
Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Different Kinds of Stimuli-Responsive Hydrogels
2.1. Thermoresponsive Hydrogels
2.2. pH-Responsive Hydrogels
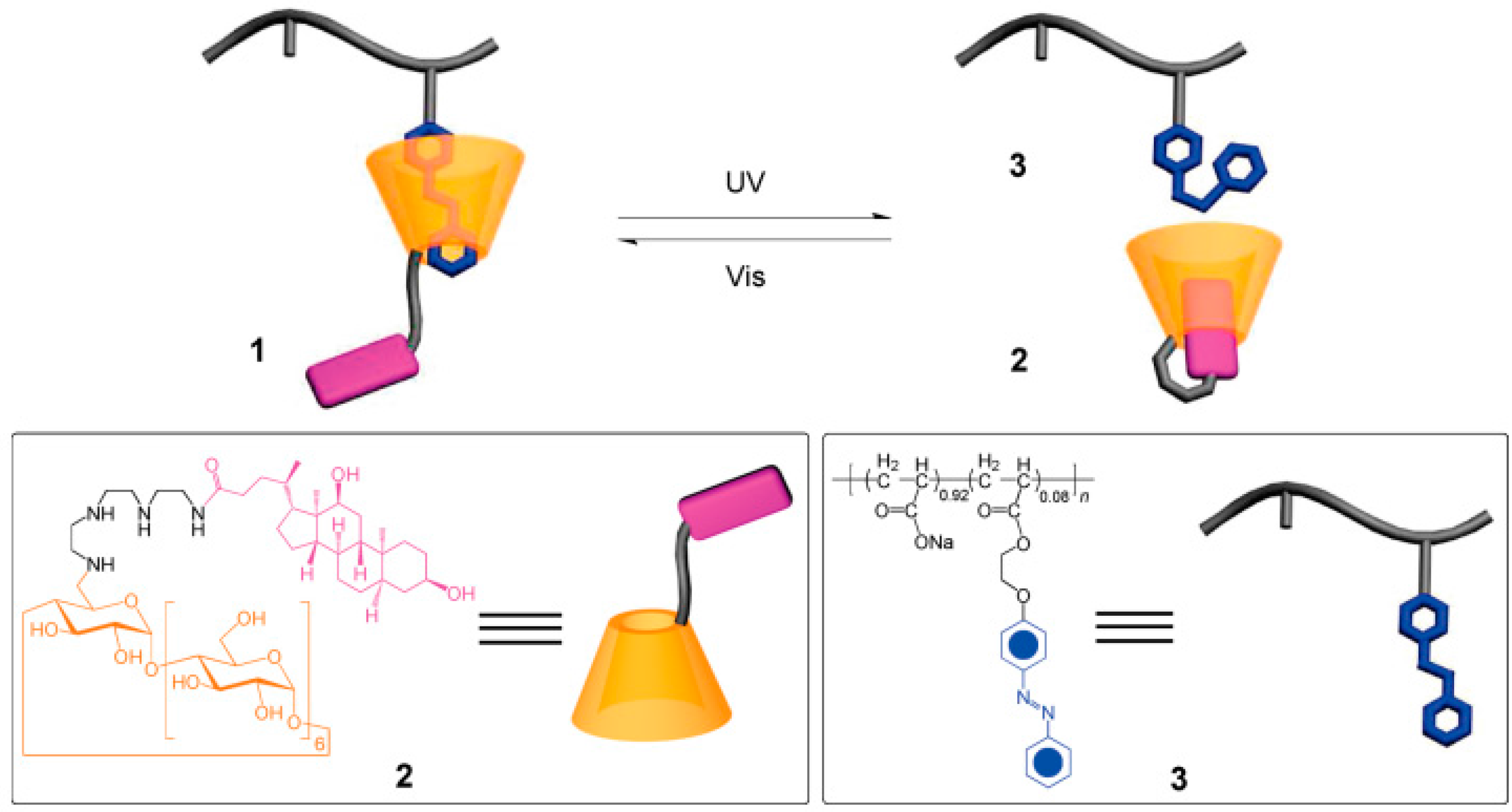
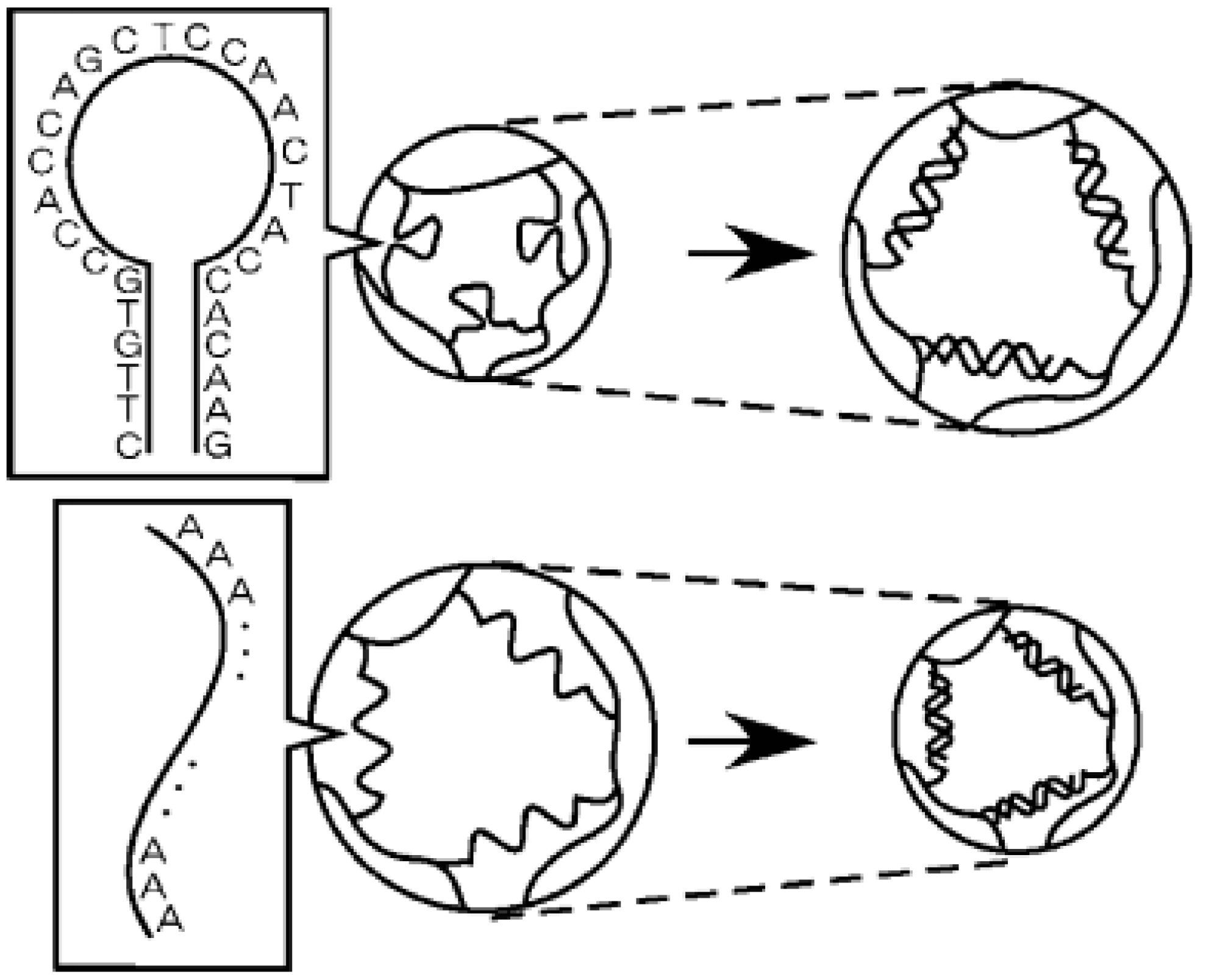
2.3. Light- and Chemical-Responsive Hydrogels
3. Different Theories behind the Hydrogel Swelling Mechanism
3.1. Equilibrium Swelling Theory
3.2. Rubber Elasticity Theory
3.3. Mechanism of Gelation
3.4. Calculation of the Mesh Size
4. Hydrogels Based on Natural Materials
4.1. Hydrogels Based on Polysaccharides
4.2. Hydrogels Based on Polypeptides
5. Synthetic Hydrogels
6. Applications in Biomedical Field
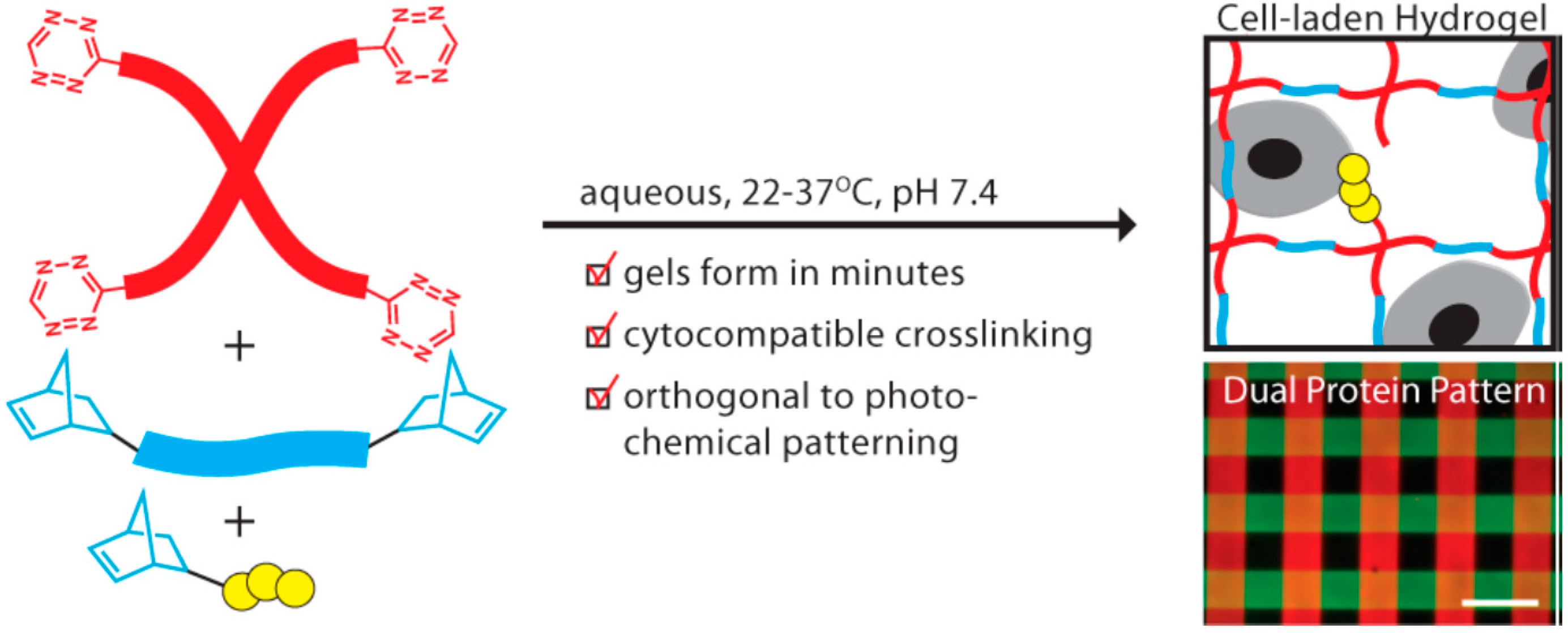
6.1. Hydrogels for Three-Dimensional Cell Culture
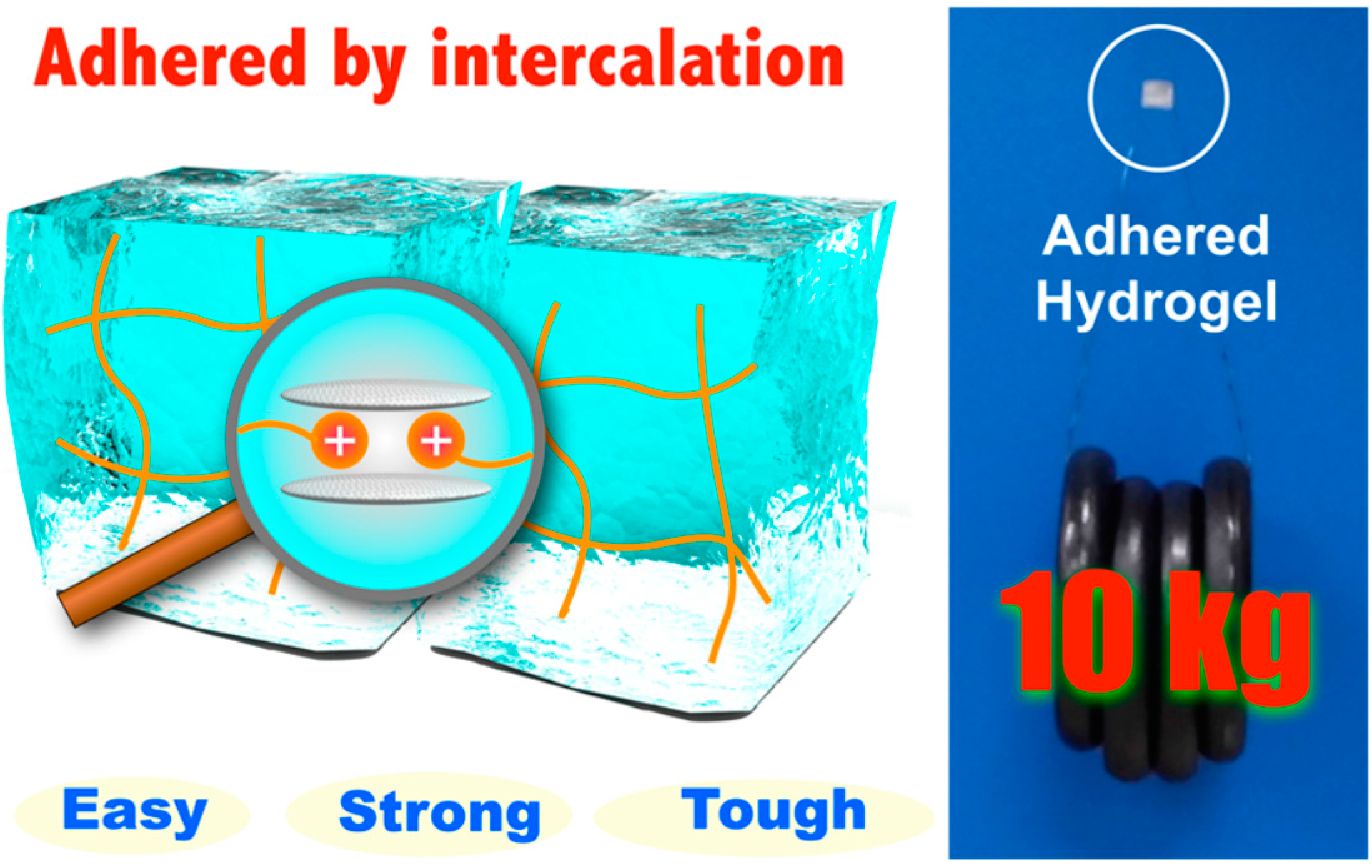
6.2. Hydrogels for Self-Healing
6.3. Hydrogels for Drug Delivery
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kaplan, J.A.; Shieh, A.; Sun, H.-L.; Croce, C.M.; Grinstaff, M.W.; Parquette, J.R. Self-assembly of a 5-fluorouracil-dipeptide hydrogel. Chem. Commun. 2016, 52, 5254–5257. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sun, Y.; Kaplan, J.A.; Grinstaff, M.W.; Parquette, J.R. Photo-crosslinking of a self-assembled coumarin-dipeptide hydrogel. New J. Chem. 2015, 39, 3225–3228. [Google Scholar] [CrossRef]
- Verhulsel, M.; Vignes, M.; Descroix, S.; Malaquin, L.; Vignjevic, D.M.; Viovy, J.L. A review of microfabrication and hydrogel engineering for micro-organs on chips. Biomaterials 2014, 35, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Daniele, M.A.; Adams, A.A.; Naciri, J.; North, S.H.; Ligler, F.S. Interpenetrating networks based on gelatin methacrylamide and PEG formed using concurrent thiol click chemistries for hydrogel tissue engineering scaffolds. Biomaterials 2014, 35, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jiao, Y.; Chai, Q. Applications of Gold Nanoparticles in Biosensors. Nano LIFE 2016, 6, 1642001. [Google Scholar] [CrossRef]
- Yu, X.; Chen, X.; Chai, Q.; Ayres, N. Synthesis of polymer organogelators using hydrogen bonding as physical cross-links. Colloid Polym. Sci. 2016, 294, 59–68. [Google Scholar] [CrossRef]
- Billiet, T.; Vandenhaute, M.; Schelfhout, J.; van Vlierberghe, S.; Dubruel, P. A review of trends and limitations in hydrogel-rapid prototyping for tissue engineering. Biomaterials 2012, 33, 6020–6041. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Wang, T.; Jiao, Y.; Chai, Q.; Yu, X. Gold Nanoparticles: Synthesis and Biological Applications. Nano LIFE 2015, 5, 1542007. [Google Scholar] [CrossRef]
- Ding, R.; Yu, X.; Wang, P.; Zhang, J.; Zhou, Y.; Cao, X.; Tang, H.; Ayres, N.; Zhang, P. Hybrid photosensitizer based on amphiphilic block copolymer stabilized silver nanoparticles for highly efficient photodynamic inactivation of bacteria. RSC Adv. 2016, 6, 20392–20398. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Yu, G.; Zhai, D.; Lee, H.R.; Zhao, W.; Liu, N.; Wang, H.; Tee, B.C.K.; Shi, Y.; Cui, Y.; et al. Hierarchical nanostructured conducting polymer hydrogel with high electrochemical activity. Proc. Natl. Acad. Sci. USA 2012, 109, 9287–9292. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Liu, B.; Shi, Y.; Pan, L.; Wang, Y.; Li, W.; Zhang, R.; Yu, G. Highly sensitive glucose sensor based on Pt nanoparticle/polyaniline hydrogel heterostructures. ACS Nano 2013, 7, 3540–3546. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y.; Pan, L.; Shi, Y.; Cheng, W.; Shi, Y.; Yu, G. A nanostructured conductive hydrogels-based biosensor platform for human metabolite detection. Nano Lett. 2015, 15, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Guan, Z.; Jia, S.; Lei, Z.; Lin, S.; Zhang, H.; Ma, Y.; Tian, Z.Q.; Yang, C.J. Au@Pt nanoparticle encapsulated target-responsive hydrogel with volumetric bar-chart chip readout for quantitative point-of-care testing. Angew. Chem. Int. Ed. 2014, 53, 12503–12507. [Google Scholar]
- Pan, G.; Guo, Q.; Ma, Y.; Yang, H.; Li, B. Thermo-responsive hydrogel layers imprinted with RGDS peptide: A system for harvesting cell sheets. Angew. Chem. Int. Ed. 2013, 52, 6907–6911. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cao, X.; Chen, X.; Ayres, N.; Zhang, P. Triplet–triplet annihilation upconversion from rationally designed polymeric emitters with tunable inter-chromophore distances. Chem. Commun. 2015, 51, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Koetting, M.C.; Guido, J.F.; Gupta, M.; Zhang, A.; Peppas, N.A. pH-responsive and enzymatically-responsive hydrogel microparticles for the oral delivery of therapeutic proteins: Effects of protein size, crosslinking density, and hydrogel degradation on protein delivery. J. Control. Release 2016, 221, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Liu, X.; Duan, S.; Yu, X.; Huang, Y.; Hayat, T.; Li, J. The adsorption of Eu(III) on carbonaceous nanofibers: Batch experiments and modeling study. J. Mol. Liq. 2016, 222, 456–462. [Google Scholar] [CrossRef]
- Peppas, N.A.; Merrill, E.W. Poly(vinyl alcohol) hydrogels: Reinforcement of radiation-crosslinked networks by crystallization. J. Polym. Sci. A Polym. Chem. 1976, 14, 441–457. [Google Scholar] [CrossRef]
- Matanović, M.R.; Kristl, J.; Grabnar, P.A. Thermoresponsive polymers: Insights into decisive hydrogel characteristics, mechanisms of gelation, and promising biomedical applications. Int. J. Pharm. 2014, 472, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N. Hydrogels of poly(vinyl alcohol) and its copolymers. Hydrogels Med. Pharm. 1986, 2, 1–48. [Google Scholar]
- Peppas, N.A.; Mongia, N.K. Ultrapure poly(vinyl alcohol) hydrogels with mucoadhesive drug delivery characteristics. Eur. J. Pharm. Biopharm. 1997, 43, 51–58. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, A.K.; Shukla, S.K.; Bhanu, S.; Kankane, S. Responsive polymers in controlled drug delivery. Prog. Polym. Sci. 2008, 33, 1088–1118. [Google Scholar] [CrossRef]
- Overstreet, D.J.; McLemore, R.Y.; Doan, B.D.; Farag, A.; Vernon, B.L. Temperature-responsive graft copolymer hydrogels for controlled swelling and drug delivery. Soft Matter 2013, 11, 294–304. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, Y.; Hu, X.; Long, Y. Temperature-responsive hydrogel with ultra-large solar modulation and high luminous transmission for “smart window” applications. J. Mater. Chem. A 2014, 2, 13550–13555. [Google Scholar] [CrossRef]
- Schoener, C.A.; Hutson, H.N.; Peppas, N.A. pH-responsive hydrogels with dispersed hydrophobic nanoparticles for the oral delivery of chemotherapeutics. J. Biomed. Mater. Res. A 2013, 101, 2229–2236. [Google Scholar] [CrossRef] [PubMed]
- De, S.K.; Aluru, N.; Johnson, B.; Crone, W.; Beebe, D.J.; Moore, J. Equilibrium swelling and kinetics of pH-responsive hydrogels: Models, experiments, and simulations. J. Microelectromech. Syst. 2002, 11, 544–555. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [Google Scholar] [PubMed]
- Dong, L.; Jiang, H. Autonomous microfluidics with stimuli-responsive hydrogels. Soft Matter 2007, 3, 1223–1230. [Google Scholar] [CrossRef]
- Holtz, J.H.; Asher, S.A. Polymerized colloidal crystal hydrogel films as intelligent chemical sensing materials. Nature 1997, 389, 829–832. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Stoddart, J.F. Azobenzene-Based Light-Responsive Hydrogel System. Langmuir 2009, 25, 8442–8446. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-W.; Zhu, D.; Jiang, H. An infrared-light responsive graphene-oxide incorporated poly(N-isopropylacrylamide) hydrogel nanocomposite. Soft Matter 2011, 7, 5604–5609. [Google Scholar] [CrossRef]
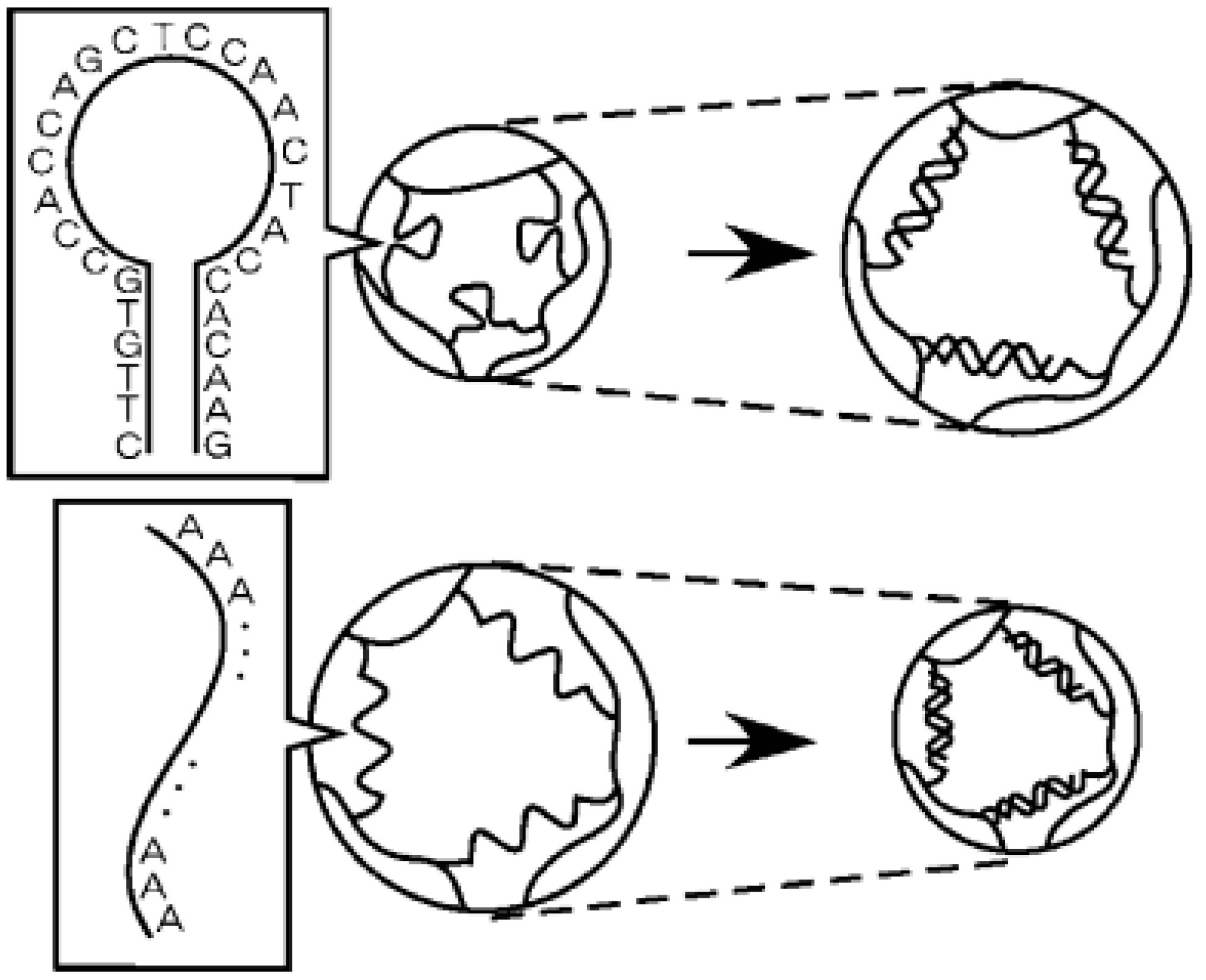
- Murakami, Y.; Maeda, M. DNA-responsive hydrogels that can shrink or swell. Biomacromolecules 2005, 6, 2927–2929. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J.; Rehner, J., Jr. Statistical mechanics of cross-linked polymer networks II. Swelling. J. Chem. Phys. 1943, 11, 521–526. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Peppas, N.A.; Merrill, E.W. Crosslinked poly(vinyl alcohol) hydrogels as swollen elastic networks. J. Appl. Polym. Sci. 1977, 21, 1763–1770. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Flory, P.J.; Rabjohn, N.; Shaffer, M.C. Dependence of elastic properties of vulcanized rubber on the degree of cross linking. J. Polym. Sci. 1949, 4, 225–245. [Google Scholar] [CrossRef]
- Peppas, N.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Lowman, A.M.; Peppas, N.A. Analysis of the complexation/decomplexation phenomena in graft copolymer networks. Macromolecules 1997, 30, 4959–4965. [Google Scholar] [CrossRef]
- Mark, J.E. Polymer Networks; Springer: Heidelberg, Germany, 1982; pp. 1–26. [Google Scholar]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv Drug Deliver Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef]
- Gao, X.; Cao, Y.; Song, X.; Zhang, Z.; Xiao, C.; He, C.; Chen, X. pH-and thermo-responsive poly(N-isopropylacrylamide-co-acrylic acid derivative) copolymers and hydrogels with LCST dependent on pH and alkyl side groups. J. Mater. Chem. B 2013, 1, 5578–5587. [Google Scholar] [CrossRef]
- Schild, H. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Ruel-Gariépy, E.; Leroux, J.-C. In situ-forming hydrogels—Review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.; Pedersen, J.S. Structural study on the micelle formation of poly(ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) triblock copolymer in aqueous solution. Macromolecules 1993, 26, 805–812. [Google Scholar] [CrossRef]
- Jaeger, J.A. Landscape division, splitting index, and effective mesh size: New measures of landscape fragmentation. Landsc. Ecol. 2000, 15, 115–130. [Google Scholar] [CrossRef]
- Canal, T.; Peppas, N.A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 1989, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, X.; Payne, G.F.; Rubloff, G.W. Biofabrication: Programmable assembly of polysaccharide hydrogels in microfluidics as biocompatible scaffolds. J. Mater. Chem. 2012, 22, 7659–7666. [Google Scholar] [CrossRef]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindström, T.; Ankerfors, M.; Gray, D.; Dorris, A. Nanocelluloses: A New Family of Nature-Based Materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Malinen, M.M.; Lauren, P.; Lou, Y.-R.; Kuisma, S.W.; Kanninen, L.; Lille, M.; Corlu, A.; GuGuen-Guillouzo, C.; Ikkala, O. Nanofibrillar cellulose hydrogel promotes three-dimensional liver cell culture. J. Control. Release 2012, 164, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Isobe, N.; Lee, D.-S.; Kwon, Y.-J.; Kimura, S.; Kuga, S.; Wada, M.; Kim, U.-J. Immobilization of protein on cellulose hydrogel. Cellulose 2011, 18, 1251–1256. [Google Scholar] [CrossRef]
- Kim, M.H.; An, S.; Won, K.; Kim, H.J.; Lee, S.H. Entrapment of enzymes into cellulose–biopolymer composite hydrogel beads using biocompatible ionic liquid. J. Mol. Catal. B Enzym. 2012, 75, 68–72. [Google Scholar] [CrossRef]
- Bott, K.; Upton, Z.; Schrobback, K.; Ehrbar, M.; Hubbell, J.A.; Lutolf, M.P.; Rizzi, S.C. The effect of matrix characteristics on fibroblast proliferation in 3D gels. Biomaterials 2010, 31, 8454–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Xu, K.; Zheng, X.; Giacomin, A.J.; Mix, A.W.; Kao, W.J. 3D cell entrapment in crosslinked thiolated gelatin-poly(ethylene glycol) diacrylate hydrogels. Biomaterials 2012, 33, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-Z.; Chen, Y.-C.; Moreno-Luna, R.; Khademhosseini, A.; Melero-Martin, J.M. Transdermal regulation of vascular network bioengineering using a photopolymerizable methacrylated gelatin hydrogel. Biomaterials 2013, 34, 6785–6796. [Google Scholar] [CrossRef] [PubMed]
- Pok, S.; Myers, J.D.; Madihally, S.V.; Jacot, J.G. A multilayered scaffold of a chitosan and gelatin hydrogel supported by a PCL core for cardiac tissue engineering. Acta Biomater. 2013, 9, 5630–5642. [Google Scholar] [CrossRef] [PubMed]
- Milašinović, N.; Kalagasidis Krušić, M.; Knežević-Jugović, Z.; Filipović, J. Hydrogels of N-isopropylacrylamide copolymers with controlled release of a model protein. Int. J. Pharm. 2010, 383, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Anghelache, A.; Teodorescu, M.; Stić, M.; Knežević-Jugović, Z.; Filipović, J. Novel crosslinked thermoresponsive hydrogels with controlled poly(ethylene glycol)—poly(propylene glycol) multiblock copolymer structure. Colloid Polym. Sci. 2014, 292, 829–838. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Naficy, S.; Razal, J.M.; Whitten, P.G.; Wallace, G.G.; Spinks, G.M. A pH-sensitive, strong double-network hydrogel: Poly(ethylene glycol) methyl ether methacrylates–poly(acrylic acid). J. Polym. Sci. B Polym. Phys. 2012, 50, 423–430. [Google Scholar] [CrossRef]
- Zhang, H.; Qadeer, A.; Chen, W. In situ gelable interpenetrating double network hydrogel formulated from binary components: Thiolated chitosan and oxidized dextran. Biomacromolecules 2011, 12, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chen, W.; Yan, L. An inorganic–organic double network hydrogel of graphene and polymer. Nanoscale 2013, 5, 6034–6039. [Google Scholar] [CrossRef] [PubMed]
- Baroli, B. Hydrogels for tissue engineering and delivery of tissue-inducing substances. J. Pharm. Sci. 2007, 96, 2197–2223. [Google Scholar] [CrossRef] [PubMed]
- Nicodemus, G.D.; Bryant, S.J. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. B Rev. 2008, 14, 149–165. [Google Scholar] [CrossRef] [PubMed]
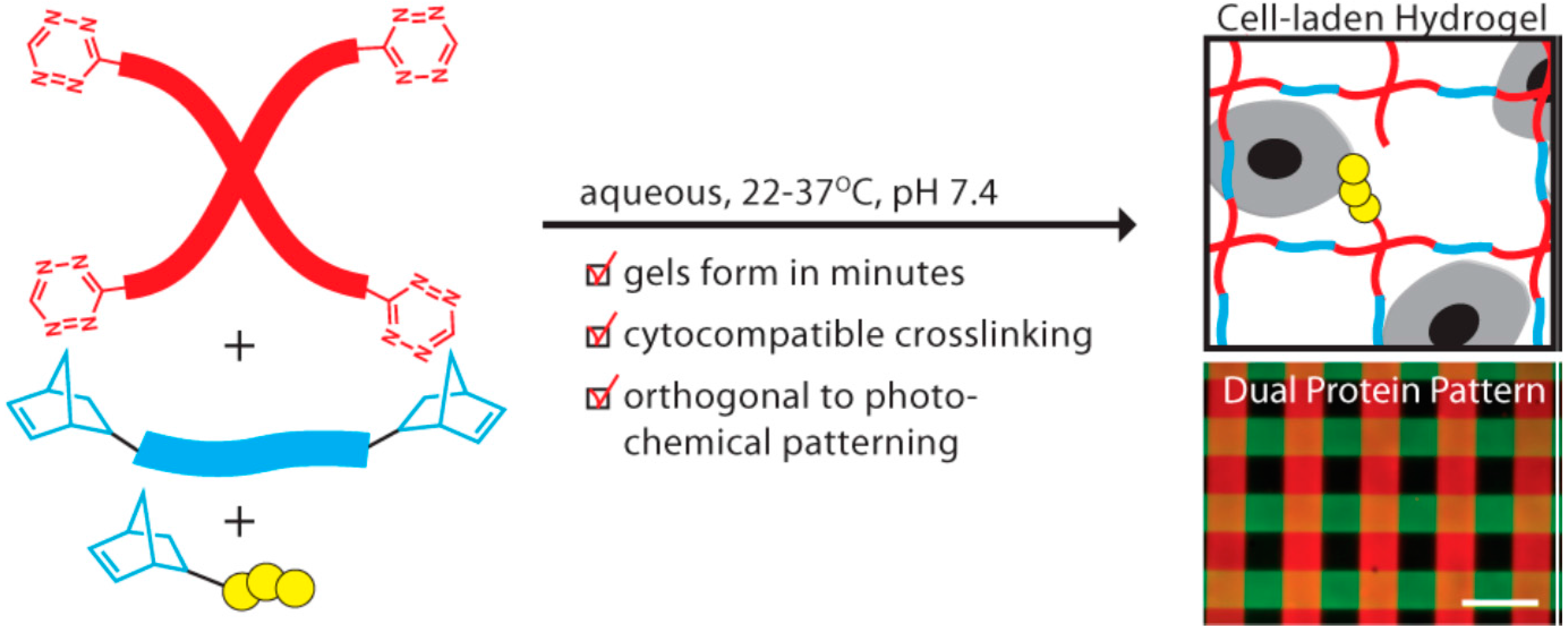
- Alge, D.L.; Azagarsamy, M.A.; Donohue, D.F.; Anseth, K.S. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine–norbornene chemistry. Biomacromolecules 2013, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, E.; Melchels, F.P.; Holzapfel, B.M.; Meckel, T.; Hutmacher, D.W.; Loessner, D. Gelatine methacrylamide-based hydrogels: An alternative three-dimensional cancer cell culture system. Acta Biomater. 2014, 10, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Meure, S.; Solomon, D. Self-healing polymeric materials: A review of recent developments. Prog. Polym. Sci. 2008, 33, 479–522. [Google Scholar] [CrossRef]
- Thakur, V.K.; Kessler, M.R. Self-healing polymer nanocomposite materials: A review. Polymer 2015, 69, 369–383. [Google Scholar] [CrossRef]
- Deng, G.; Li, F.; Yu, H.; Liu, F.; Liu, C.; Sun, W.; Jiang, H.; Chen, Y. Dynamic hydrogels with an environmental adaptive self-healing ability and dual responsive sol–gel transitions. ACS Macro Lett. 2012, 1, 275–279. [Google Scholar] [CrossRef]
- Amiri, S.; Rahimi, A. Hybrid nanocomposite coating by sol–gel method: A review. Iran Polym. J. 2016, 25, 559–577. [Google Scholar] [CrossRef]
- Bode, S.; Bose, R.; Matthes, S.; Ehrhardt, M.; Seifert, A.; Schacher, F.; Paulus, R.; Stumpf, S.; Sandmann, B.; Vitz, J. Self-healing metallopolymers based on cadmium bis(terpyridine) complex containing polymer networks. Polym. Chem. 2013, 4, 4966–4973. [Google Scholar] [CrossRef]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.; Hayes, W.; Mackay, M.E.; Rowan, S.J. A Self-repairing, supramolecular polymer system: Healability as a consequence of donor–acceptor π–π stacking interactions. Chem. Commun. 2009, 6717–6719. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Hill, M.R.; Sumerlin, B.S. Self-healing hydrogels containing reversible oxime crosslinks. Soft Matter 2015, 11, 6152–6161. [Google Scholar] [CrossRef] [PubMed]
- Tuncaboylu, D.C.; Sari, M.; Oppermann, W.; Okay, O. Tough and self-healing hydrogels formed via hydrophobic interactions. Macromolecules 2011, 44, 4997–5005. [Google Scholar] [CrossRef]
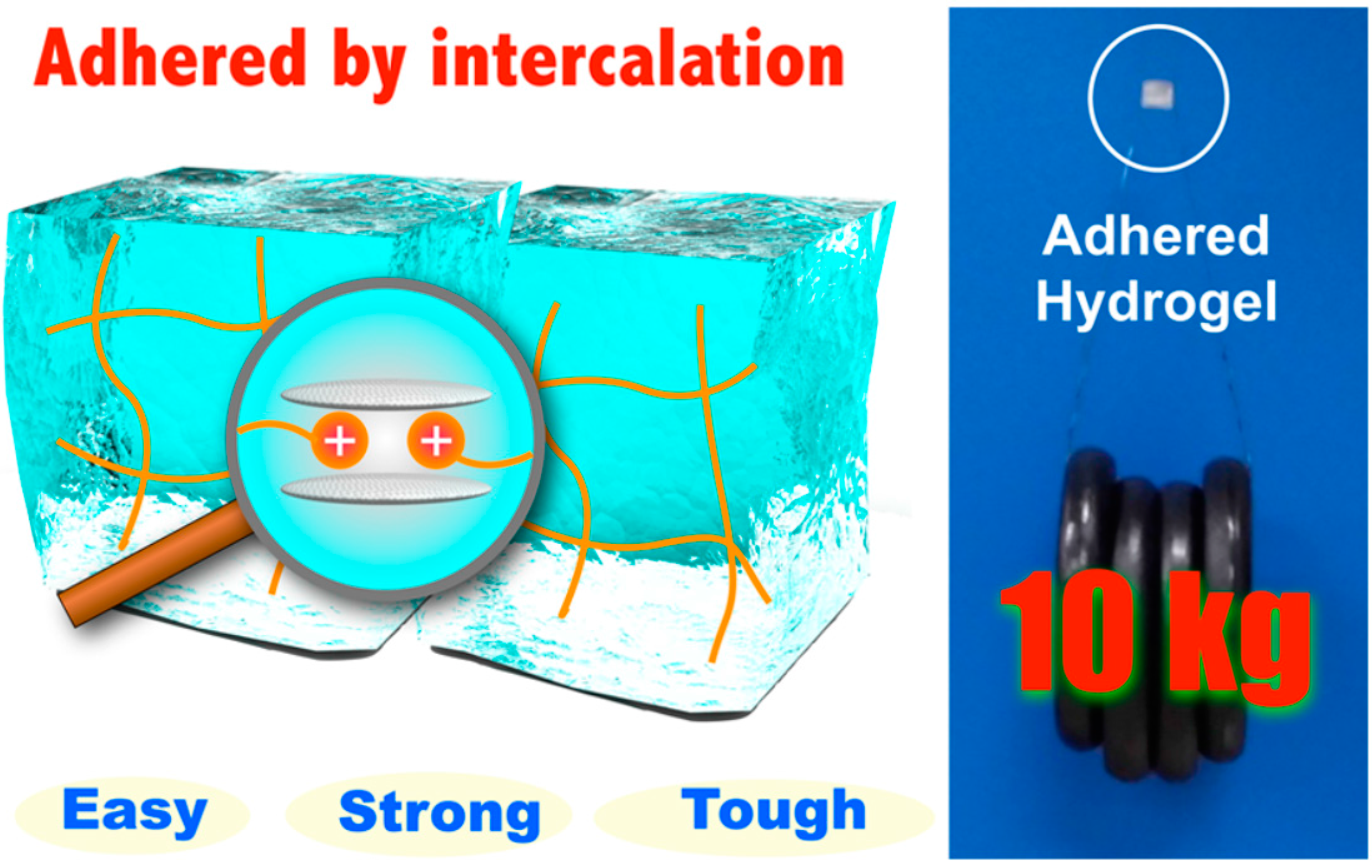
- Tamesue, S.; Yasuda, K.; Noguchi, S.; Mitsumata, T.; Yamauchi, T. Highly Tolerant and Durable Adhesion between Hydrogels Utilizing Intercalation of Cationic Substituents into Layered Inorganic Compounds. ACS Macro Lett. 2016, 5, 704–708. [Google Scholar] [CrossRef]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Paavola, A.; Yliruusi, J.; Kajimoto, Y.; Kalso, E.; Wahlström, T.; Rosenberg, P. Controlled release of lidocaine from injectable gels and efficacy in rat sciatic nerve block. Pharm. Res. 1995, 12, 1997–2002. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Cohn, D. Ethoxysilane-capped PEO–PPO–PEO triblocks: A new family of reverse thermo-responsive polymers. Biomaterials 2004, 25, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.Y.; Chung, T.W.; Kim, B.C.; Kim, M.K.; Lee, J.H.; Wee, W.R.; Cho, C.S. Release of ciprofloxacin from poloxamer-graft-hyaluronic acid hydrogels in vitro. Int. J. Pharm. 2003, 260, 83–91. [Google Scholar] [CrossRef]
- Kim, M.R.; Park, T.G. Temperature-responsive and degradable hyaluronic acid/Pluronic composite hydrogels for controlled release of human growth hormone. J. Control. Release 2002, 80, 69–77. [Google Scholar] [CrossRef]
- Xiong, L.; Luo, Q.; Wang, Y.; Li, X.; Shen, Z.; Zhu, W. An injectable drug-loaded hydrogel based on a supramolecular polymeric prodrug. Chem. Commun. 2015, 51, 14644–14647. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. https://doi.org/10.3390/gels3010006
Chai Q, Jiao Y, Yu X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels. 2017; 3(1):6. https://doi.org/10.3390/gels3010006
Chicago/Turabian StyleChai, Qinyuan, Yang Jiao, and Xinjun Yu. 2017. "Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them" Gels 3, no. 1: 6. https://doi.org/10.3390/gels3010006