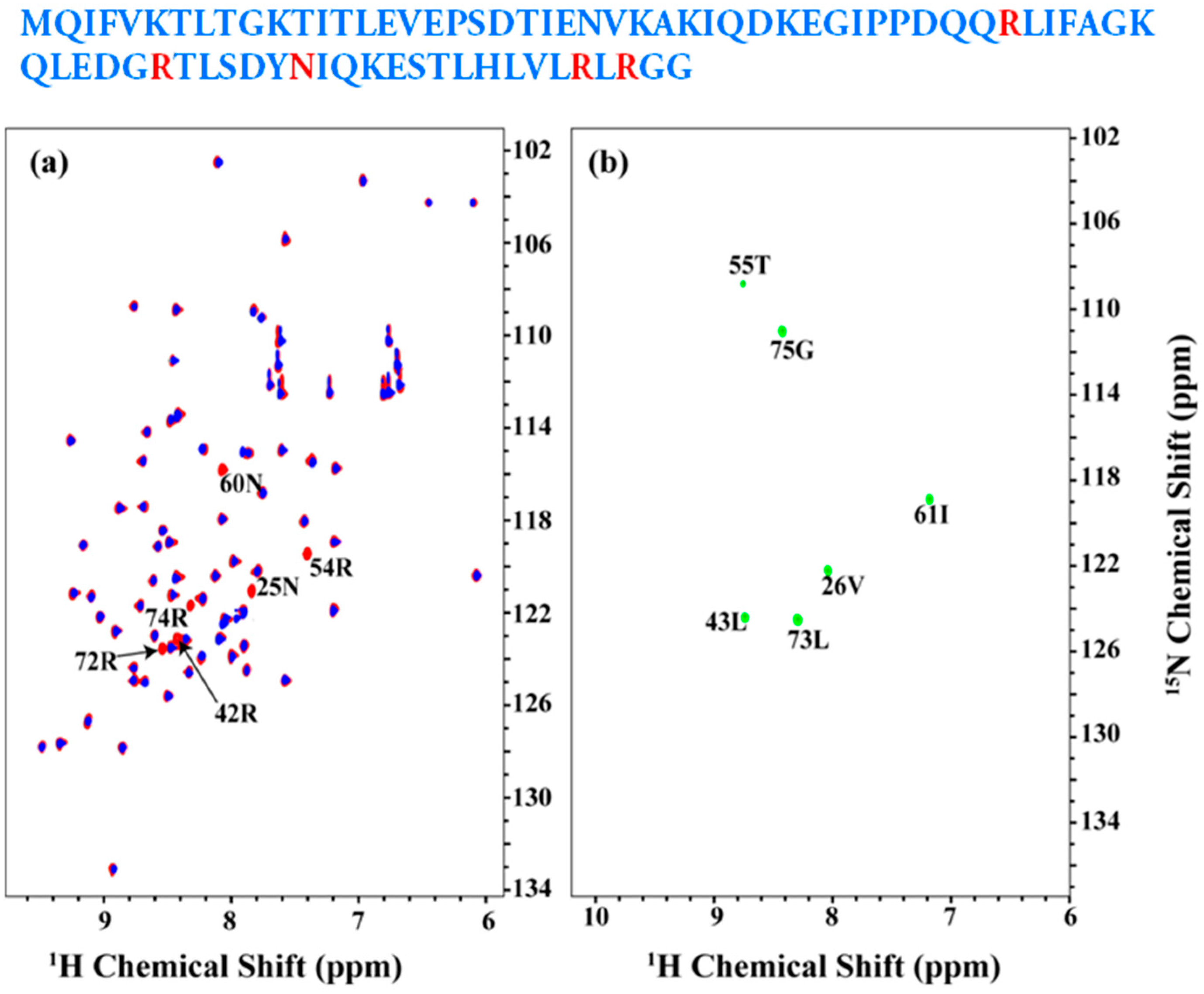
2.2. Resonance Assignment Strategy
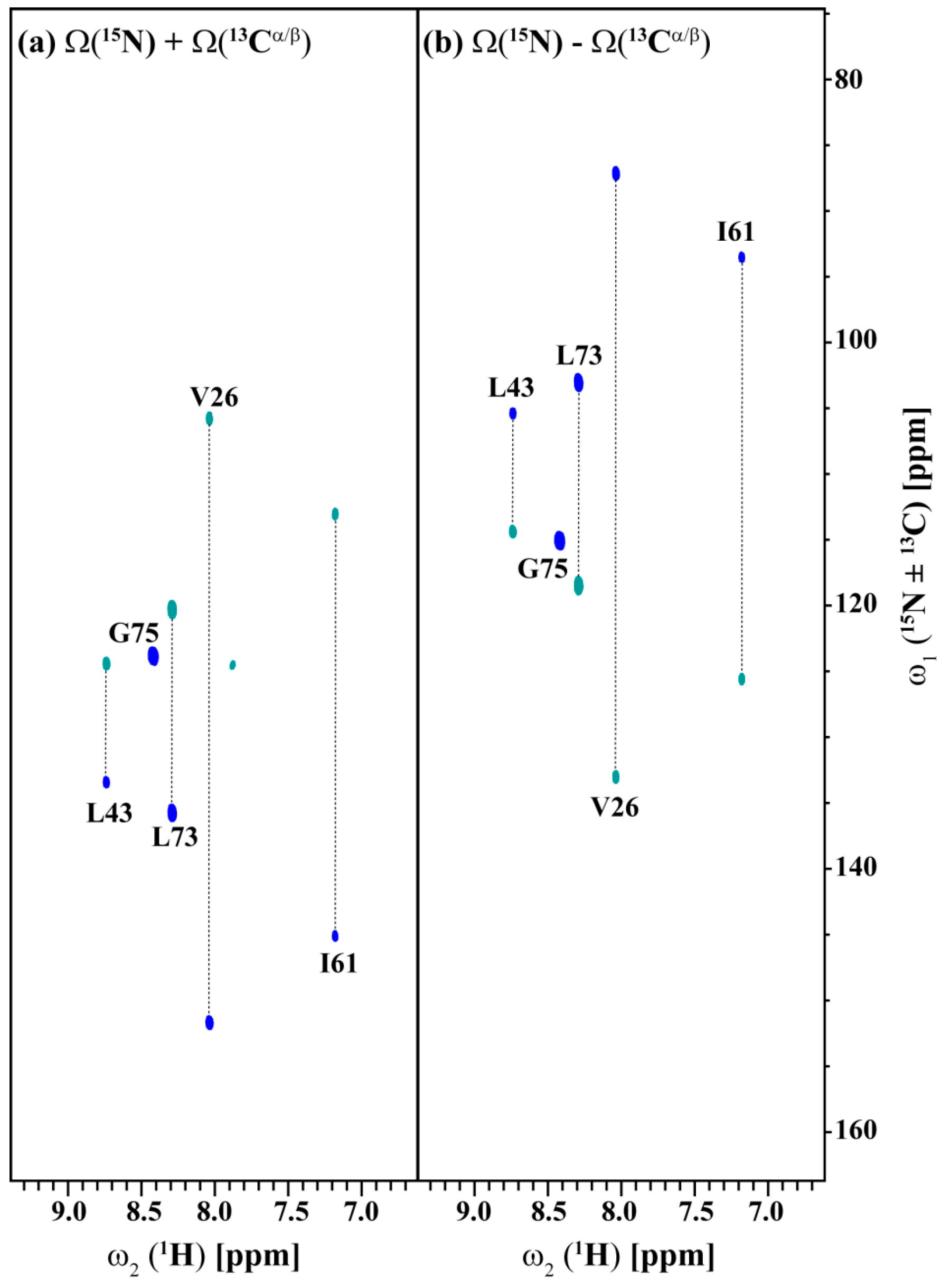
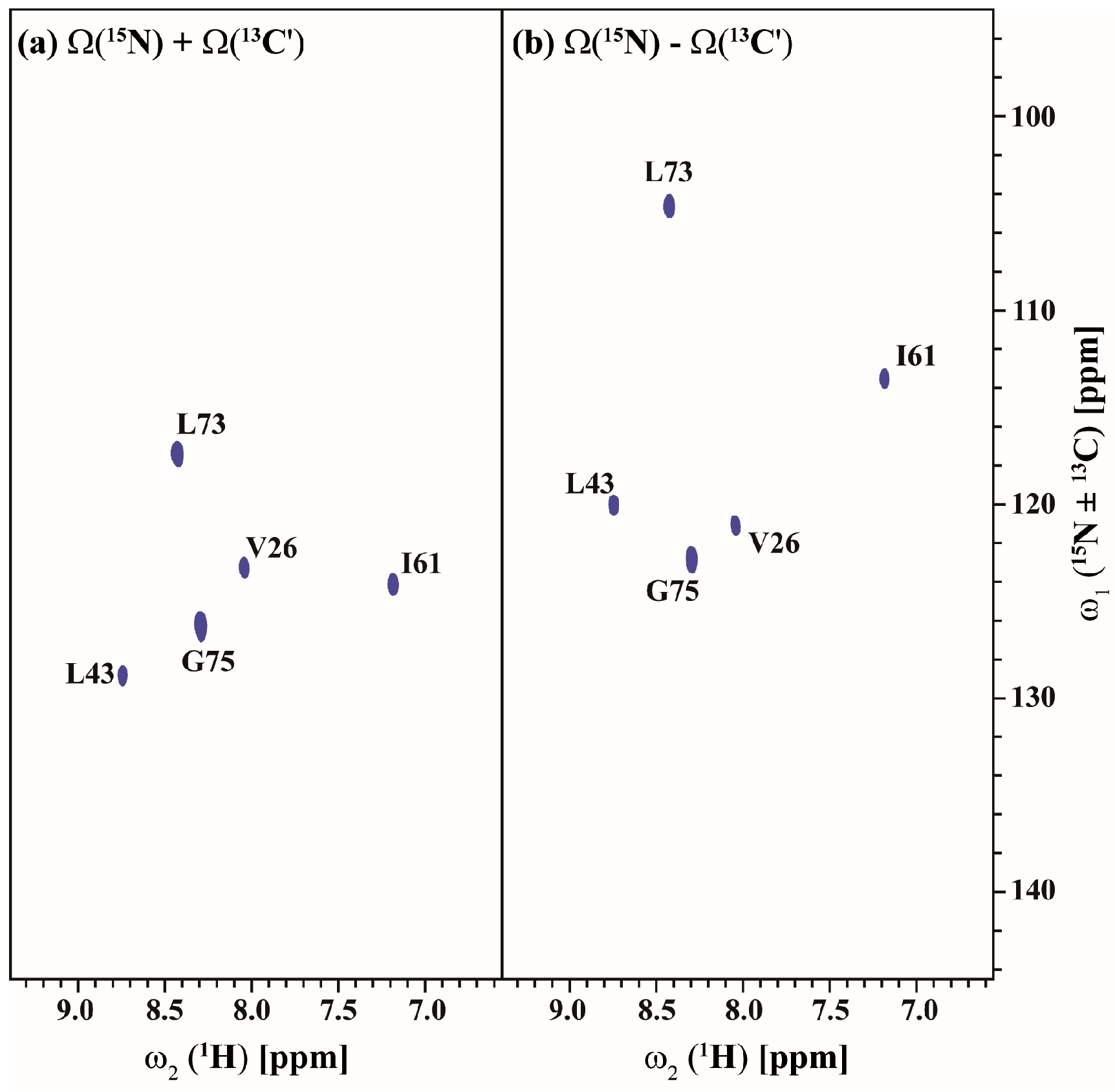
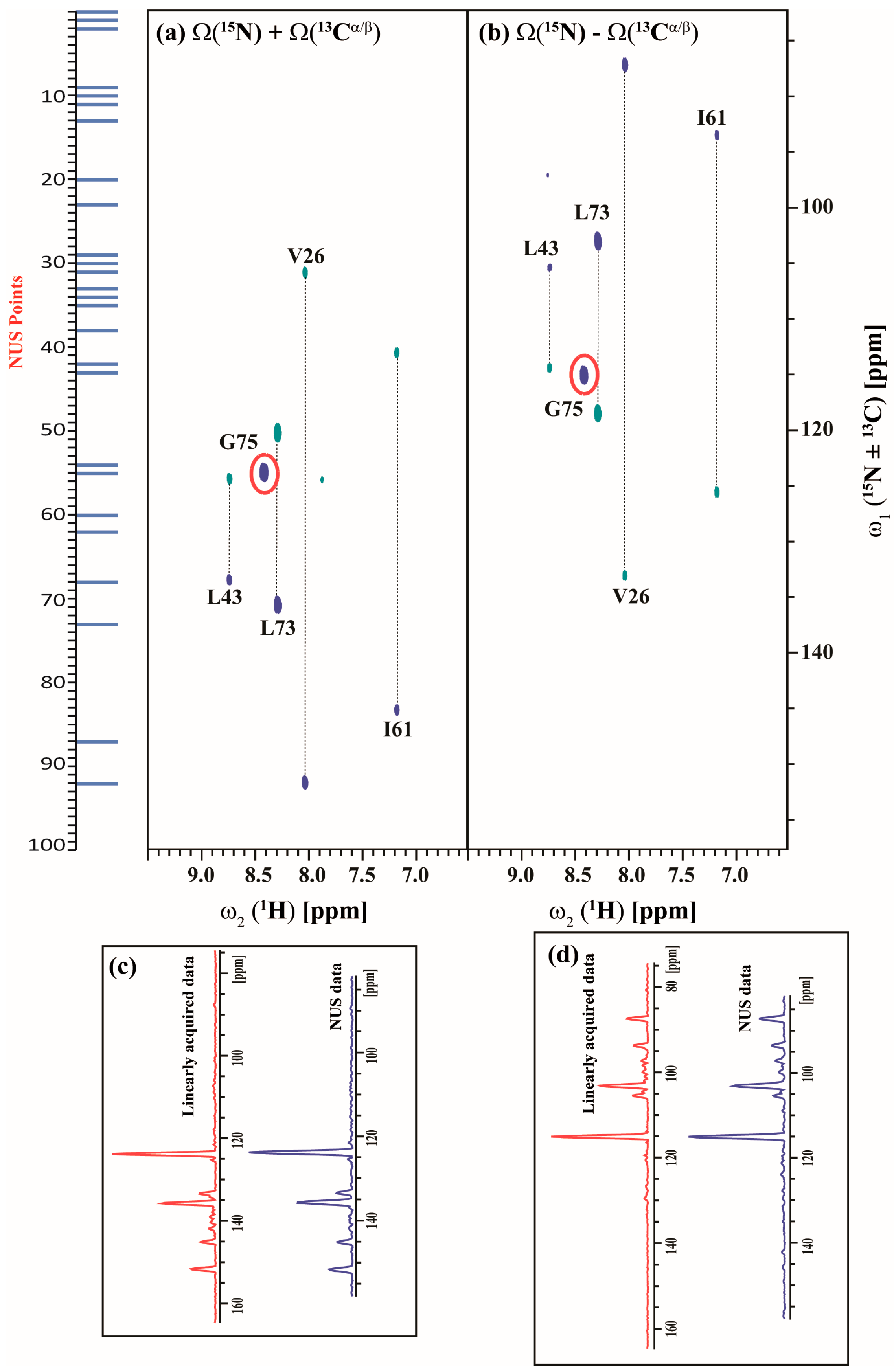
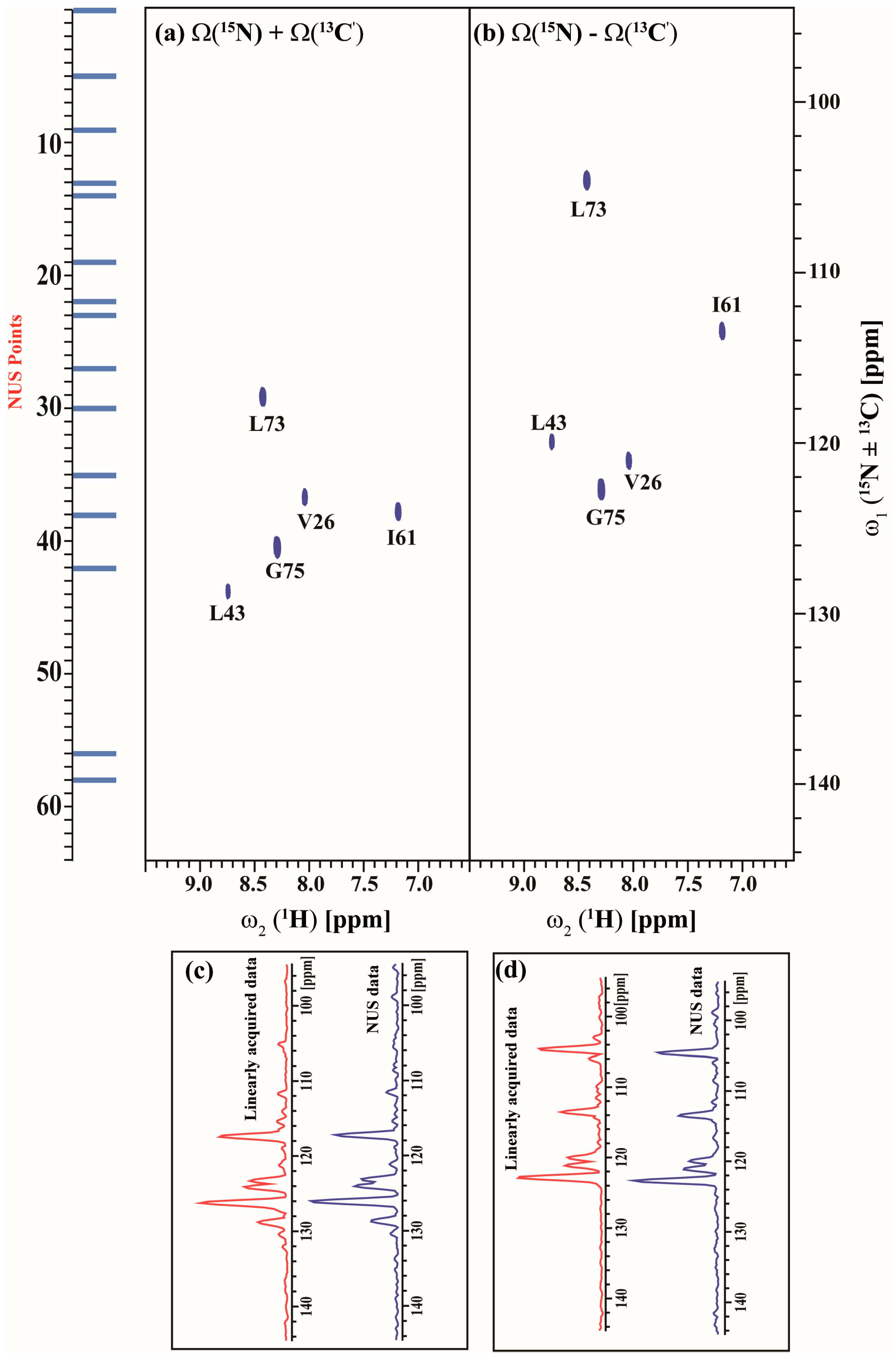
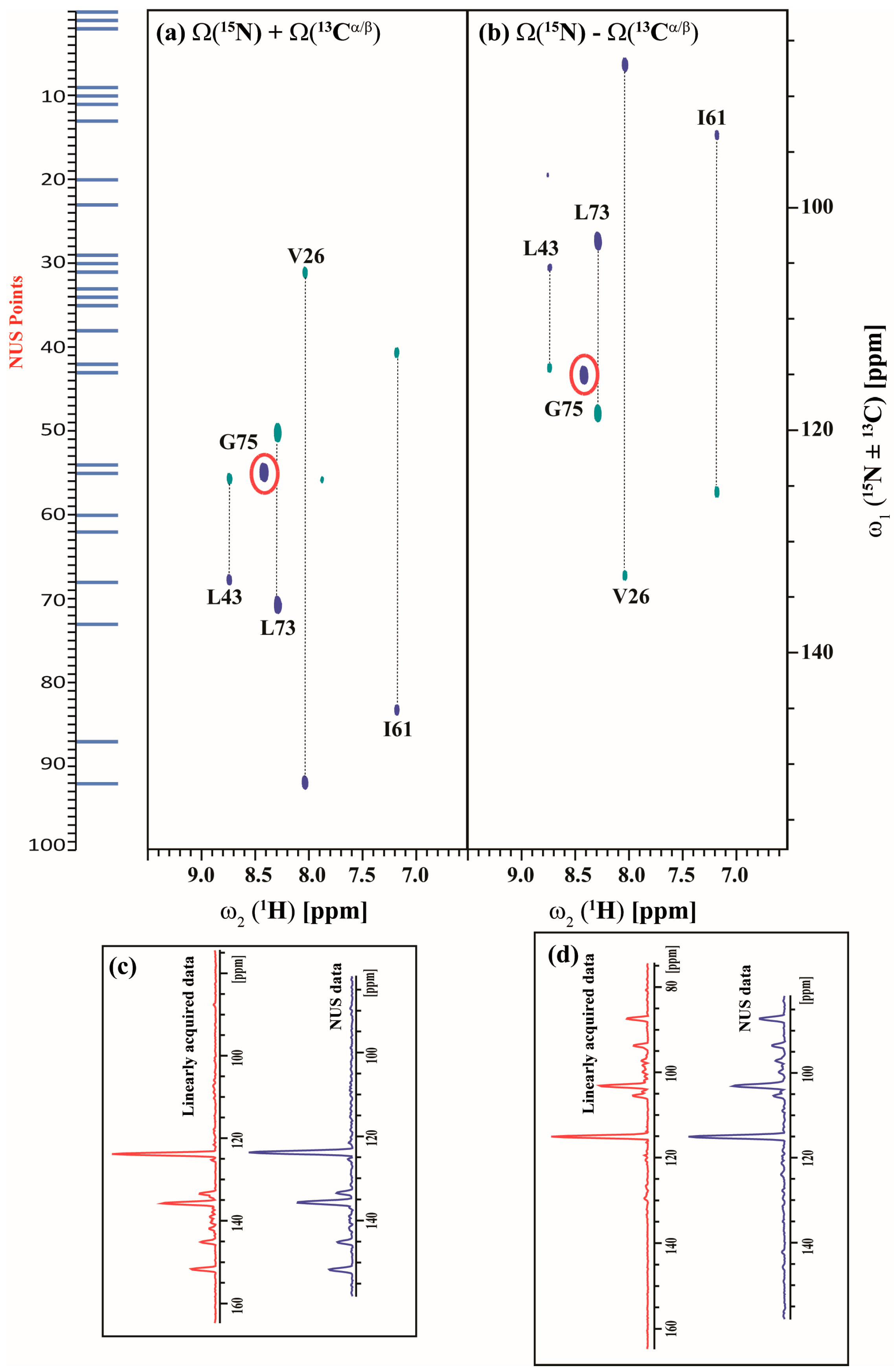
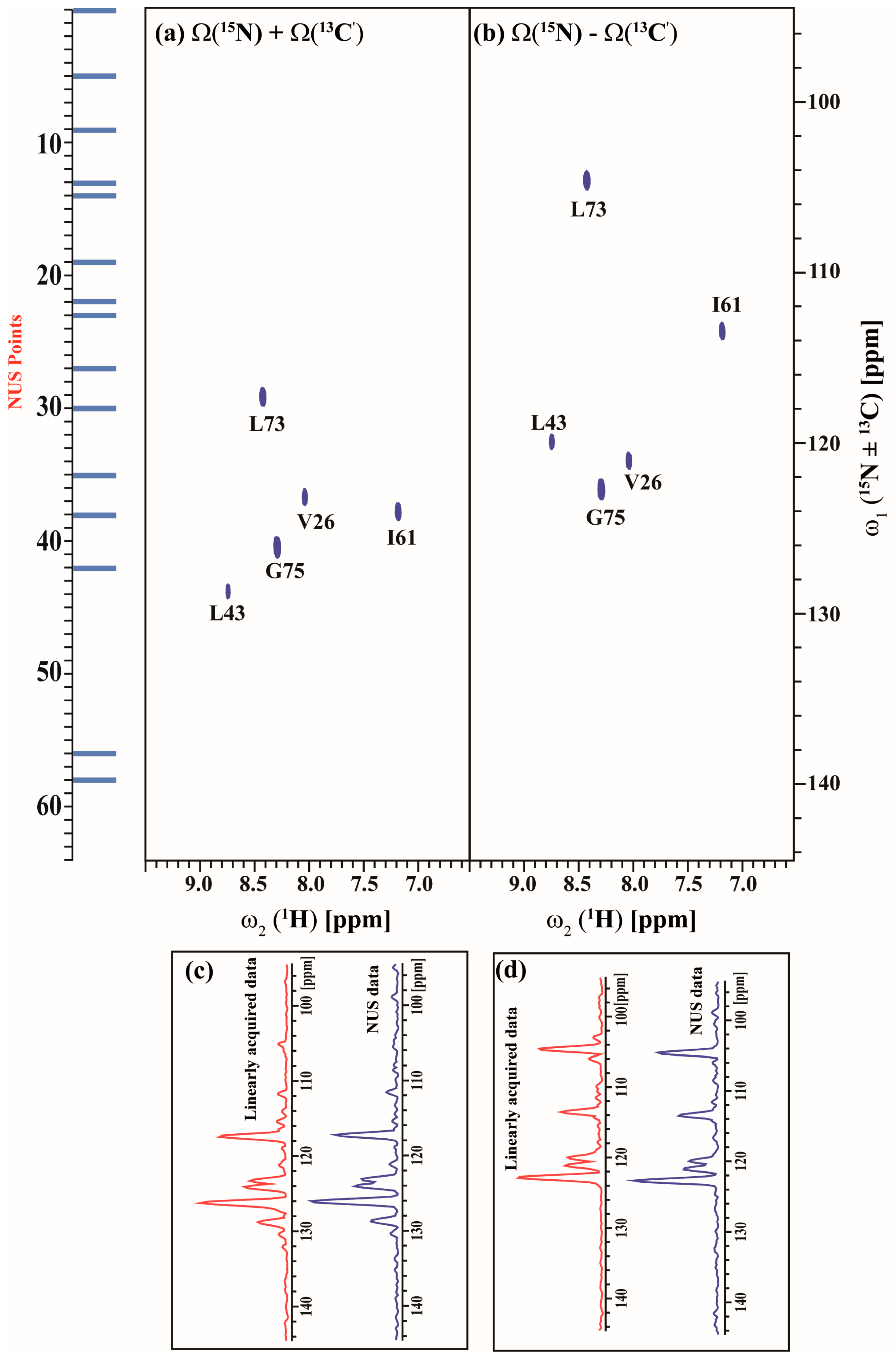
2.3. Data Acquisition Using Non-Uniform Sampling (NUS)
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. NMR Data Collection
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Atreya, H.S.; Szyperski, T. Rapid NMR data collection. Methods Enzymol. 2005, 394, 78–108. [Google Scholar] [PubMed]
- Szyperski, T.; Atreya, H.S. Principles and applications of GFT projection NMR spectroscopy. Magn. Reson. Chem. 2006, 44, S51–S60. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P. Fast-pulsing longitudinal relaxation optimized techniques: Enriching the toolbox of fast biomolecular NMR spectroscopy. Prog. NMR Spectrosc. 2009, 55, 238–265. [Google Scholar] [CrossRef]
- Felli, I.C.; Brutscher, B. Recent Advances in Solution NMR: Fast Methods and Heteronuclear Direct Detection. ChemPhysChem 2009, 10, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.; Fairbrother, W.J.; Palmer, A.G.; Skelton, N.J. Protein NMR Spectroscopy; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Pudakalakatti, S.; Chandra, K.; Thirupathi, R.; Atreya, H.S. Rapid Characterization of Molecular Diffusion by NMR. Chem. Eur. J. 2014, 20, 15719–15722. [Google Scholar] [CrossRef] [PubMed]
- Mulleti, S.; Singh, A.; Brahmkhatri, V.P.; Chandra, K.; Raza, T.; Mukherjee, S.P.; Seelamantula, C.S.; Atreya, H.S. Super-resolved nuclear magnetic resonance spectroscopy. Sci. Rep. 2017, 7, 9651. [Google Scholar]
- Khaneja, N.; Dubey, A.; Atreya, H.S. Ultra broadband NMR spectroscopy using multiple rotating frame technique. J. Magn. Reson. 2016, 265 (Suppl. C), 117–128. [Google Scholar] [CrossRef] [PubMed]
- Atreya, H.S. Isotope labeling in Biomolecular NMR. In Advances in Experimental Medicine and Biology; Springer: Amsterdam, The Netherlands, 2012; pp. 1–219. [Google Scholar]
- Krishnarjuna, B.; Jaipuria, G.; Thakur, A.; D’Silva, P.; Atreya, H.S. Amino acid selective unlabeling for sequence specific resonance assignments in proteins. J. Biomol. NMR 2011, 49, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, C.; Dubey, A.; Atreya, H.S. Amino acid selective unlabeling in protein nmr spectroscopy. In Methods in Enzymology; Kelman, Z., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 167–189. [Google Scholar]
- Vuister, G.W.; Kim, S.J.; Wu, C.; Bax, A. 2D and 3D NMR-Study of Phenylalanine Residues in Proteins by Reverse Isotopic Labeling. J. Am. Chem. Soc. 1994, 116, 9206–9210. [Google Scholar] [CrossRef]
- Shortle, D. Assignment of amino acid type in 1H-15N correlation spectra by labeling with 14N-amino acids. J. Magn. Reson. 1994, 105, 88–90. [Google Scholar] [CrossRef]
- Atreya, H.S.; Chary, K.V.R. Amino acid selective ‘unlabelling’ for residue-specific NMR assignments in proteins. Curr. Sci. 2000, 79, 504–507. [Google Scholar]
- Atreya, H.S.; Chary, K.V.R. Selective ‘unlabeling’ of amino acids in fractionally 13C labeled proteins: An approach for stereospecific NMR assignments of CH3 groups in Val and Leu residues. J. Biomol. NMR 2001, 19, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Kadumuri, R.V.; Jaipuria, G.; Vadrevu, R.; Atreya, H.S. Rapid NMR assignments of proteins by using optimized combinatorial selective unlabeling. ChemBioChem 2016, 17, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Szyperski, T. GFT NMR, a new approach to rapidly obtain precise high-dimensional NMR spectral information. J. Am. Chem. Soc. 2003, 125, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Atreya, H.S.; Szyperski, T. G-matrix Fourier transform NMR spectroscopy for complete protein resonance assignment. Proc. Natl. Acad. Sci. USA 2004, 101, 9642–9647. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, V.Y.; Ibraghimov, I.V.; Billeter, M. MUNIN: A new approach to multi-dimensional NMR spectra interpretation. J. Biomol. NMR 2001, 20, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, V.Y.; Ibraghimov, I.; Billeter, M. Optimizing resolution in multidimensional NMR by three-way decomposition. J. Biomol. NMR 2003, 27, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Rovnyak, D.; Frueh, D.P.; Sastry, M.; Sun, Z.Y.J.; Stern, A.S.; Hoch, J.C.; Wagner, G. Accelerated acquisition of high resolution triple-resonance spectra using non-uniform sampling and maximum entropy reconstruction. J. Magn. Reson. 2004, 170, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hoch, J.C.; Stern, A.S. Maximum entropy reconstruction, spectrum analysis and deconvolution in multidimensional nuclear magnetic resonance. In Nuclear Magnetic Resonance of Biologica Macromolecules; Academic Press: Cambridge, MA, USA, 2001; Part A; pp. 159–178. [Google Scholar]
- Hoch, J.C.; Maciejewski, M.W.; Filipovic, B. Randomization improves sparse sampling in multidimensional NMR. J. Magn. Reson. 2008, 193, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.A.; Orekhov, V.Y. Targeted acquisition for real-time NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Kay, L.E.; Ibraghimov, I.; Orekhov, V.Y. High-resolution four-dimensional H-1-C-13 NOE spectroscopy using methyl-TROSY, sparse data acquisition, and multidimensional decomposition. J. Am. Chem. Soc. 2005, 127, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Rasia, R.M.; Brutscher, B.; Plevin, M.J. Selective isotopic unlabeling of proteins using metabolic precursors: Application to NMR assignment of intrinsically disordered proteins. ChemBioChem 2012, 13, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.S.; Krieger, C.; Ball, L.J.; Yu, Y.; Richter, G.; Schmieder, P.; Bacher, A.; Oschkinat, H. Application of amino acid type-specific 1H and 14N labeling in a 2H-, 15N-labeled background to a 47 kDa homodimer: Potential for NMR structure determination of large proteins. J. Biomol. NMR 1999, 14, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Mohan, P.M.K.; Barve, M.A.; Chatteljee, A.; Ghosh-Roy, R.V. Hosur, NMR comparison of the native energy landscapes of DLC8 dimer and monomer. Biophys. Chem. 2008, 134, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Kay, L.E. Stereospecific NMR assignments of prochiral methyls, rotameric states and dynamics of valine residues in malate synthase G. J. Am. Chem. Soc. 2004, 126, 9827–9836. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mustafi, S.M.; Atreya, H.S.; Chary, K.V.R. Measurement of 1J(Ni,Cαi), 1J(Ni,C′i−1), 2J(Ni,Cαi−1), 2J(HNi,C′i−1) and 2J(HNi,Cαi) values in 13C/15N-labeled proteins. Magn. Reson. Chem. 2005, 43, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.; Mishra, P.; Atreya, H.S.; Hosur, R.V. Reduced dimensionality 3D HNCAN for unambiguous HN, CA and N assignments in proteins. J. Magn. Reson. 2012, 216, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Chandra, K.; Jaipuria, G.; Shet, D.; Atreya, H.S. Efficient sequential assignments in proteins with reduced dimensionality 3D HN(CA)NH. J. Biomol. NMR 2011, 52, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Franks, W.T.; Atreya, H.S.; Szyperski, T.; Rienstra, C.M. GFT projection NMR spectroscopy for proteins in the solid state. J. Biomol. NMR 2010, 48, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Jaipuria, G.; Thakur, A.; D’Silva, P.; Atreya, H.S. High-resolution methyl edited GFT NMR experiments for protein resonance assignments and structure determination. J. Biomol. NMR 2010, 48, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Swain, M.; Atreya, H.S. CSSI-PRO: A method for secondary structure type editing, assignment and estimation in proteins using linear combination of backbone chemical shifts. J. Biomol. NMR 2009, 44, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, R.P.; Atreya, H.S.; Chary, K.V.R. Chemical shift based editing of CH3 groups in fractionally C-13-labelled proteins using GFT (3,2)D CT-HCCH-COSY: Stereospecific assignments of CH3 groups of Val and Leu residues. J. Biomol. NMR 2008, 42, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, R.P.; Rout, A.K.; Atreya, H.S.; Chary, K.V.R. Identification of C-terminal neighbours of amino acid residues without an aliphatic C-13(gamma) supercript stop as an aid to NMR assignments in proteins. J. Biomol. NMR 2008, 41, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, R.P.; Rout, A.K.; Chary, K.V.R.; Atreya, H.S. Rapid measurement of pseudocontact shifts in paramagnetic proteins by GFT NMR spectroscopy. Open Magn. Reson. J. 2008, 1, 16–28. [Google Scholar] [CrossRef]
- Zhang, Q.; Atreya, H.S.; Kamen, D.E.; Girvin, M.E.; Szyperski, T. GFT projection NMR based resonance assignment of membrane proteins: Application to subunit c of E. coli F1F0 ATP synthase in LPPG micelles. J. Biomol. NMR 2008, 40, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Atreya, H.S.; Garcia, E.; Shen, Y.; Szyperski, T. J-GFT NMR for precise measurement of mutually correlated nuclear spin-spin couplings. J. Am. Chem. Soc. 2007, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, R.P.; Rout, A.K.; Chary, K.V.R.; Atreya, H.S. Rapid measurement of 3J(HN-Hα) and 3J(N-Hβ) coupling constants in polypeptides. J. Biomol. NMR 2007, 39, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Atreya, H.S.; Eletsky, A.; Szyperski, T. Resonance assignment of proteins with high shift degeneracy based on 5D spectral information encoded in G2FT NMR experiments. J. Am. Chem. Soc. 2005, 127, 4554–4555. [Google Scholar] [CrossRef] [PubMed]
- Eletsky, A.; Atreya, H.S.; Liu, G.H.; Szyperski, T. Probing structure and functional dynamics of (large) proteins with aromatic rings: L-GFT-TROSY (4,3)D HCCHNMR spectroscopy. J. Am. Chem. Soc. 2005, 127, 14578–14579. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Aramini, J.; Atreya, H.S.; Eletsky, A.; Xiao, R.; Acton, T.; Ma, L.C.; Montelione, G.T.; Szyperski, T. GFT NMR based resonance assignment for the 21 kDa human protein UFC1. J. Biomol. NMR 2005, 32, 261. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Shen, Y.; Atreya, H.S.; Parish, D.; Shao, Y.; Sukumaran, D.K.; Xiao, R.; Yee, A.; Lemak, A.; Bhattacharya, A.; et al. NMR data collection and analysis protocol for high-throughput protein structure determination. Proc. Natl. Acad. Sci. USA 2005, 102, 10487–10492. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Atreya, H.S.; Liu, G.H.; Szyperski, T. G-matrix Fourier transform NOESY-based protocol for high-quality protein structure determination. J. Am. Chem. Soc. 2005, 127, 9085–9099. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRpipe—A Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Bartels, C.; Xia, T.H.; Billeter, M.; Güntert, P.; Wüthrich, K. The Program Xeasy for Computer-Supported NMR Spectral-Analysis of Biological Macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnarjuna, B.; Chandra, K.; Atreya, H.S. Accelerating NMR-Based Structural Studies of Proteins by Combining Amino Acid Selective Unlabeling and Fast NMR Methods. Magnetochemistry 2018, 4, 2. https://doi.org/10.3390/magnetochemistry4010002
Krishnarjuna B, Chandra K, Atreya HS. Accelerating NMR-Based Structural Studies of Proteins by Combining Amino Acid Selective Unlabeling and Fast NMR Methods. Magnetochemistry. 2018; 4(1):2. https://doi.org/10.3390/magnetochemistry4010002
Chicago/Turabian StyleKrishnarjuna, Bankala, Kousik Chandra, and Hanudatta S. Atreya. 2018. "Accelerating NMR-Based Structural Studies of Proteins by Combining Amino Acid Selective Unlabeling and Fast NMR Methods" Magnetochemistry 4, no. 1: 2. https://doi.org/10.3390/magnetochemistry4010002