
Synthesis of 4-Hydroxy-2,6-di(pyrazol-1-yl)pyridine, and the Spin State Behaviour of Its Iron(II) Complex Salts


Abstract
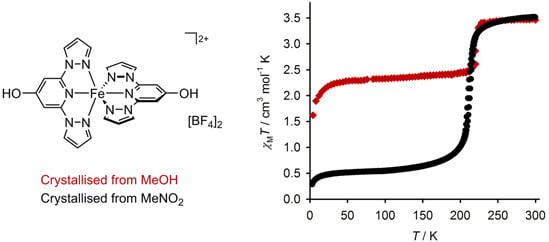
:
1. Introduction
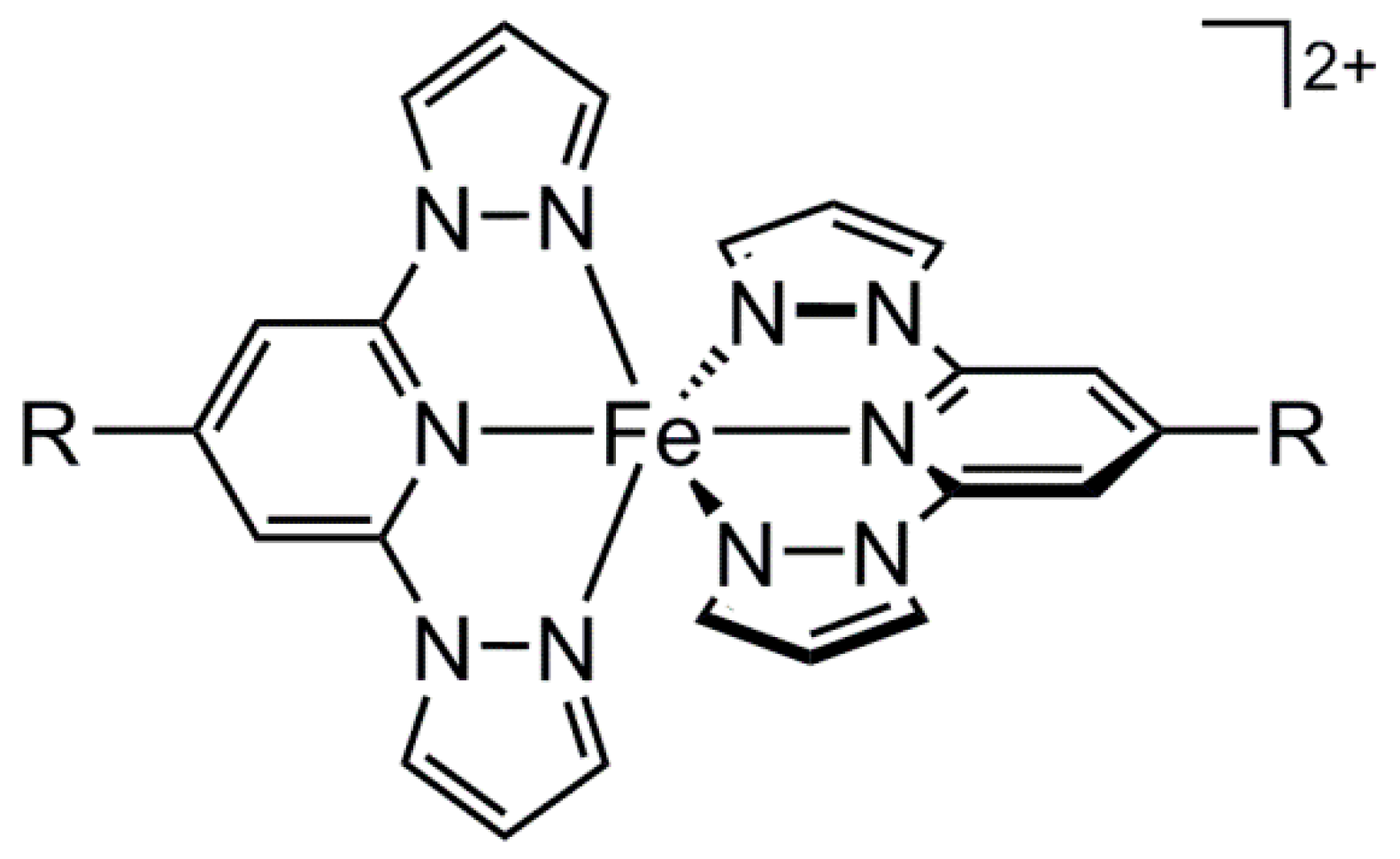
2. Results and Discussion
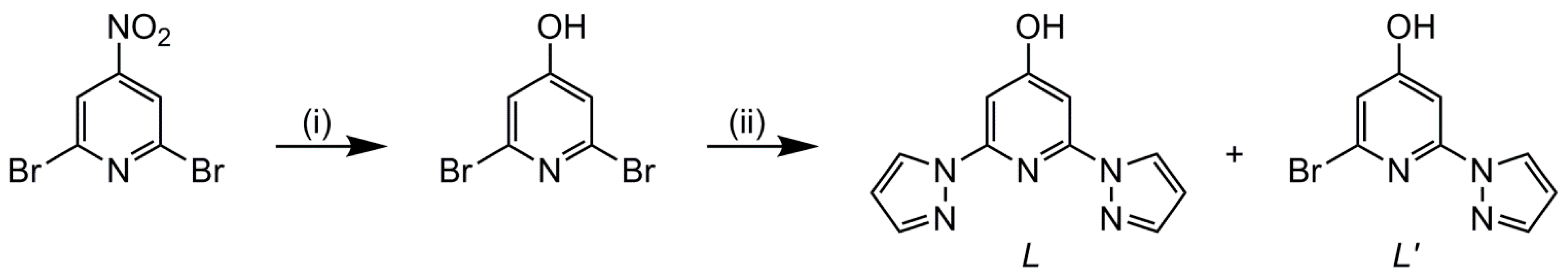
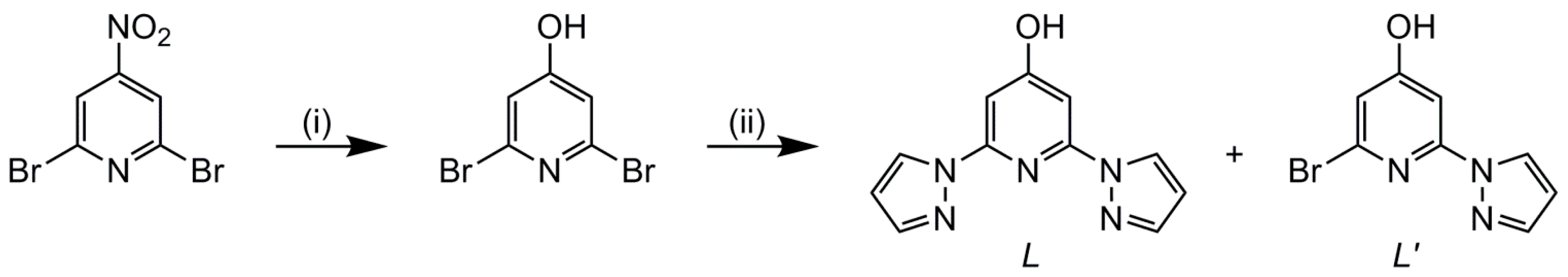
2.1. Synthesis
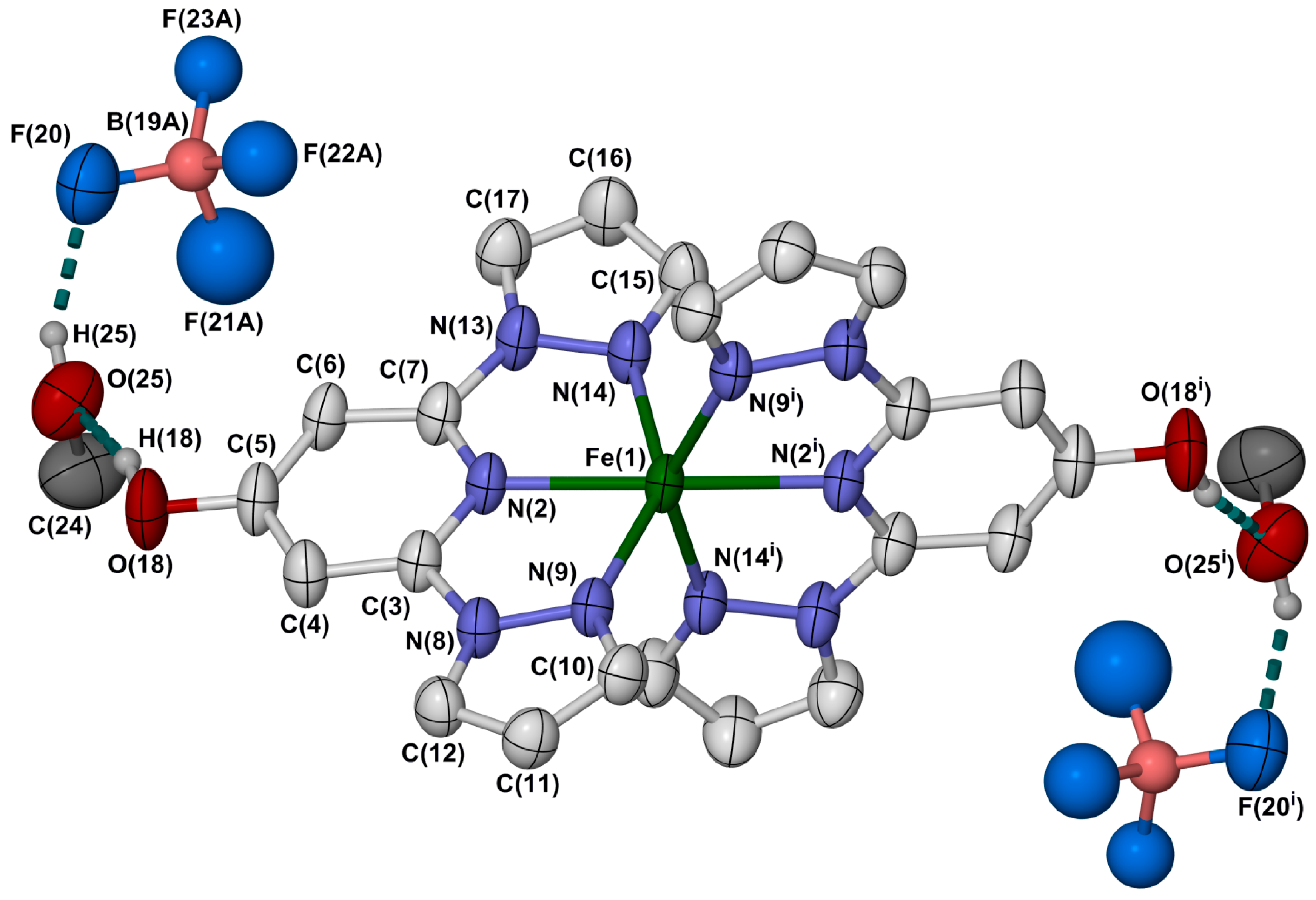
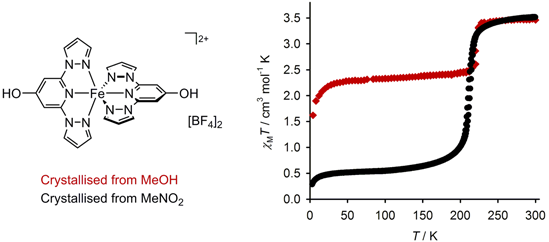
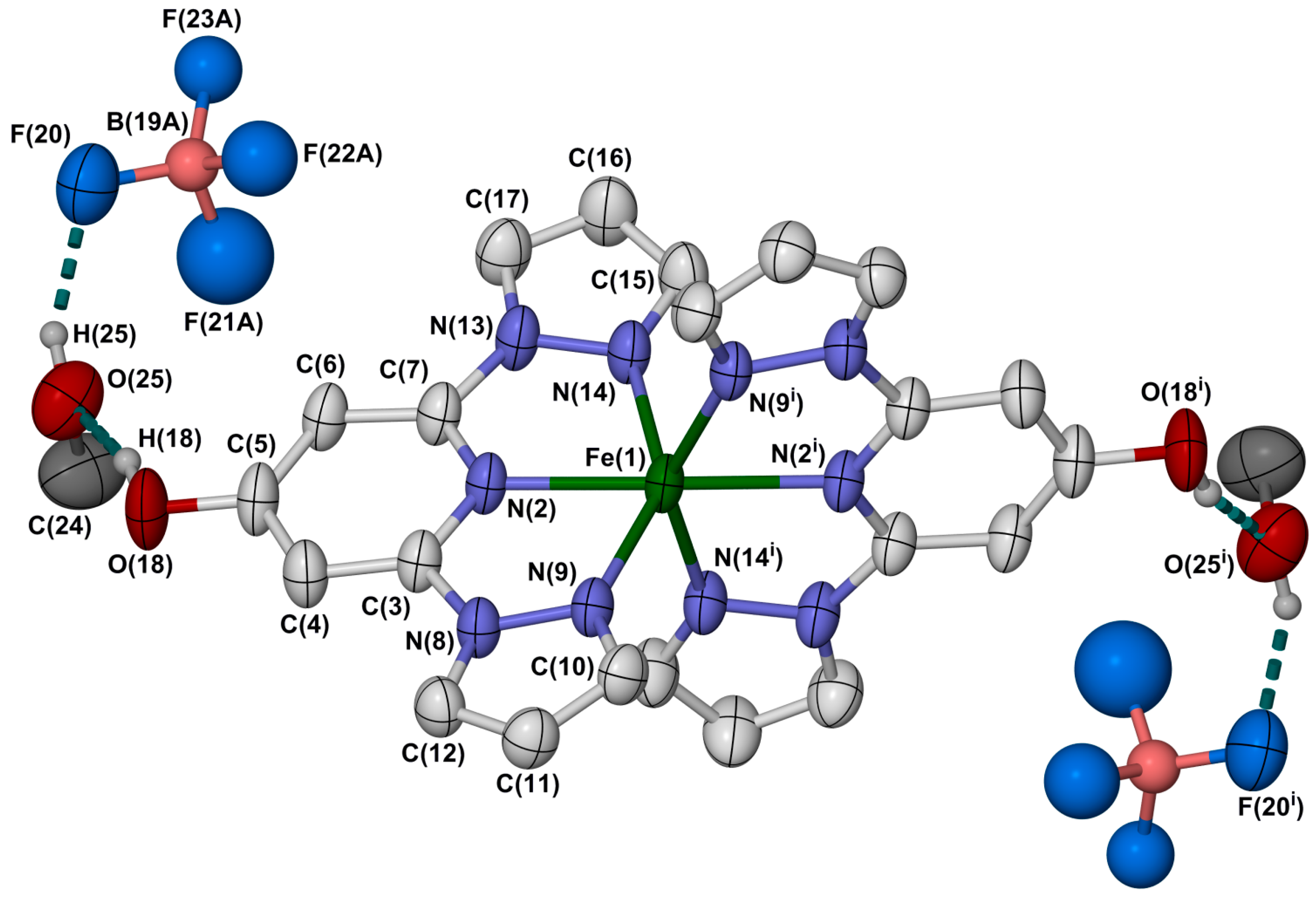
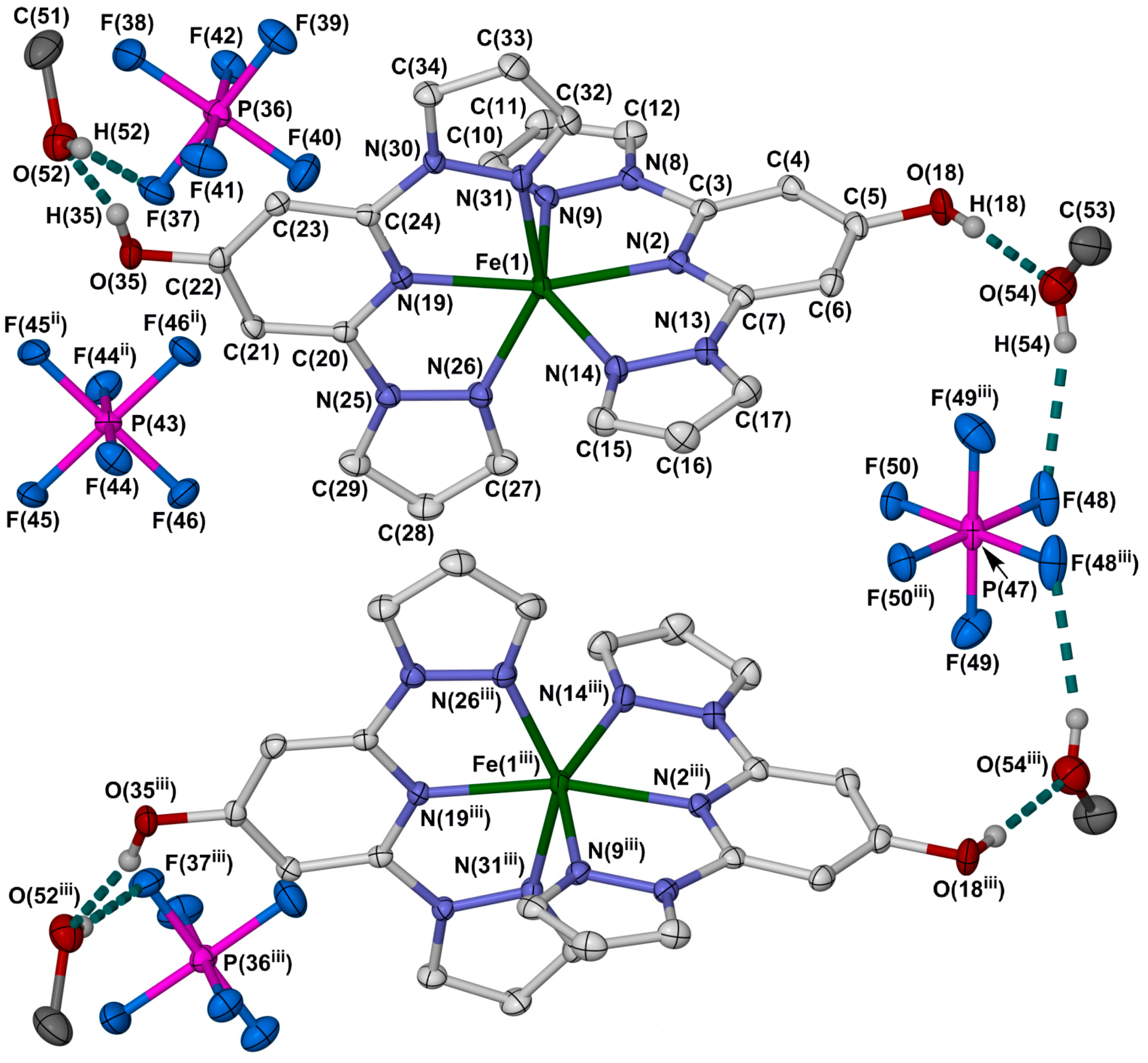
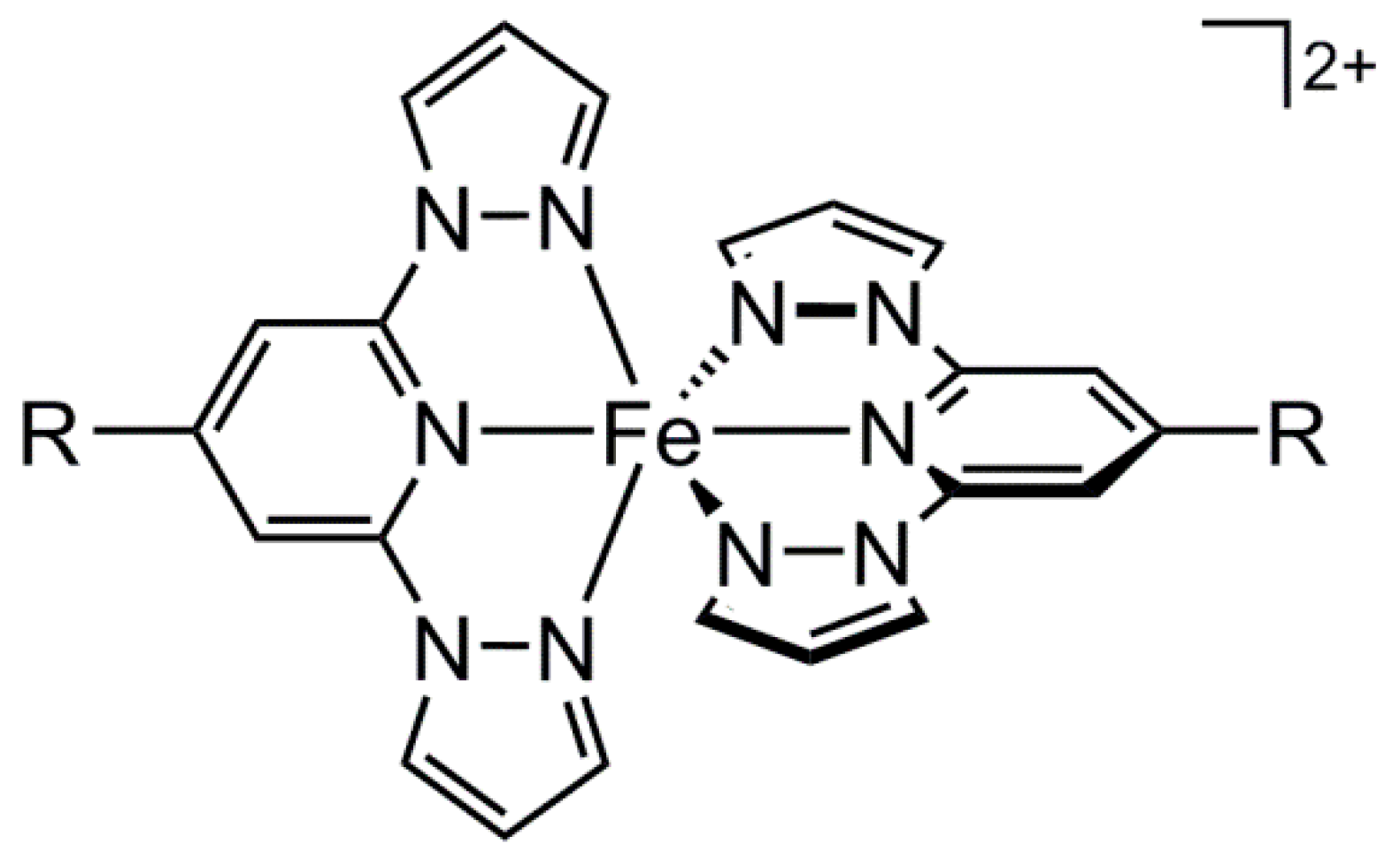
2.2. Crystallographic Characterisation
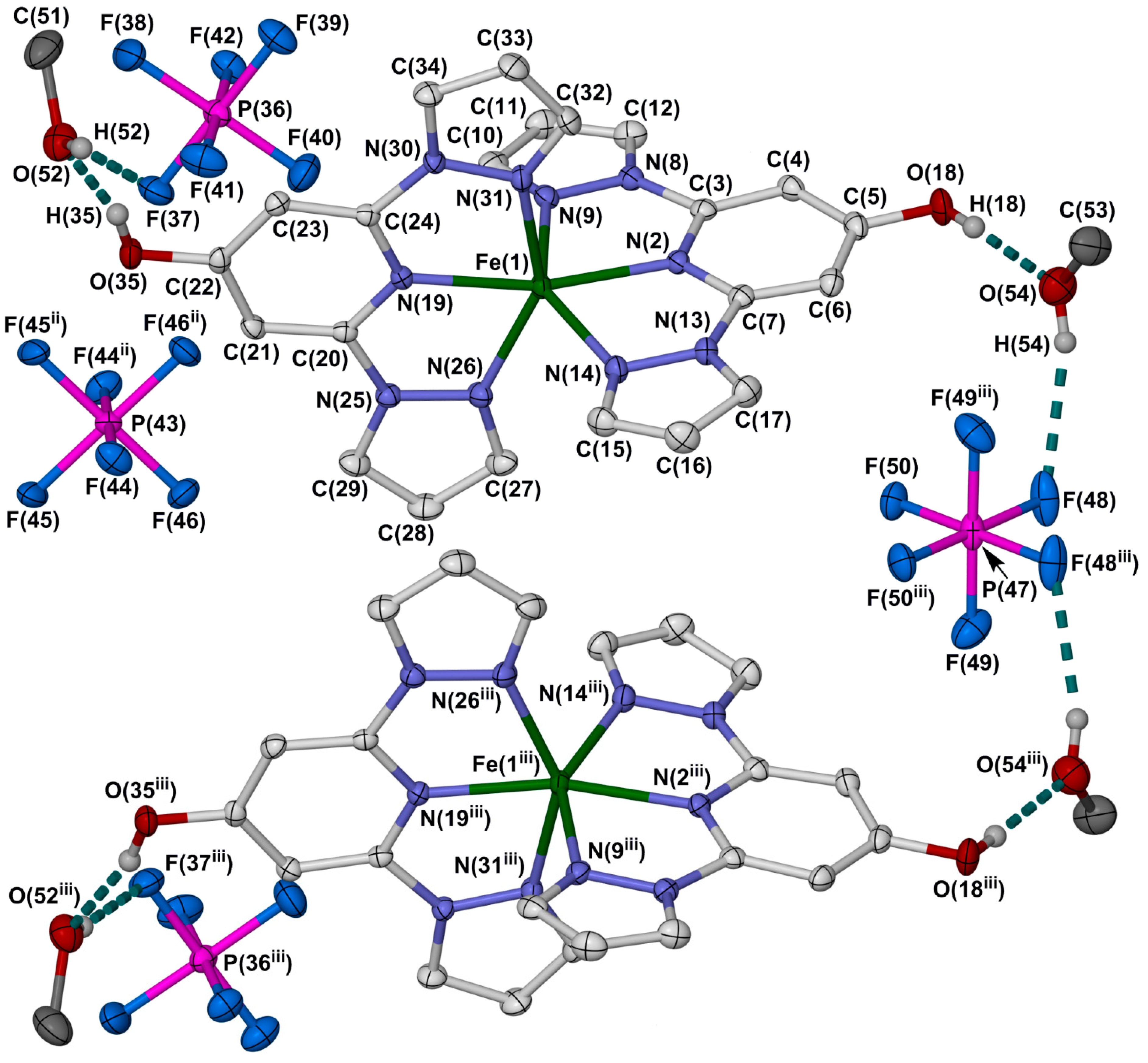

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1·2MeOH | 2·1.75MeOH·0.25H2O | 3·2MeOH |
|---|---|---|---|
| T (K) | 150 | 150 | 100 |
| Fe(1)−N(2) | 2.147(3) | 2.0665(19) | 2.1204(16) |
| Fe(1)−N(9) | 2.217(3) | 2.138(3) | 2.1936(17) |
| Fe(1)−N(14) | 2.208(3) | 2.137(2) | 2.1860(17) |
| Fe(1)−N(19) | - | 2.069(2) | 2.1237(16) |
| Fe(1)−N(26) | - | 2.116(3) | 2.1630(17) |
| Fe(1)−N(31) | - | 2.135(2) | 2.1852(17) |
| α | 73.22(15) | 75.51(16) | 73.35(12) |
| Σ | 153.7(4) | 130.7(3) | 153.0(2) |
| Θ | 481 | 414 | 477 |
| ϕ | 165.55(15) | 169.06(7) | 166.68(6) |
| θ | 85.17(3) | 86.67(3) | 87.36(1) |
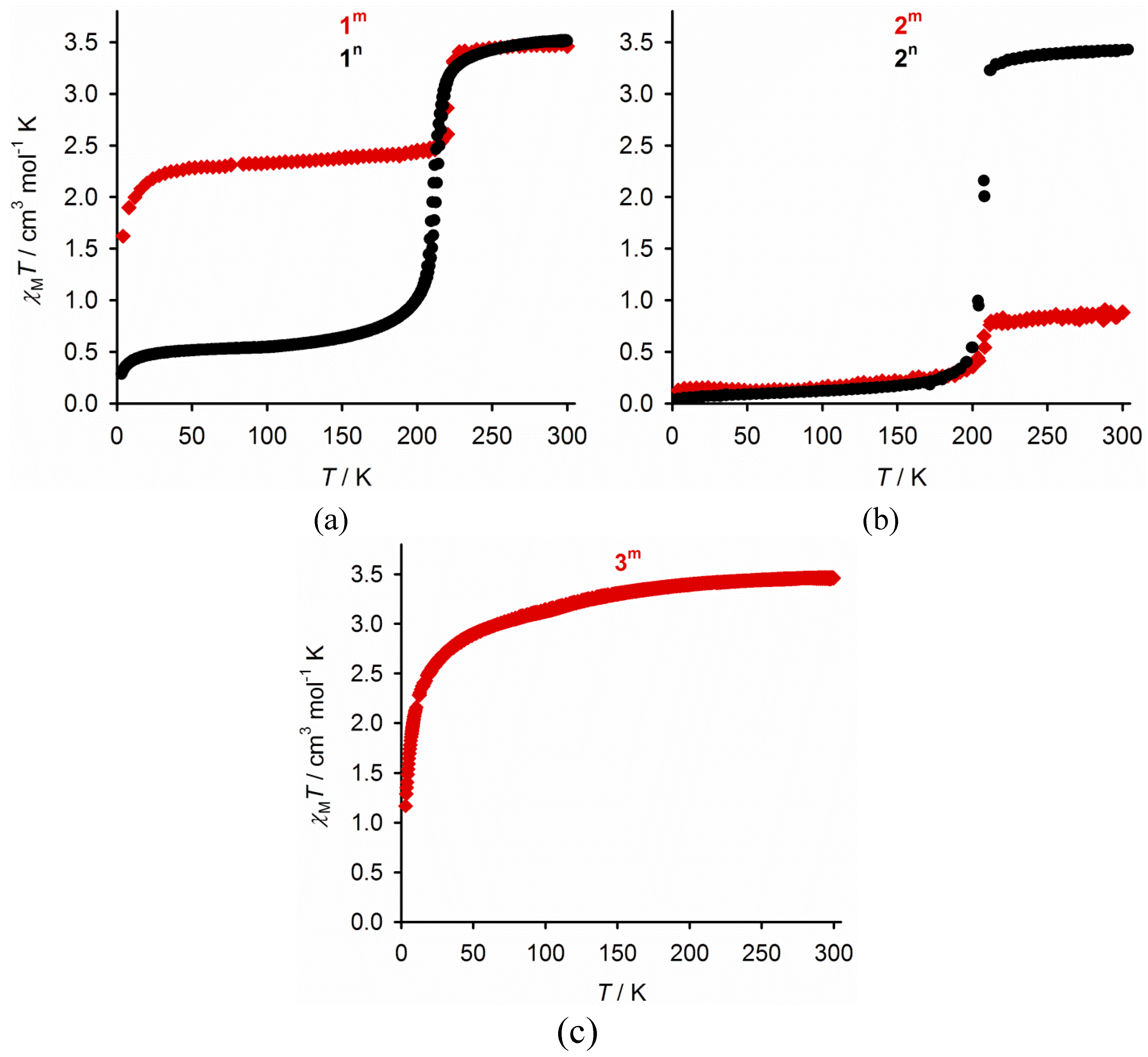
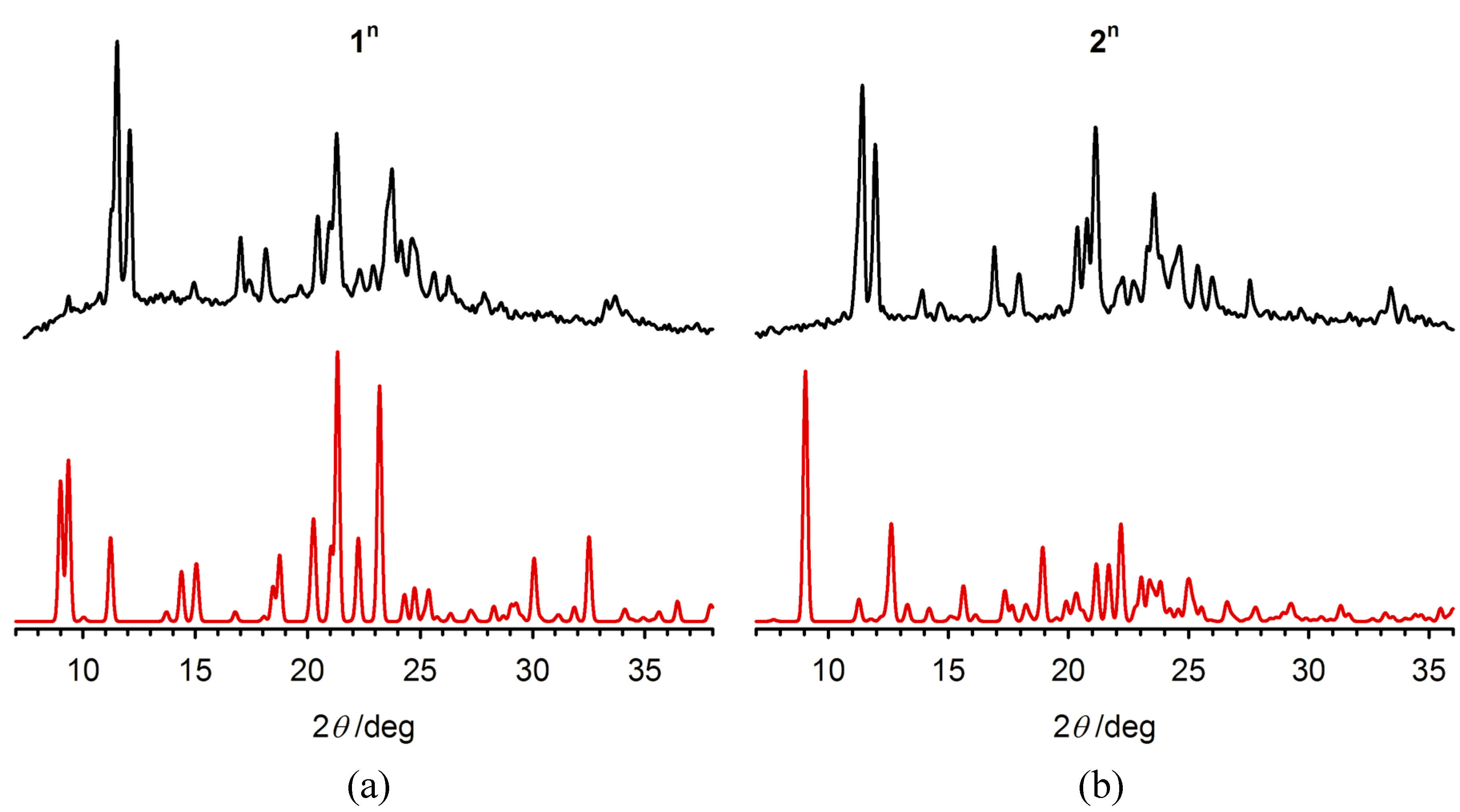
2.3. Characterisation of the Bulk Materials

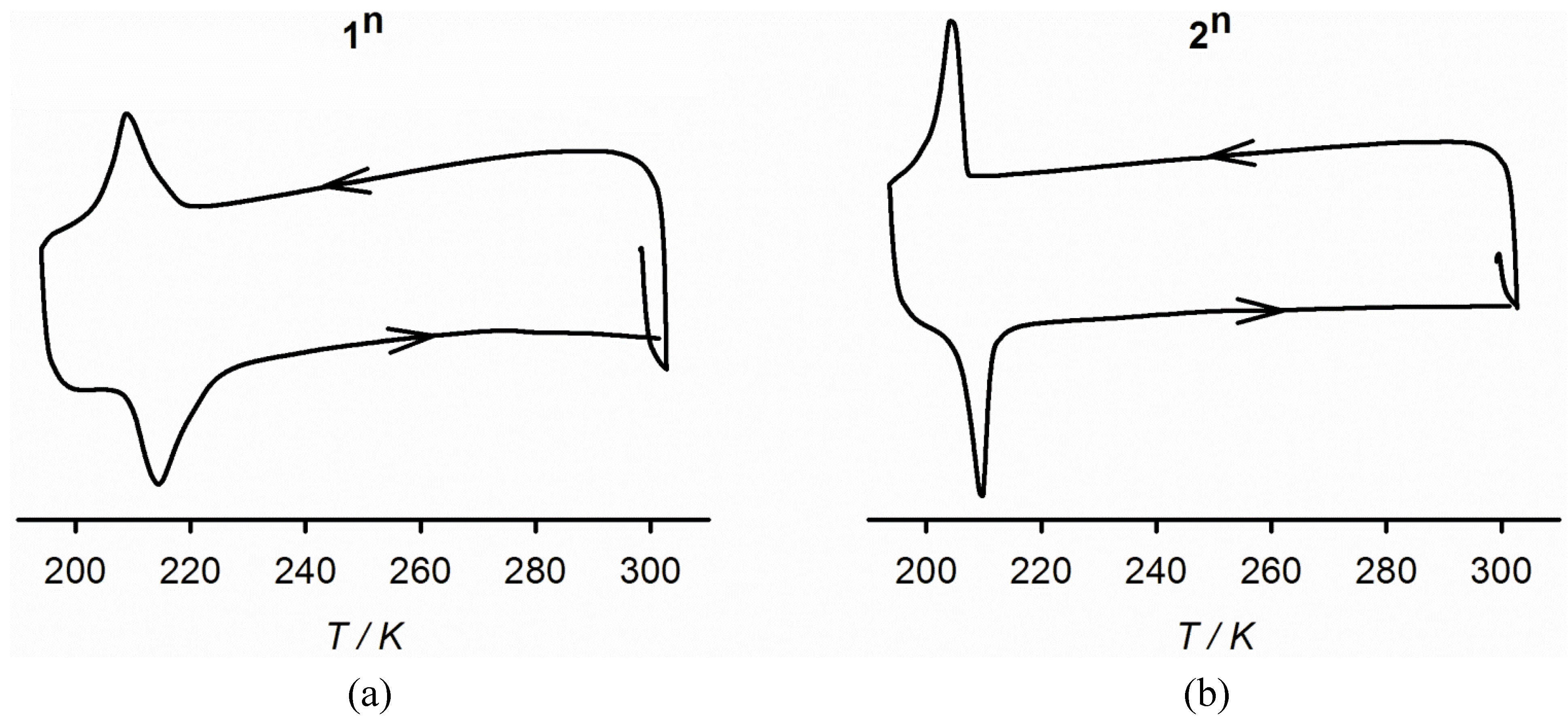
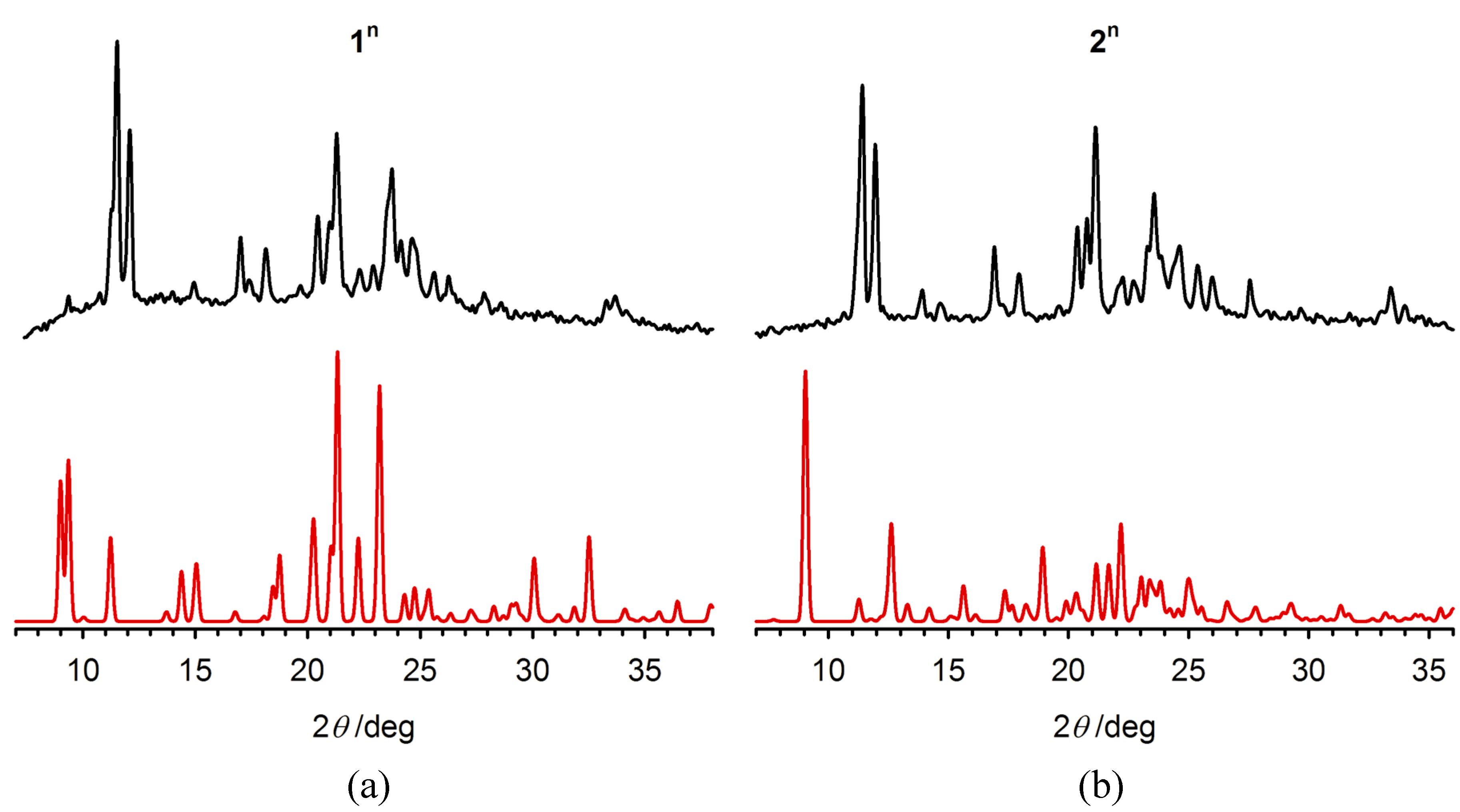
3. Experimental Section
3.1. Instrumentation
3.2. Synthesis
3.2.1. Synthesis of 4-Hydroxy-2,6-di(pyrazol-1-yl)pyridine (L) and 2-Bromo-4-Hydroxy-6-(pyrazol-1-yl)pyridine (L′)
3.2.2. Synthesis of the Complexes
3.3. Crystal Structure Determinations
| Compound | 1·2MeOH | 2·1.75MeOH·0.25H2O | 3·2MeOH |
|---|---|---|---|
| formula | C24H26B2F8FeN10O4 | C23.75H25.5Cl2FeN10O12 | C24H26F12FeN10O2P2 |
| Mr | 748.02 | 769.79 | 864.34 |
| crystal class | monoclinic | orthorhombic | orthorhombic |
| space group | C2/c | Pccn | Pccn |
| a/Å | 23.635(3) | 12.478(3) | 12.4620(2) |
| b/Å | 11.8299(12) | 28.434(6) | 28.4031(4) |
| c/Å | 15.7821(19) | 18.806(4) | 18.7480(3) |
| β/° | 131.720(7) | - | - |
| V/Å3 | 3293.6(7) | 6672(3) | 6636.03(18) |
| Z | 4 | 8 | 8 |
| T/K | 150(2) | 150(2) | 100(2) |
| Dcalcd/Mg·m−3 | 1.508 | 1.533 | 1.730 |
| μ/mm−1 | 0.551 a | 0.686 a | 5.649 b |
| measured reflections | 30968 | 214641 | 17673 |
| unique reflections | 3980 | 11663 | 6582 |
| observed reflections | 3382 | 8429 | 5707 |
| Rint | 0.066 | 320.080 | 0.025 |
| R1 [Fo > 4σ(Fo)] c | 0.076 | 0.059 | 0.033 |
| wR2 [all data] d | 0.230 | 0.168 | 0.083 |
| GoF | 1.065 | 1.051 | 1.040 |
| Δρmax, Δρmin/eÅ−3 | 1.43, −0.65 | 0.75, −0.56 | 1.40, −0.47 |
3.3.1. Structure Refinement of 1·2MeOH.
3.3.2. Structure Refinement of 2·1.75MeOH·0.25H2O
3.3.3. Structure Refinement of 3·2MeOH
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Olguín, J.; Brooker, S. Spin crossover active iron(II) complexes of selected pyrazole-pyridine/pyrazine ligands. Coord. Chem. Rev. 2011, 255, 203–240. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin state behaviour of iron(II)/dipyrazolylpyridine complexes. new insights from crystallographic and solution measurements. Coord. Chem. Rev. 2015, 289–290, 2–12. [Google Scholar] [CrossRef]
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds I-III. Topics in Current Chemistry Vols. 233–235; Springer: Berlin, Germany, 2004.
- Halcrow, M.A. (Ed.) Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Chichester, UK, 2013; p. 568.
- Halcrow, M.A. Recent advances in the synthesis and applications of 2,6-dipyrazolylpyridine derivatives and their complexes. New J. Chem. 2014, 38, 1868–1882. [Google Scholar]
- González-Prieto, R.; Fleury, B.; Schramm, F.; Zoppellaro, G.; Chandrasekar, R.; Fuhr, O.; Lebedkin, S.; Kappes, M.; Ruben, M. Tuning the spin-transition properties of pyrene-decorated 2,6-bispyrazolylpyridine based Fe(II) complexes. Dalton Trans. 2011, 40, 7564–7570. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Han, L.; Oshio, H. Magnetic bistability and single-crystal-to-single-crystal transformation induced by guest desorption. J. Am. Chem. Soc. 2007, 129, 5312–5313. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Sakamoto, R.; Takahashi, K.; Nishihara, H. Solid-state ligand-driven light-induced spin change at ambient temperatures in bis(dipyrazolylstyrylpyridine)iron(II) complexes. Inorg. Chem. 2013, 52, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Stocker, M.; Gieb, K.; Müller, P.; Haryono, M.; Student, K.; Grohmann, A. Spin-state patterns in surface-grafted beads of iron(II) complexes. Angew. Chem. Int. Ed. 2010, 49, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Devid, E.J.; Martinho, P.N.; Kamalakar, M.V.; Šalitroš, I.; Prendergast, U.; Dayen, J.-F.; Meded, V.; Lemma, T.; González-Prieto, R.; Evers, F.; et al. Spin transition in arrays of gold nanoparticles and spin crossover molecules. ACS Nano 2015, 9, 4496–4507. [Google Scholar] [CrossRef] [PubMed]
- Pukenas, L.; Benn, F.; Lovell, E.; Santoro, A.; Kershaw Cook, L.J.; Halcrow, M.A.; Evans, S.D. Bead-like structures and self-assembled monolayers from 2,6-dipyrazolylpyridines and their iron(II) complexes. J. Mater. Chem. C 2015, 3, 7890–7896. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Fisher, J.; Harding, L.P.; Halcrow, M.A. An iron(II) spin-crossover metallacycle from a back-to-back bis-[dipyrazolylpyridine]. Dalton Trans. 2015, 44, 9417–9425. [Google Scholar] [CrossRef] [PubMed]
- Kershaw Cook, L.J.; Shepherd, H.J.; Comyn, T.P.; Baldé, C.; Cespedes, O.; Chastanet, G.; Halcrow, M.A. Decoupled spin-crossover and structural phase transition in a molecular iron(II) complex. Chem. Eur. J. 2015, 21, 4805–4816. [Google Scholar] [CrossRef] [PubMed]
- Kershaw Cook, L.J.; Kulmaczewski, R.; Barrett, S.A.; Halcrow, M.A. Iron(II) complexes of 4-sulfanyl-, 4-sulfinyl- and 4-sulfonyl-2,6-dipyrazolylpyridine ligands. A subtle interplay between spin-crossover and crystallographic phase changes. Inorg. Chem. Front. 2015, 2, 662–670. [Google Scholar] [CrossRef]
- Gaspar, A.B.; Muñoz, M.C.; Niel, V.; Real, J.A. [CoII(4-terpyridone)2]X2: A novel cobalt(II) spin crossover system [4-terpyridone = 2,6-bis(2-pyridyl)-4(1H)-pyridone]. Inorg. Chem. 2001, 40, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Galet, A.; Gaspar, A.B.; Muñoz, M.C.; Real, J.A. Influence of the counterion and the solvent molecules in the spin crossover system [Co(4-terpyridone)2]Xp·nH2O. Inorg. Chem. 2006, 45, 4413–4422. [Google Scholar] [CrossRef] [PubMed]
- Agustí, G.; Bartual, C.; Martínez, V.; Muñoz-Lara, F.J.; Gaspar, A.B.; Muñoz, M.C.; Real, J.A. Polymorphism and “reverse” spin transition in the spin crossover system [Co(4-terpyridone)2](CF3SO3)2·1H2O. New J. Chem. 2009, 33, 1262–1267. [Google Scholar] [CrossRef]
- Holland, J.M.; Kilner, C.A.; Thornton-Pett, M.; Halcrow, M.A. Steric effects on the electronic and molecular structures of nickel(II) and cobalt(II) 2,6-dipyrazol-1-ylpyridine complexes. Polyhedron 2001, 20, 2829–2840. [Google Scholar] [CrossRef]
- Jameson, D.L.; Goldsby, K.A. 2,6-Bis(N-pyrazolyl)pyridines: The convenient synthesis of a family of planar tridentate N3 ligands that are terpyridine analogues. J. Org. Chem. 1990, 55, 4992–4994. [Google Scholar] [CrossRef]
- Vidonne, A.; Philp, D. Integrating replication processes with mechanically interlocked molecular architectures. Tetrahedron 2008, 64, 8464–8475. [Google Scholar] [CrossRef]
- Rodruígez-Ubis, J.C.; Sedano, R.; Barrosso, G.; Juanes, O.; Brunet, E. Lanthanide complexes of polyacid ligands derived from 2,6-bis(pyrazol-l-yl)pyridine, pyrazine, and 6,6′-bis(pyrazol-l-y1)-2,2′-bipyridine: Synthesis and luminescence properties. Helv. Chim. Acta 1997, 80, 86–96. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, G.; Majima, K.; Matsumoto, K. Synthesis of a terbium fluorescent chelate and its application to time-resolved fluoroimmunoassay. Anal. Chem. 2001, 73, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Elhaïk, J.; Pask, C.M.; Kilner, C.A.; Halcrow, M.A. Synthesis of 2,6-di(pyrazol-1-yl)-4-bromomethylpyridine, and its conversion to other 2,6-di(pyrazol-1-yl)pyridines substituted at the pyridine ring. Tetrahedron 2007, 63, 291–298. [Google Scholar] [CrossRef]
- Murguly, E.; Norsten, T.B.; Branda, N. Tautomerism of 4-hydroxyterpyridine in the solid, solution and gas phases: An X-ray, FT-IR and NMR study. J. Chem. Soc. Perkin Trans. 1999, 2, 2789–2794. [Google Scholar] [CrossRef]
- McCusker, J.K.; Rheingold, A.L.; Hendrickson, D.N. Variable-temperature studies of laser-initiated 5T2 → 1A1 intersystem crossing in spin-crossover complexes: Empirical correlations between activation parameters and ligand structure in a series of polypyridyl ferrous complexes. Inorg. Chem. 1996, 35, 2100–2112. [Google Scholar] [CrossRef]
- Guionneau, P.; Marchivie, M.; Bravic, G.; Létard, J.-F.; Chasseau, D. Structural aspects of spin crossover. Example of the [FeIILn(NCS)2] complexes. Top. Curr. Chem. 2004, 234, 97–128. [Google Scholar]
- Holland, J.M.; McAllister, J.A.; Kilner, C.A.; Thornton-Pett, M.; Bridgeman, A.J.; Halcrow, M.A. Stereochemical effects on the spin-state transition shown by salts of [FeL2]2+ [L = 2,6-di(pyrazol-1-yl)pyridine]. J. Chem. Soc. Dalton Trans. 2002, 548–554. [Google Scholar] [CrossRef]
- Clemente-León, M.; Coronado, E.; Giménez-López, M.C.; Romero, F.M. Structural, thermal, and magnetic study of solvation processes in spin-crossover [Fe(bpp)2][Cr(L)(ox)2]2·nH2O complexes. Inorg. Chem. 2007, 46, 11266–11276. [Google Scholar] [CrossRef] [PubMed]
- Amoore, J.J.M.; Kepert, C.J.; Cashion, J.D.; Moubaraki, B.; Neville, S.M.; Murray, K.S. Structural and magnetic resolution of a two-step full spin-crossover transition in a dinuclear iron(II) pyridyl-bridged compound. Chem. Eur. J. 2006, 12, 8220–8227. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, R.; Kilner, C.A.; Halcrow, M.A. Iron(II) complexes with a terpyridine embrace packing motif show remarkably consistent cooperative spin-transitions. Chem. Commun. 2007, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, R.; Chastanet, G.; Tuna, F.; Malkin, T.L.; Barrett, S.A.; Kilner, C.A.; Létard, J.-F.; Halcrow, M.A. The synthesis of new 2,6-di(pyrazol-1-yl)pyrazine derivatives, and the spin state behavior of their iron(II) complexes. Eur. J. Inorg. Chem. 2013, 819–831. [Google Scholar] [CrossRef]
- Brooker, S. Spin crossover with thermal hysteresis: Practicalities and lessons learnt. Chem. Soc. Rev. 2015, 44, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.J. Magnetochemistry—Theory and experimentation. Prog. Inorg. Chem. 1982, 29, 203–283. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cook, L.J.K.; Halcrow, M.A. Synthesis of 4-Hydroxy-2,6-di(pyrazol-1-yl)pyridine, and the Spin State Behaviour of Its Iron(II) Complex Salts. Magnetochemistry 2015, 1, 3-16. https://doi.org/10.3390/magnetochemistry1010003
Cook LJK, Halcrow MA. Synthesis of 4-Hydroxy-2,6-di(pyrazol-1-yl)pyridine, and the Spin State Behaviour of Its Iron(II) Complex Salts. Magnetochemistry. 2015; 1(1):3-16. https://doi.org/10.3390/magnetochemistry1010003
Chicago/Turabian StyleCook, Laurence J. Kershaw, and Malcolm A. Halcrow. 2015. "Synthesis of 4-Hydroxy-2,6-di(pyrazol-1-yl)pyridine, and the Spin State Behaviour of Its Iron(II) Complex Salts" Magnetochemistry 1, no. 1: 3-16. https://doi.org/10.3390/magnetochemistry1010003