Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata


1
Department of Biology, William Paterson University, Wayne, NJ 07470, USA
2
Public Health Research Institute, New Jersey Medical School, Rutgers Biomedical and Health Sciences, Newark, NJ 07103, USA
*
Authors to whom correspondence should be addressed.
J. Fungi 2018, 4(3), 105; https://doi.org/10.3390/jof4030105
Submission received: 16 August 2018
/
Revised: 28 August 2018
/
Accepted: 30 August 2018
/
Published: 1 September 2018
(This article belongs to the Special Issue Treatments for Fungal Infections)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Candida glabrata has thoroughly adapted to successfully colonize human mucosal membranes and survive in vivo pressures. prior to and during antifungal treatment. Out of all the medically relevant Candida species, C. glabrata has emerged as a leading cause of azole, echinocandin, and multidrug (MDR: azole + echinocandin) adaptive resistance. Neither mechanism of resistance is intrinsic to C. glabrata, since stable genetic resistance depends on mutation of drug target genes, FKS1 and FKS2 (echinocandin resistance), and a transcription factor, PDR1, which controls expression of major drug transporters, such as CDR1 (azole resistance). However, another hallmark of C. glabrata is the ability to withstand drug pressure both in vitro and in vivo prior to stable “genetic escape”. Additionally, these resistance events can arise within individual patients, which underscores the importance of understanding how this fungus is adapting to its environment and to drug exposure in vivo. Here, we explore the evolution of echinocandin resistance as a multistep model that includes general cell stress, drug adaptation (tolerance), and genetic escape. The extensive genetic diversity reported in C. glabrata is highlighted.
1. Epidemiology and Mechanisms of Resistance
Invasive fungal infections are a major cause of global morbidity and mortality, accounting for nearly 1.4 million deaths a year [1]. Fungal populations colonize the human host at multiple body sites and represent the majority of eukaryotes in the human gut microbiome, with most organisms having a potential to act as opportunistic pathogens during immunosuppression or when natural barriers are disrupted [2]. Bloodstream fungal infections, largely caused by yeasts of the Candida genus, are associated with high mortality rates (45–75%) and pose a serious threat to immunocompromised individuals, including cancer and AIDS patients, organ transplant recipients, and premature infants. The increasing burden of fungal infections has led to a rise in the use of antifungal agents for their treatment and prevention. Unfortunately, treatment options for invasive fungal infections are extremely limited, as there are few antifungal drug classes. For decades, the azole antifungals (e.g., fluconazole), which are fungistatic drugs targeting membrane sterol biosynthesis, were used as primary prophylaxis/therapy to prevent/treat Candida infections, with C. albicans as the predominant infecting species. But epidemiological shifts in infecting organisms toward non-albicans Candida species, which are inherently azole resistant (e.g., C. krusei) or rapidly acquire resistance (e.g., C. glabrata), has led to the widespread use of echinocandin antifungal drugs.
In most clinical settings, C. albicans is the predominant bloodstream pathogen. Yet, the prevalence of C. glabrata infections has been rising for several decades and, at 18–25% of Candida isolates, it is the second most common Candida bloodstream infection in North America. In some settings, such as patients with hematological malignancies, it is the principal bloodstream fungal pathogen [3]. Due to the widespread use of azole antifungals for prophylaxis/therapy, global azole resistance among C. glabrata isolates is around 8% [4], while some centers have rates exceeding 20% [5]. Echinocandin therapy is highly efficacious, but emerging echinocandin drug resistance is a growing threat to successful clinical management. Among C. albicans and other Candida species, the frequency of echinocandin resistance remains relatively low (1–3%) [6,7], but this is not true for C. glabrata, where resistance is more severe and often presents as multidrug (MDR) resistance [8,9]. While echinocandin resistance among C. glabrata isolates ranges from 3–5% in population-based studies [10], some centers report rates of 10–15% [3,11]. Strains with MDR phenotypes (azole and echinocandin, and sometime polyene resistance) are increasingly encountered with some centers. Nearly one-third of echinocandin resistant isolates are also resistant to azoles [12].
While multiple mechanisms of azole resistance have been reported for Candida species [13], the overwhelming singular mechanism of resistance identified in clinical isolates of C. glabrata is mutation of the transcription factor PDR1, which leads to increased expression of multidrug transporters that act as efflux pumps [14,15]. Unlike azoles, multidrug transporters do not appear to play a role in echinocandin resistance, as echinocandins are not substrates for transport [16]. As such, echinocandins are fully active against azole resistant Candida [17].
The echinocandin drugs (caspofungin, micafungin and anidulafungin), which were first approved for clinical use in 2001, target and inhibit the membrane-associated (and fungal specific) β-1-3-d-glucan synthase and block the biosynthesis of β-1,3-glucan, a major structural component of the fungal cell wall. They are broadly active against Candida species, in which they are considered fungicidal (more on this later). The enzyme complex consists of a structural/catalytic subunit encoded by FKS genes; and its activity is regulated by Rho, a GTP-binding protein [18]. Clinical resistance involves modification of the Fks subunits [19]. In C. glabrata, two functionally redundant genes, FKS1 and FKS2, encode glucan synthase catalytic subunits [20]. In most Candida species mutations occur in two highly conserved “hot-spot” regions of FKS1 and, in C. glabrata, FKS2. Resistance-conferring amino acid substitutions induce elevated MIC values [21] and the most prominent mutations can reduce the sensitivity of glucan synthase (IC50) to drug by >3,000 fold [22]. In the 16 years following FDA approval of caspofungin, FKS mutations are still the only mechanism associated with clinical failures [10,23]. Given a long clinical history of safe and efficacious therapy, echinocandins are now the IDSA recommended preferred antifungal agent for treatment of candidiasis among high-risk patient populations [24].
Echinocandin resistance always arises during therapy and is associated with repeated or chronic drug exposure, although resistance can also follow brief drug exposure [25]. Thus, C. glabrata has an elevated potential relative to other Candida species to develop echinocandin resistance, for reasons that are currently not understood. The global resistance problem is expected to grow more severe as expanding numbers of patients are exposed to antifungal prophylaxis and echinocandin drugs like caspofungin are now generic. Given the importance of this drug class as a first-line agent, there is an urgent need to better understand factors that contribute to and limit the emergence of echinocandin resistance among patients with C. glabrata infections.
2. Evolution of Echinocandin Resistance
Clinical antifungal treatment failure is most often a combination of microbial factors, host factors, drug pharmacokinetics (PK)/pharmacodynamics (PD), and drug distribution at the site of infections. All of these factors contribute to therapeutic efficacy and resistance development, although this review will primarily focus on microbial genetic factors contributing to echinocandin resistance. While the terminal step of echinocandin resistance (FKS mutation) has been well defined, mechanisms used by Candida to survive as both a commensal and an opportunistic pathogen within a harsh environment consisting of bacterial microbiota and host immune factors are less well characterized. All colonizing strains of Candida employ mechanisms of adaptation, but C. glabrata has a prominent ability to adapt and survive antifungal pressure in vivo, resulting in drug resistance. The emerging pathogen C. auris has arisen as a considerable public health concern following reports of elevated rates of antifungal resistance and horizontal transmission within healthcare centers [26]. Conversely, like other Candida species, transmission of C. glabrata between patients has rarely been reported, suggesting independent development of antifungal resistance within most patients. Unlike C. albicans, C. glabrata does not normally form hyphae or secrete hydrolytic enzymes, and therefore, elicits a lesser immune response [27]. Despite this apparent lack of virulence factors, C. glabrata can robustly replicate and disseminate upon host immunosuppression. The following sections will explore factors that allow C. glabrata to adapt to its environment and develop antifungal resistance at higher rates than other species.
2.1. Drug Adaptation Is a Key Intermediate Leading to Echinocandin Resistance
Although echinocandins are considered fungicidal drugs in Candida species, careful examination of their effect on C. glabrata both in vitro and in vivo shows that while the vast majority of cells die upon echinocandin exposure, roughly one in 104–5 of cells survive and demonstrate “drug adaptation” over a wide range of drug exposures (Figure 1). Similarly in an in vivo infection, echinocandin tolerance is manifested as a decline in target organ fungal burdens (e.g., from 109 to 104 cells), but not true sterilization, as fungal stasis is achieved (i.e., no net change cell counts) [28]. Cells that survive echinocandin action (without forming FKS mutations) are defined as drug tolerant (or adapted), as they are fully sensitive to drug when re-cultured. They may display higher MIC values but respond to drug in pharmacodynamic models [29]. Ultimately, such adapted cells can persist long enough to give rise to FKS mutants, which escape drug action and result in clinical failure (Figure 2). Despite this key role of drug adaptation in development of drug resistance, the factors underlying echinocandin adaptation in C. glabrata have not been well defined, particularly in vivo.
One factor that may aid C. glabrata in echinocandin adaptation is poor drug penetration into sites of colonization or infection. The echinocandins are intravenously administered drugs that appear to distribute weakly in the GI tract [30]. Some echinocandins, like micafungin, penetrate intraabdominal abscesses of murine models at considerably lower concentrations than what is measured in the blood [31]. Following echinocandin treatment, fungal clearance may be observed in the bloodstream, although cells located at sites of colonization or deep tissue infection have been exposed to lower levels of drug, resulting in a potential reservoir of FKS mediated resistance. Subsequent or repeated treatment with an echinocandin can lead to rapid breakthrough [32]. This clinical scenario has been modeled in mice as repeated treatments of caspofungin at 4× the humanized dose increased the frequency of FKS mutants formed within the GI tract in a model of colonization [30]. Drug penetration can also be hindered by the formation of a biofilm matrix by Candida species [33]. The new echinocandin rezafungin (formerly CD101; Cidara, San Diego, CA, USA) can be administered safely at a considerably higher level and can achieve favorable probabilities of PK-PD target attainment [34], which results in increased efficacy and reduced burden/sterilization at the site of intraabdominal abscesses [31]. Ultimately, a balance between drug concentration and mutant prevention would be best, and targeting drug adaptation mechanisms (see below) in combination with an echinocandin may prove beneficial. These are questions researchers should consider when studying echinocandin adaptation and resistance.
2.2. Cellular Drivers of Echinocandin Adaptation
Stress tolerance, including antifungal drug tolerance, has been attributed to the activation of multiple stress response pathways within the yeast cell, including the cell wall integrity pathway/Protein Kinase C (PKC)/mitogen activated protein kinase (MAPK) cascade signaling, Hsp90-dependent calcium/calcineurin signaling, high osmolarity glycerol (HOG) signaling, and the cyclic AMP/Protein Kinase A (PKA) signaling pathway [13]. While these responses have been extensively studied in the model fungus S. cerevisiae [35], to which C. glabrata is closely related, C. glabrata, unlike S. cerevisiae, has evolved to survive within the human host. Thus, stress tolerance pathways in C. glabrata likely have key differences from those in S. cerevisiae to reflect the very different challenges of their environments, and should be validated in animal models of colonization and infection. In general, these stress response pathways seem to be involved in the response to multiple antifungal classes and are sometimes, but not always, conserved across fungi. While stress-triggered changes in transcriptional profiles have been reported in S. cerevisiae [36], C. albicans [37] and C. glabrata [38], the roles of these signaling pathways in C. glabrata antifungal drug tolerance have not been systematically investigated.
As detailed above, echinocandin adaptation in C. glabrata is a key step towards development of FKS escape mutations (Figure 2). Echinocandins target the fungal cell wall. It has been well established that in response to cell wall damage, fungi upregulate a number of stress responses and cell wall maintenance pathways that help the cells tolerate and survive the stress [39]. Of particular importance upon echinocandin exposure is the cell wall integrity pathway which regulates glucan synthesis through Rho1 and cell wall repair. Rho1 activation leads to upregulation of the FKS genes and activation of PKC. Cells with decreased PKC activity or those lacking activated MAP kinases (e.g., ScBCK1, ScSLT2, CaMKC1) are hypersensitive to the echinocandins [40,41,42].
2.3. C. glabrata Specific Echinocandin Adaptation
Some of the stress induced mechanisms mentioned above, such as the cell wall integrity pathway (e.g., WSC1, MKK1, BCK1, SLT2) [43,44,45], Hsp90 and calcineurin signaling [46,47], and chromatin remodeling [48,49], have been shown to abrogate echinocandin tolerance or adaptation when disrupted or targeted in C. glabrata. In S. cerevisiae and C. albicans, echinocandin-induced PKC1 expression has been linked to increased production of cell wall components chitin and mannan, potentially compensating for the loss of β-glucans [40,50,51]. In C. glabrata, the significance of chitin during echinocandin exposure seems to be more complicated. While one study reported that an increase in chitin led to incomplete killing of C. glabrata by caspofungin [45], another reported that there were no significant increases in chitin production upon caspofungin exposure in vitro [52]. A more recent study noted an increase in C. glabrata chitin levels upon murine GI tract colonization [53]. We have shown that treatment of colonized mice with a combination of caspofungin and the chitin synthase inhibitor Nikkomycin Z caused an increase in killing of C. glabrata within the murine GI tract and a decrease of dissemination upon immunosuppression [30]. In addition to the apparent compensation for β-glucan loss, recent studies have also noted the elevated expression of specific genes (e.g., BGL2, XOG1, GAS2) that are related to the replacement of β-1,3-glucans in the biofilm matrix following echinocandin exposure [54,55], which may also influence adaptation.
In a comprehensive study by Schwarzmuller and colleagues [44], a partial C. glabrata gene knockout library was constructed and screened for increased susceptibilities to antifungals, including caspofungin. Multiple gene knockouts, including those involved in cell wall organization, chromatin assembly, transcriptional regulation, and signal transduction, were associated with caspofungin hypersensitivity [44]. Many of these genes have not been linked to echinocandin hypersensitivity in S. cerevisiae or C. albicans, although for most, it remains to be shown if targeting these cellular pathways/components would negate echinocandin adaptation in vivo. Another important study analyzed genome mutations throughout the echinocandin treatment course of a patient with recurrent C. glabrata candidemia [46]. Tracking the progression of Candida prior to the acquisition of an FKS mutation will begin to shed light on factors essential for echinocandin adaptation.
2.4. Echinocandin- and FKS Gene- Specific Effects
Different echinocandins may elicit varying or different fungal adaptive responses. For example, targeting specific sphingolipid biosynthesis genes or chemically altering the sphingolipid cellular makeup led to a differential echinocandin susceptibility pattern in Candida species, including C. glabrata [56,57]. Although, this differential activity may be due to the physical interaction between the echinocandins and the target Fks proteins within the membrane, potential echinocandin-specific effects should be considered when attempting to “target” an adaptive response mechanism. New glucan synthase targeting echinocandins that are in development may also produce differing cellular responses. As stated above, rezufungin can reportedly penetrate into deep tissue lesions better than micafungin [31] and exhibits a long half-life in PK studies [58,59]. An orally-active glucan synthase inhibitor, SCY-078 (Scynexis, Jersey City, NJ, USA), exhibits activity against some otherwise-resistant FKS mutants [60], likely a result of a slightly different binding spot on the Fks protein [61].
As detailed above, genetic resistance to echinocandins requires the formation of mutations within “hotspot” regions of glucan synthase subunits, encoded by FKS genes. Most Candida species rely on one essential FKS gene (FKS1), while FKS2 and FKS3 are expressed at lower levels and have yet to be fully characterized in C. albicans. In S. cerevisiae, FKS2 and FKS3 are important during sporulation and mating [62,63]. Interestingly, a recent study demonstrated that expression of FKS2 and FKS3 in C. albicans can influence overall drug sensitivity [64]. C. glabrata is the only Candida species that has two seemingly redundant, yet differentially regulated, FKS subunits: FKS1 and FKS2 (this is also true in S. cerevisiae). Unlike S. cerevisiae, sporulation and mating have not been observed in C. glabrata yeast. FKS2 expression is dependent upon the calcium/calcineurin/Hsp90 signaling pathway, and targeting of this pathway either genetically or chemically results in a reversal of Fks2-mediated resistance in C. glabrata [20,46]. While FKS2 expression was increased following caspofungin or calcium exposure, the authors concluded that transcriptional control was not the only mechanism of Fks2 modulation in C. glabrata [20]. Gaining a better understanding of how each FKS gene is controlled, transcriptionally and otherwise, will help tease out one more unique property of C. glabrata and the response to echinocandins.
2.5. MDR, PDR1 and Adhesins
Candida glabrata readily forms MDR phenotypes, which involves separate resistance mechanisms for each drug class (modification of drug target site for echinocandins versus expression of drug efflux transporters for azoles). Despite the apparent lack of mechanistic overlap, a nexus may exist. The presence of a PDR1 mutation appears to increase the ability of C. glabrata to adapt to other stressors, including echinocandin exposure. Specific PDR1 mutations in C. glabrata not only confer azole resistance, but can also enhance adhesion to epithelial cells through increased expression of the epithelial adhesin gene EPA1 [65,66,67,68]. The genome of C. glabrata carries a large number of EPA (epithelial adhesin) genes that encode for adhesin proteins [69,70,71]. Interestingly, a recent study found that separate clinical isolates expressed a unique variety of adhesins and other cell wall proteins [72], most likely due to the subtelomeric positions of adhesin genes and the unusually high genomic plasticity of C. glabrata [70,73] (see more below). PDR1-mediated increased expression of EPA1 increased organ colonization in a mouse UTI model [66] and virulence in a model of hematogenous disseminated candidiasis [74,75]. An increase in adhesion that aids in colonization of mucosal membranes may also increase echinocandin tolerance through common cellular pathways (Figure 2). Again, the expansion and dissemination of C. glabrata is dependent upon the host’s immune response, and this is highlighted by the ability of natural killer (NK) cells to recognize C. glabrata through binding of Epa proteins [76].
2.6. Exploiting Genetic Diversity
According to classical evolution, random mutations arise in microbial populations, whereupon a change in conditions (e.g., exposure to antifungal drug) favors pre-existing mutants that are more fit under the new conditions (i.e., resistant to the drug). However, an extensive body of work in bacteria [77,78,79,80] and S. cerevisiae [81], as well as computational models of mutation rates [82], indicates that in stressed cells genome maintenance and repair mechanisms are altered, promoting mutability and increasing the pool of genetic diversity from which drug-resistant mutations can emerge. Furthermore, heteroresistance may play a vital role in cellular adaptation during stress, and epigenetic and post-translational modification mechanisms are emerging [83,84]. Such mechanisms may be particularly important for haploid organisms like C. glabrata that have extremely limited ability to generate genetic diversity via meiosis and recombination [85]. Thus, the probability that a tolerant C. glabrata cell will genetically escape drug action is a function of its mutagenic potential (Figure 2). However, the mechanisms of mutagenesis operating in drug-tolerant C. glabrata cells are not fully known.
The ability to increase genetic diversity within a C. glabrata population would help the yeast survive as a commensal and transition into a pathogen. Several studies, including ours, have shown that clinical isolates of C. glabrata show astounding genetic diversity both in terms of nucleotide sequence and chromosome structure [86,87,88,89]. C. glabrata can seemingly duplicate and reorganize chromosomes at high frequencies generating changes in size and variation of chromosomes [88,90]. As a result, studies have identified gene duplications in C. glabrata to include that of cell wall proteins, such as mannosyltransferases, aspartyl proteases, phospholipases, the ABC transporter PDH1, and the sterol transporter AUS1 [88,90]. Additionally, as mentioned earlier, EPA adhesin genes important for mucosal colonization have also been heavily duplicated within C. glabrata genomes [72,90]. It is not clear whether these rearrangements occur acutely in response to treatment and/or represent divergent sub-species best adapted for colonization. Variations in karyotypes were identified in clinical isolates taken from the same patients over the course of antifungal treatment [86,91,92]; however, we have also found that that different sequence types (STs), or clades, are characterized not only by different single nucleotide polymorphisms (SNPs) but also by varying chromosomal configurations [86]. While there is a high correlation between chromosomal configurations and STs, it is not absolute.
Are chromosomal integrity components in C. glabrata missing or downregulated? According to Polakova and colleagues [88], homologs of two proteins (Ten1 and Rif2) that function in S. cerevisiae telomere length end protection and length regulation are absent from the C. glabrata genome, although additional homologs with similar functions, such as Rap1, Sir3, and Rif1, have been characterized in C. glabrata [73,93]. Expression of the adhesin genes is regulated by several subtelomeric silencing complexes (see [72] for review). The extensive chromosomal rearrangements between strains have been a partial barrier to rapid Illumina whole genome sequencing of C. glabrata clinical isolates because the reference strain ATCC 2001, which belongs to ST15, cannot serve as an appropriate template for assembly of genomes of many other STs. Overcoming these technical difficulties will aid in the understanding of C. glabrata drug adaptation through chromosomal rearrangement.
Fungi contain multiple mechanisms that regulate mutagenesis, including several highly-conserved DNA repair systems, such as double-strand break repair (DSBR), base-excision repair (BER), nucleotide-excision repair (NER), post-replication repair (PRR), and mismatch repair (MMR). DNA polymerases, including several error-prone polymerases, also impinge on mutation rates [94,95,96]. Defects or programmed changes (e.g., as during stress-induced mutagenesis [77]) in these mechanisms are often associated with increased mutation rates [97]. These pathways have been well studied in vitro in the model fungus S. cerevisiae. We have previously evaluated the role of MMR in C. glabrata and shown that active MMR suppresses emergence of drug-resistant mutants and that naturally occurring variants in C. glabrata MMR gene MSH2 may promote development of resistance in some clades [87,98].
Importantly, we and others have found that different MSH2 genotypes are characteristic of distinct STs/clades, suggesting that different STs may have different propensity towards mutability and acquiring drug resistant gene variants [86,98,99,100]. This is significant because the distribution of C. glabrata STs varies both by geography and over time. For instance, C. glabrata ST distribution in Atlanta area hospitals changed significantly between 1992 and 2008, the time period that includes the introduction of echinocandins [101]. One significant change is the increased prevalence of ST16, which carries a msh2 variant associated with increased echinocandin resistance frequencies in vitro [87] and was shown to be more prevalent among drug-resistant clinical isolates [102], suggesting that this ST may have an increased capacity for drug escape. However, there are also expanding STs (e.g., ST3) that do not carry specific msh2 alterations, emphasizing that there are additional factors at play.
Specific MSH2 alleles most likely diversify populations of C. glabrata to better survive in vivo, and upon prolonged antifungal exposure, may aid in drug target mutation. Multiple clinical studies performed at non-U.S. clinics have reported no correlation between MSH2 genotype and clinical resistance frequencies in populations with limited drug exposure and/or very low levels of drug resistance [98,99,100,103]. DNA repair alterations may be more relevant in certain populations where antifungals are routinely used for prophylaxis and treatment, and where a higher prevalence of MDR phenotypes are observed [87]. It should be noted that not all MSH2 mutations lead to significant increases in mutants in vitro; for example, alleles encoding for P208S/N890I and E231G/L269F produced greater frequencies of resistant mutants in vitro, while others produced smaller or no increases in frequencies [98,103].
Additional mechanisms at play within individual isolates exhibiting the same MSH2 genotype also likely affect the mutagenic properties. For example, when we expressed a wild type copy of MSH2 in several strains that contained deficient MSH2 alleles, an increase in FKS mutagenesis was complemented in some strains, but not in others [104], indicative of additional mechanisms of mutagenesis at play. Importantly, MSH2 likely represents one piece in a multifaceted and complex puzzle that makes up drug escape. Our preliminary studies also show that disruption of other genes involved in MMR, such as PMS1 and MSH6, produce greater frequencies of antifungal-resistance and FKS mutagenesis in vitro (Figure 3). How sequence polymorphisms or transcriptional control of these genes affects C. glabrata is unknown. As listed above, additional cellular mechanisms may also influence mutagenesis in C. glabrata and ultimately affect its ability to colonize, disseminate, and develop resistance. In a broader context, defects or changes in DNA repair may be an evolutionarily adaptive mechanism(s) of C. glabrata to generate greater genetic diversity among colonizing strains in order to better adapt to its environment, and introduction of antifungal drug into that environment is just one more factor in a slew of others. Notably, the consequences of this genetic diversity on colonization, infection, and drug resistance are not fully understood. A more dynamic view of cellular mutagenic potential may be more relevant that individual components. This will require new tools and approaches.
3. Conclusions
Rates of acquired resistance to azoles and echinocandins are substantially higher among strains of Candida glabrata compared to other Candida species. The ability of C. glabrata to survive antifungal pressure at high rates within individual patients highlights its astounding adaptive flexibility. This flexibility is likely due to a myriad of factors, including strong general cell stress responses (e.g., cell wall integrity pathway and regulation of associated genes) and multiple mechanisms of drug adaptation (e.g., HSP90/calcineurin, chitin synthesis, adhesion, genetic diversity). The combination of these cellular mechanisms (and other factors such as host immune status and drug penetration and pharmacokinetics) ultimately permit or enhance genetic escape (PDR1 and FKS mutations) and stable resistance, which can result in clinical failure. Importantly, how many of these factors influence colonization, infection, and drug resistance in vivo have not been fully determined.
Author Contributions
K.R.H. and D.S.P. performed experiments and wrote the manuscript.
Funding
This research was supported by the NIH grant AI109025 to D.S.P. and by an Arnold O. Beckman postdoctoral fellowship from the Arnold and Mabel Beckman Foundation to K.R.H.
Acknowledgments
This research was supported by the NIH grant AI109025 to D.S.P. and by an Arnold O. Beckman postdoctoral fellowship from the Arnold and Mabel Beckman Foundation to K.R.H.
Conflicts of Interest
D.S.P. receives funding from US National Institutes of Health and contracts from the CDC, Astellas, Scynexis, Cidara and Amplyx. He serves on advisory boards for Astellas, Cidara, Amplyx, Scynexis, and Matinas. In addition, D.S.P. has an issued US patent concerning echinocandin resistance. K.R.H. declares no competing financial interests. The authors alone are responsible for the content and writing of the paper.
References
- Brown, G.D.; Denning, D.W.; Levitz, S.M. Tackling human fungal infections. Science 2012, 336, 647. [Google Scholar] [CrossRef] [PubMed]
- Huseyin, C.E.; O’Toole, P.W.; Cotter, P.D.; Scanlan, P.D. Forgotten fungi-the gut mycobiome in human health and disease. FEMS Microbiol. Rev. 2017, 41, 479–511. [Google Scholar] [CrossRef] [PubMed]
- Farmakiotis, D.; Tarrand, J.J.; Kontoyiannis, D.P. Drug-resistant Candida glabrata infection in cancer patients. Emerg. Infect. Dis. 2014, 20, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Deshpande, L.M.; Davis, A.P.; Rhomberg, P.R.; Pfaller, M.A. Monitoring antifungal resistance in a global collection of invasive yeasts and molds: Application of clsi epidemiological cutoff values and whole-genome sequencing analysis for detection of azole resistance in Candida albicans. Antimicrob. Agents Chemother. 2017, 61, e00906-17. [Google Scholar] [CrossRef] [PubMed]
- Farmakiotis, D.; Kontoyiannis, D.P. Epidemiology of antifungal resistance in human pathogenic yeasts: Current viewpoint and practical recommendations for management. Int. J. Antimicrob. Agents 2017, 50, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Dannaoui, E.; Desnos-Ollivier, M.; Garcia-Hermoso, D.; Grenouillet, F.; Cassaing, S.; Baixench, M.T.; Bretagne, S.; Dromer, F.; Lortholary, O. Candida spp. With acquired echinocandin resistance, france, 2004–2010(1). Emerg. Infect. Dis. 2012, 18, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Woosley, L.N.; Diekema, D.J.; Messer, S.A.; Jones, R.N.; Pfaller, M.A. Low prevalence of FKS1 hot spot 1 mutations in a worldwide collection of Candida strains. Antimicrob. Agents Chemother. 2010, 54, 2655–2659. [Google Scholar] [CrossRef] [PubMed]
- Ostrosky-Zeichner, L. Candida glabrata and FKS mutations: Witnessing the emergence of the true multidrug-resistant Candida. Clin. Infect. Dis. 2013, 56, 1733–1734. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and management. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Perlin, D.S. Mechanisms of echinocandin antifungal drug resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jimenez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing echinocandin resistance in Candida glabrata: Clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 2013, 56, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.D.; Iqbal, N.; Bolden, C.B.; Kuykendall, R.J.; Harrison, L.H.; Farley, M.M.; Schaffner, W.; Beldavs, Z.G.; Chiller, T.M.; Park, B.J.; et al. Role of FKS mutations in Candida glabrata: Mic values, echinocandin resistance, and multidrug resistance. Antimicrob. Agents Chemother. 2014, 58, 4690–4696. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Sanglard, D.; Howard, S.J.; Rogers, P.D.; Perlin, D.S. Mechanisms of antifungal drug resistance. Cold Spring Harb. Perspect. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.F.; Krol, A.A.; Sarti, K.E.; Bennett, J.E. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 2006, 50, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Vermitsky, J.P.; Earhart, K.D.; Smith, W.L.; Homayouni, R.; Edlind, T.D.; Rogers, P.D. PDR1 regulates multidrug resistance in Candida glabrata: Gene disruption and genome-wide expression studies. Mol. Microbiol. 2006, 61, 704–722. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Maki, K.; Ikeda, F.; Holmes, A.R.; Lamping, E.; Niimi, M.; Monk, B.C.; Cannon, R.D. Overexpression of Candida albicans cdr1, cdr2, or mdr1 does not produce significant changes in echinocandin susceptibility. Antimicrob. Agents Chemother. 2006, 50, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Current perspectives on echinocandin class drugs. Future Microbiol. 2011, 6, 441–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur, P.; Baginsky, W. In vitro activity of 1,3-β-d-glucan synthase requires the GTP-binding protein rho1. J. Biol. Chem. 1996, 271, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kelly, R.; Kahn, J.N.; Robles, J.; Hsu, M.J.; Register, E.; Li, W.; Vyas, V.; Fan, H.; Abruzzo, G.; et al. Specific substitutions in the echinocandin target fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. Isolates. Antimicrob. Agents Chemother. 2005, 49, 3264–3273. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Alastruey-Izquierdo, A.; Healey, K.R.; Johnson, M.E.; Perlin, D.S.; Edlind, T.D. FkS1 and FkS2 are functionally redundant but differentially regulated in Candida glabrata: Implications for echinocandin resistance. Antimicrob. Agents Chemother. 2012, 56, 6304–6309. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Perlin, D.S. Echinocandin resistance: An emerging clinical problem? Curr. Opin. Infect. Dis. 2014, 27, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Effron, G.; Park, S.; Perlin, D.S. Correlating echinocandin mic and kinetic inhibition of FKS1 mutant glucan synthases for Candida albicans: Implications for interpretive breakpoints. Antimicrob. Agents Chemother. 2009, 53, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Nguyen, M.H.; Press, E.G.; Kwa, A.L.; Cheng, S.; Du, C.; Clancy, C.J. The presence of an fks mutation rather than mic is an independent risk factor for failure of echinocandin therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob. Agents Chemother. 2012, 56, 4862–4869. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S., 2nd; Wiederhold, N.P.; Wickes, B.L.; Patterson, T.F.; Jorgensen, J.H. Rapid emergence of echinocandin resistance in Candida glabrata resulting in clinical and microbiologic failure. Antimicrob. Agents Chemother. 2013, 57, 4559–4561. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, S.R.; Berkow, E.L.; Chow, N.; Welsh, R.M. Candida auris for the clinical microbiology laboratory: Not your grandfather’s Candida species. Clin. Microbiol. Newsl. 2017, 39, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Brunke, S.; Hube, B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell. Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Livermore, J.; Sharp, A.; Goodwin, J.; Gregson, L.; Alastruey-Izquierdo, A.; Perlin, D.S.; Warn, P.A.; Hope, W.W. Pharmacodynamics of echinocandins against Candida glabrata: Requirement for dosage escalation to achieve maximal antifungal activity in neutropenic hosts. Antimicrob. Agents Chemother. 2011, 55, 4880–4887. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.; Castanheira, M.; Diekema, D.; Pfaller, M.; Andes, D. Optimizing echinocandin dosing and susceptibility breakpoint determination via in vivo pharmacodynamic evaluation against Candida glabrata with and without FKS mutations. Antimicrob. Agents Chemother. 2012, 56, 5875–5882. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Nagasaki, Y.; Zimmerman, M.; Kordalewska, M.; Park, S.; Zhao, Y.; Perlin, D.S. The gastrointestinal tract is a major source of echinocandin drug resistance in a murine model of Candida glabrata colonization and systemic dissemination. Antimicrob. Agents Chemother. 2017, 61, e01412-17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Prideaux, B.; Nagasaki, Y.; Lee, M.H.; Chen, P.Y.; Blanc, L.; Ho, H.; Clancy, C.J.; Nguyen, M.H.; Dartois, V.; et al. Unraveling drug penetration of echinocandin antifungals at the site of infection in an intra-abdominal abscess model. Antimicrob. Agents Chemother. 2017, 61, e01009-17. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.D.; Garcia-Effron, G.; Zaas, A.K.; Perfect, J.R.; Perlin, D.S.; Alexander, B.D. Breakthrough invasive candidiasis in patients on micafungin. J. Clin. Microbiol. 2010, 48, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Rodrigues, C.F.; Araujo, D.; Rodrigues, M.E.; Henriques, M. Candida species biofilms’ antifungal resistance. J. Fungi 2017, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.C.; Lakota, E.A.; Flanagan, S.; Ong, V.; Sandison, T.; Rubino, C.M.; Bhavnani, S.M.; Ambrose, P.G. Overcoming the resistance hurdle: Pharmacokinetic-pharmacodynamic target attainment analyses for rezafungin (cd101) against Candida albicans and Candida glabrata. Antimicrob. Agents Chemother. 2018, 62, e02614-17. [Google Scholar] [CrossRef] [PubMed]
- Lesage, G.; Bussey, H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006, 70, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Rogers, P.D.; Baerson, S.R.; Jacob, M.R.; Barker, K.S.; Cleary, J.D.; Walker, L.A.; Nagle, D.G.; Clark, A.M. Genome-wide expression profiling of the response to polyene, pyrimidine, azole, and echinocandin antifungal agents in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 34998–35015. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.T.; Lee, R.E.; Barker, K.S.; Wei, L.; Homayouni, R.; Rogers, P.D. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob. Agents Chemother. 2005, 49, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.G.; Arora, G.; Ferrandino, R.; Gerace, E.L.; Mohammednetej, M.; Nosair, W.; Rattila, S.; Subic, A.Z.; Rolfes, R. Identification of genes in Candida glabrata conferring altered responses to caspofungin, a cell wall synthesis inhibitor. G3 2016, 6, 2893–2907. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Steinbach, W.J. Stress, drugs, and evolution: The role of cellular signaling in fungal drug resistance. Eukaryot. Cell 2008, 7, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Markovich, S.; Yekutiel, A.; Shalit, I.; Shadkchan, Y.; Osherov, N. Genomic approach to identification of mutations affecting caspofungin susceptibility in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2004, 48, 3871–3876. [Google Scholar] [CrossRef] [PubMed]
- Reinoso-Martin, C.; Schuller, C.; Schuetzer-Muehlbauer, M.; Kuchler, K. The yeast protein kinase C cell integrity pathway mediates tolerance to the antifungal drug caspofungin through activation of slt2p mitogen-activated protein kinase signaling. Eukaryot. Cell 2003, 2, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P.; Kontoyiannis, D.P.; Prince, R.A.; Lewis, R.E. Attenuation of the activity of caspofungin at high concentrations against Candida albicans: Possible role of cell wall integrity and calcineurin pathways. Antimicrob. Agents Chemother. 2005, 49, 5146–5148. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Inamine, T.; Yamauchi, S.; Nagayoshi, Y.; Saijo, T.; Izumikawa, K.; Seki, M.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; et al. Role of the slt2 mitogen-activated protein kinase pathway in cell wall integrity and virulence in Candida glabrata. FEMS Yeast Res. 2010, 10, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Schwarzmuller, T.; Ma, B.; Hiller, E.; Istel, F.; Tscherner, M.; Brunke, S.; Ames, L.; Firon, A.; Green, B.; Cabral, V.; et al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 2014, 10, e1004211. [Google Scholar] [CrossRef] [PubMed]
- Cota, J.M.; Grabinski, J.L.; Talbert, R.L.; Burgess, D.S.; Rogers, P.D.; Edlind, T.D.; Wiederhold, N.P. Increases in slt2 expression and chitin content are associated with incomplete killing of Candida glabrata by caspofungin. Antimicrob. Agents Chemother. 2008, 52, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Singh-Babak, S.D.; Babak, T.; Diezmann, S.; Hill, J.A.; Xie, J.L.; Chen, Y.L.; Poutanen, S.M.; Rennie, R.P.; Heitman, J.; Cowen, L.E. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog. 2012, 8, e1002718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Yamauchi, S.; Inamine, T.; Nagayoshi, Y.; Saijo, T.; Izumikawa, K.; Seki, M.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; et al. Roles of calcineurin and crz1 in antifungal susceptibility and virulence of Candida glabrata. Antimicrob. Agents Chemother. 2010, 54, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.N.; Balusu, S.; Gorityala, N.; Dandu, L.; Kaur, R. Functional genomic analysis of Candida glabrata-macrophage interaction: Role of chromatin remodeling in virulence. PLoS Pathog. 2012, 8, e1002863. [Google Scholar] [CrossRef] [PubMed]
- Garnaud, C.; Champleboux, M.; Maubon, D.; Cornet, M.; Govin, J. Histone deacetylases and their inhibition in Candida species. Front. Microbiol. 2016, 7, 1238. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.A.; Ichinomiya, M.; Koshi, Y.; Horiuchi, H. Escape of Candida from caspofungin inhibition at concentrations above the mic (paradoxical effect) accomplished by increased cell wall chitin; evidence for β-1,6-glucan synthesis inhibition by caspofungin. Antimicrob. Agents Chemother. 2006, 50, 3160–3161. [Google Scholar] [CrossRef] [PubMed]
- Munro, C.A.; Selvaggini, S.; de Bruijn, I.; Walker, L.; Lenardon, M.D.; Gerssen, B.; Milne, S.; Brown, A.J.; Gow, N.A. The PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin synthesis in Candida albicans. Mol. Microbiol. 2007, 63, 1399–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.A.; Gow, N.A.; Munro, C.A. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob. Agents Chemother. 2013, 57, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Charlet, R.; Pruvost, Y.; Tumba, G.; Istel, F.; Poulain, D.; Kuchler, K.; Sendid, B.; Jawhara, S. Remodeling of the Candida glabrata cell wall in the gastrointestinal tract affects the gut microbiota and the immune response. Sci. Rep. 2018, 8, 3316. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.F.; Henriques, M. Portrait of matrix gene expression in Candida glabrata biofilms with stress induced by different drugs. Genes 2018, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.F.; Rodrigues, M.E.; Henriques, M. Susceptibility of Candida glabrata biofilms to echinocandins: Alterations in the matrix composition. Biofouling 2018, 34, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Challa, K.K.; Edlind, T.D.; Katiyar, S.K. Sphingolipids mediate differential echinocandin susceptibility in Candida albicans and Aspergillus nidulans. Antimicrob. Agents Chemother. 2015, 59, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Katiyar, S.K.; Raj, S.; Edlind, T.D. Crs-mis in Candida glabrata: Sphingolipids modulate echinocandin-fks interaction. Mol. Microbiol. 2012, 86, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ong, V.; James, K.D.; Smith, S.; Krishnan, B.R. Pharmacokinetics of the novel echinocandin cd101 in multiple animal species. Antimicrob. Agents Chemother. 2017, 61, e01626-16. [Google Scholar] [CrossRef] [PubMed]
- Sandison, T.; Ong, V.; Lee, J.; Thye, D. Safety and pharmacokinetics of cd101 IV, a novel echinocandin, in healthy adults. Antimicrob. Agents Chemother. 2017, 61, e01627-16. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Messer, S.A.; Rhomberg, P.R.; Borroto-Esoda, K.; Castanheira, M. Differential activity of the oral glucan synthase inhibitor scy-078 against wild-type and echinocandin-resistant strains of Candida species. Antimicrob. Agents Chemother. 2017, 61, e00161-17. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Ortigosa, C.; Perez, W.B.; Angulo, D.; Borroto-Esoda, K.; Perlin, D.S. De novo acquisition of resistance to scy-078 in Candida glabrata involves FKS mutations that both overlap and are distinct from those conferring echinocandin resistance. Antimicrob. Agents Chemother. 2017, 61, e00833-17. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.; Morin, N.; Baginsky, W.; el-Sherbeini, M.; Clemas, J.A.; Nielsen, J.B.; Foor, F. Differential expression and function of two homologous subunits of yeast 1,3-β-d-glucan synthase. Mol. Cell. Biol. 1995, 15, 5671–5681. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Hirata, A.; Nogami, S.; Beauvais, A.; Latge, J.P.; Ohya, Y. Homologous subunits of 1,3-β-glucan synthase are important for spore wall assembly in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Suwunnakorn, S.; Wakabayashi, H.; Kordalewska, M.; Perlin, D.S.; Rustchenko, E. Fks2 and fks3 genes of opportunistic human pathogen Candida albicans influence echinocandin susceptibility. Antimicrob. Agents Chemother. 2018, 62, e02299-17. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.; Ischer, F.; Leibundgut-Landmann, S.; Sanglard, D. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect. Immun. 2013, 81, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.A.; Moeckli, B.; Torelli, R.; Posteraro, B.; Sanguinetti, M.; Sanglard, D. Upregulation of the adhesin gene epa1 mediated by PDR1 in Candida glabrata leads to enhanced host colonization. mSphere 2016, 1, e00065-15. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Wang, C.; Tian, Y.; Dong, D.; Jiang, C.; Mao, E.; Peng, Y. CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 2018, 61, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Salazar, S.B.; Wang, C.; Munsterkotter, M.; Okamoto, M.; Takahashi-Nakaguchi, A.; Chibana, H.; Lopes, M.M.; Guldener, U.; Butler, G.; Mira, N.P. Comparative genomic and transcriptomic analyses unveil novel features of azole resistance and adaptation to the human host in Candida glabrata. FEMS Yeast Res. 2018, 18, fox079. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Ghori, N.; Falkow, S. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 1999, 285, 578–582. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.W.; Kraneveld, E.A.; Yin, Q.Y.; Dekker, H.L.; Gross, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The cell wall of the human pathogen Candida glabrata: Differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.; Beaudoing, E.; Tran, V.D.T.; Sanglard, D. Comparative genomics of two sequential Candida glabrata clinical isolates. G3 2017, 7, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fuentes, E.; Gutierrez-Escobedo, G.; Timmermans, B.; Van Dijck, P.; De Las Penas, A.; Castano, I. Candida glabrata’s genome plasticity confers a unique pattern of expressed cell wall proteins. J. Fungi 2018, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Castano, I.; Pan, S.J.; Zupancic, M.; Hennequin, C.; Dujon, B.; Cormack, B.P. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 2005, 55, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Sanguinetti, M.; Torelli, R.; Posteraro, B.; Sanglard, D. Contribution of cgpdr1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS ONE 2011, 6, e17589. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of function mutations in cgpdr1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed]
- Vitenshtein, A.; Charpak-Amikam, Y.; Yamin, R.; Bauman, Y.; Isaacson, B.; Stein, N.; Berhani, O.; Dassa, L.; Gamliel, M.; Gur, C.; et al. NK cell recognition of Candida glabrata through binding of nkp46 and ncr1 to fungal ligands epa1, epa6, and epa7. Cell Host Microbe 2016, 20, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Galhardo, R.S.; Hastings, P.J.; Rosenberg, S.M. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 399–435. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.J.; Aponyi, I.; Rosenberg, S.M. General stress response regulator rpos in adaptive mutation and amplification in Escherichia coli. Genetics 2004, 166, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Ponder, R.G.; Fonville, N.C.; Rosenberg, S.M. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 2005, 19, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Shee, C.; Gibson, J.L.; Darrow, M.C.; Gonzalez, C.; Rosenberg, S.M. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 13659–13664. [Google Scholar] [CrossRef] [PubMed]
- Shor, E.; Fox, C.A.; Broach, J.R. The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS Genet. 2013, 9, e1003680. [Google Scholar] [CrossRef] [PubMed]
- MacLean, R.C.; Torres-Barcelo, C.; Moxon, R. Evaluating evolutionary models of stress-induced mutagenesis in bacteria. Nat. Rev. Genet. 2013, 14, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, R.; Zimmerman, O.; Finn, T.; Amit, S.; Novikov, A.; Wertheimer, N.; Lurie-Weinberger, M.; Berman, J. Heteroresistance to fluconazole is a continuously distributed phenotype among Candida glabrata clinical strains associated with in vivo persistence. mBio 2016, 7, e00655-16. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.; Alves, R.; Carneiro, C.; Silva, S.; Brown, A.J.; Istel, F.; Kuchler, K.; Sampaio, P.; Casal, M.; Henriques, M.; et al. Candida glabrata susceptibility to antifungals and phagocytosis is modulated by acetate. Front. Microbiol. 2015, 6, 919. [Google Scholar] [CrossRef] [PubMed]
- Lott, T.J.; Frade, J.P.; Lockhart, S.R. Multilocus sequence type analysis reveals both clonality and recombination in populations of Candida glabrata bloodstream isolates from U.S. Surveillance studies. Eukaryot. Cell 2010, 9, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Jimenez Ortigosa, C.; Shor, E.; Perlin, D.S. Genetic drivers of multidrug resistance in Candida glabrata. Front. Microbiol. 2016, 7, 1995. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Zhao, Y.; Perez, W.B.; Lockhart, S.R.; Sobel, J.D.; Farmakiotis, D.; Kontoyiannis, D.P.; Sanglard, D.; Taj-Aldeen, S.J.; Alexander, B.D.; et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 2016, 7, 11128. [Google Scholar] [CrossRef] [PubMed]
- Polakova, S.; Blume, C.; Zarate, J.A.; Mentel, M.; Jorck-Ramberg, D.; Stenderup, J.; Piskur, J. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc. Natl. Acad. Sci. USA 2009, 106, 2688–2693. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, A.R.; Pujol, C.; Denning, D.W.; Soll, D.R.; Fox, A.J. Multilocus sequence typing of Candida glabrata reveals geographically enriched clades. J. Clin. Microbiol. 2003, 41, 5709–5717. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.; Thierry, A.; Coppee, J.Y.; Gouyette, C.; Hennequin, C.; Sismeiro, O.; Talla, E.; Dujon, B.; Fairhead, C. Genomic polymorphism in the population of Candida glabrata: Gene copy-number variation and chromosomal translocations. Fungal Genet. Biol. 2009, 46, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Chen, Y.C.; Lo, H.J.; Chen, K.W.; Li, S.Y. Assessment of Candida glabrata strain relatedness by pulsed-field gel electrophoresis and multilocus sequence typing. J. Clin. Microbiol. 2007, 45, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Chae, M.J.; Song, J.W.; Jung, S.I.; Cho, D.; Kee, S.J.; Kim, S.H.; Shin, M.G.; Suh, S.P.; Ryang, D.W. Changes in karyotype and azole susceptibility of sequential bloodstream isolates from patients with Candida glabrata candidemia. J. Clin. Microbiol. 2007, 45, 2385–2391. [Google Scholar] [CrossRef] [PubMed]
- Haw, R.; Yarragudi, A.D.; Uemura, H. Isolation of a Candida glabrata homologue of rap1, a regulator of transcription and telomere function in Saccharomyces cerevisiae. Yeast 2001, 18, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Haber, J.E. Mutations arising during repair of chromosome breaks. Annu. Rev. Genet. 2012, 46, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Jinks-Robertson, S. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics 2013, 193, 1025–1064. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delliere, S.; Healey, K.; Gits-Muselli, M.; Carrara, B.; Barbaro, A.; Guigue, N.; Lecefel, C.; Touratier, S.; Desnos-Ollivier, M.; Perlin, D.S.; et al. Fluconazole and echinocandin resistance of Candida glabrata correlates better with antifungal drug exposure rather than with MSH2 mutator genotype in a french cohort of patients harboring low rates of resistance. Front. Microbiol. 2016, 7, 2038. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.A.; Won, E.J.; Kim, M.N.; Lee, W.G.; Lee, K.; Lee, H.S.; Uh, Y.; Healey, K.R.; Perlin, D.S.; Choi, M.J.; et al. Multilocus sequence typing (mlst) genotypes of Candida glabrata bloodstream isolates in Korea: Association with antifungal resistance, mutations in mismatch repair gene (MSH2), and clinical outcomes. Front. Microbiol. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xiao, M.; Wang, H.; Yu, S.Y.; Zhang, G.; Zhao, Y.; Xu, Y.C. Profiling of PDR1 and MSH2 in Candida glabrata bloodstream isolates from a multicenter study in China. Antimicrob. Agents Chemother. 2018, 62, e00153-18. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, A.A.; Farley, M.M.; Harrison, L.H.; Stein, B.; Hollick, R.; Lockhart, S.R.; Magill, S.S.; Derado, G.; Park, B.J.; Chiller, T.M. Changes in incidence and antifungal drug resistance in candidemia: Results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin. Infect. Dis. 2012, 55, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Lott, T.J.; Frade, J.P.; Lyon, G.M.; Iqbal, N.; Lockhart, S.R. Bloodstream and non-invasive isolates of candida glabrata have similar population structures and fluconazole susceptibilities. Med Mycol 2012, 50, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Healey, K.R.; Yadav, P.; Upadhyaya, G.; Sachdeva, N.; Sarma, S.; Kumar, A.; Tarai, B.; Perlin, D.S.; Chowdhary, A. Absence of azole or echinocandin resistance in Candida glabrata isolates in India despite background prevalence of strains with defects in the DNA mismatch repair pathway. Antimicrob. Agents Chemother. 2018, 62, e00195-18. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.; Shor, E.; Perlin, D. Antifungal resistant isolates of Candida glabrata from the united states are enriched for specific sequence types with distinct MSH2 alleles. In Proceedings of the 20th Congress of the International Society for Human and Animal Mycology, ISHAM, Amsterdam, The Netherlands, 30 June–4 July 2018. Poster: P475. [Google Scholar]
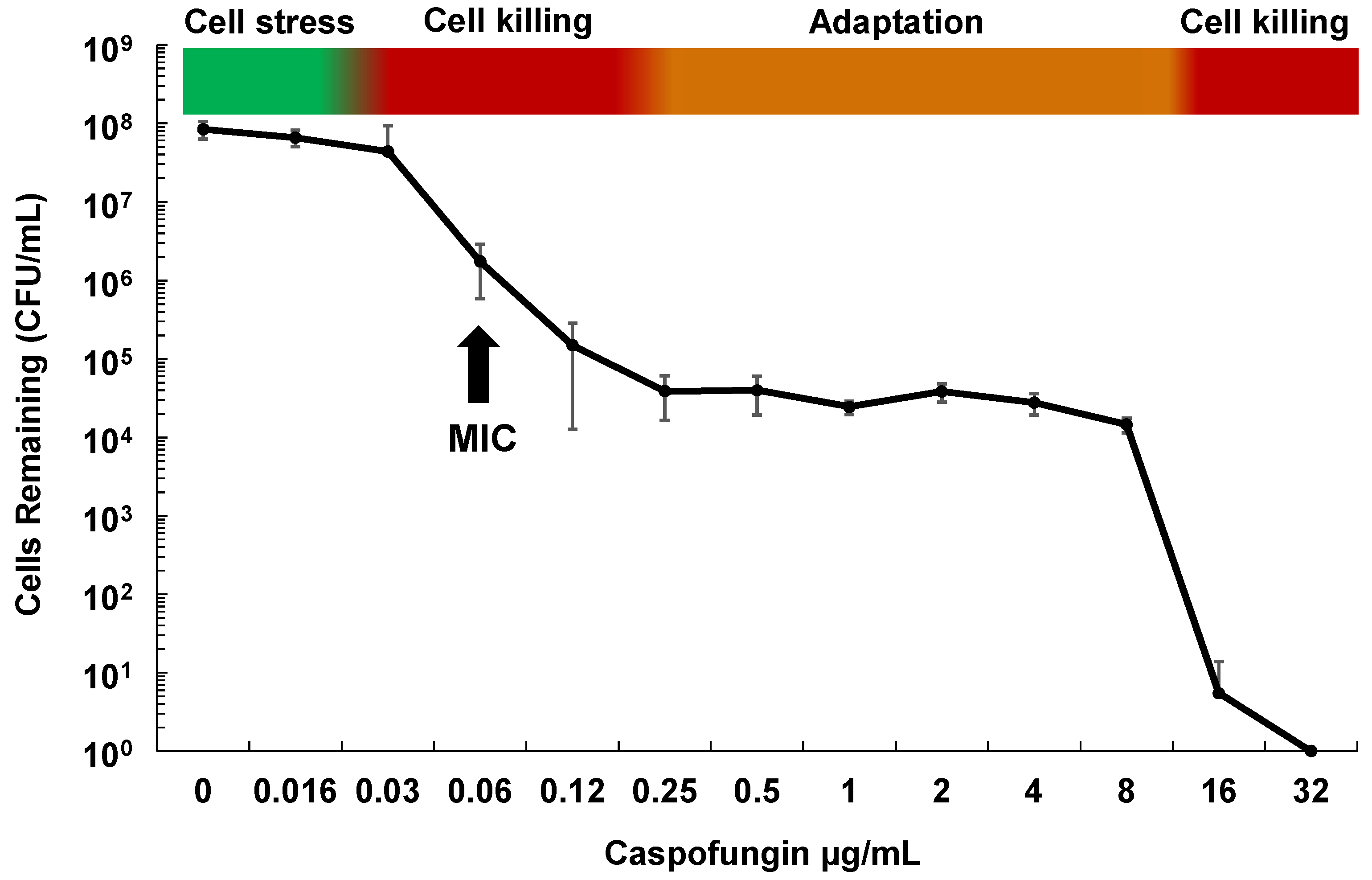
Figure 1.
Phases of in vitro cell killing and adaptation with echinocandins and Candida glabrata. Cells (1×107) of C. glabrata ATCC 2001 were grown in RPMI medium containing caspofungin at the indicated concentrations for 20 h. Dilutions were then plated onto drug free agar-containing plates to determine surviving cell counts. Shown is the average of 4 independent experiments ± standard deviations. The minimum inhibitory concentration (MIC) is indicated for reference.
Figure 1.
Phases of in vitro cell killing and adaptation with echinocandins and Candida glabrata. Cells (1×107) of C. glabrata ATCC 2001 were grown in RPMI medium containing caspofungin at the indicated concentrations for 20 h. Dilutions were then plated onto drug free agar-containing plates to determine surviving cell counts. Shown is the average of 4 independent experiments ± standard deviations. The minimum inhibitory concentration (MIC) is indicated for reference.
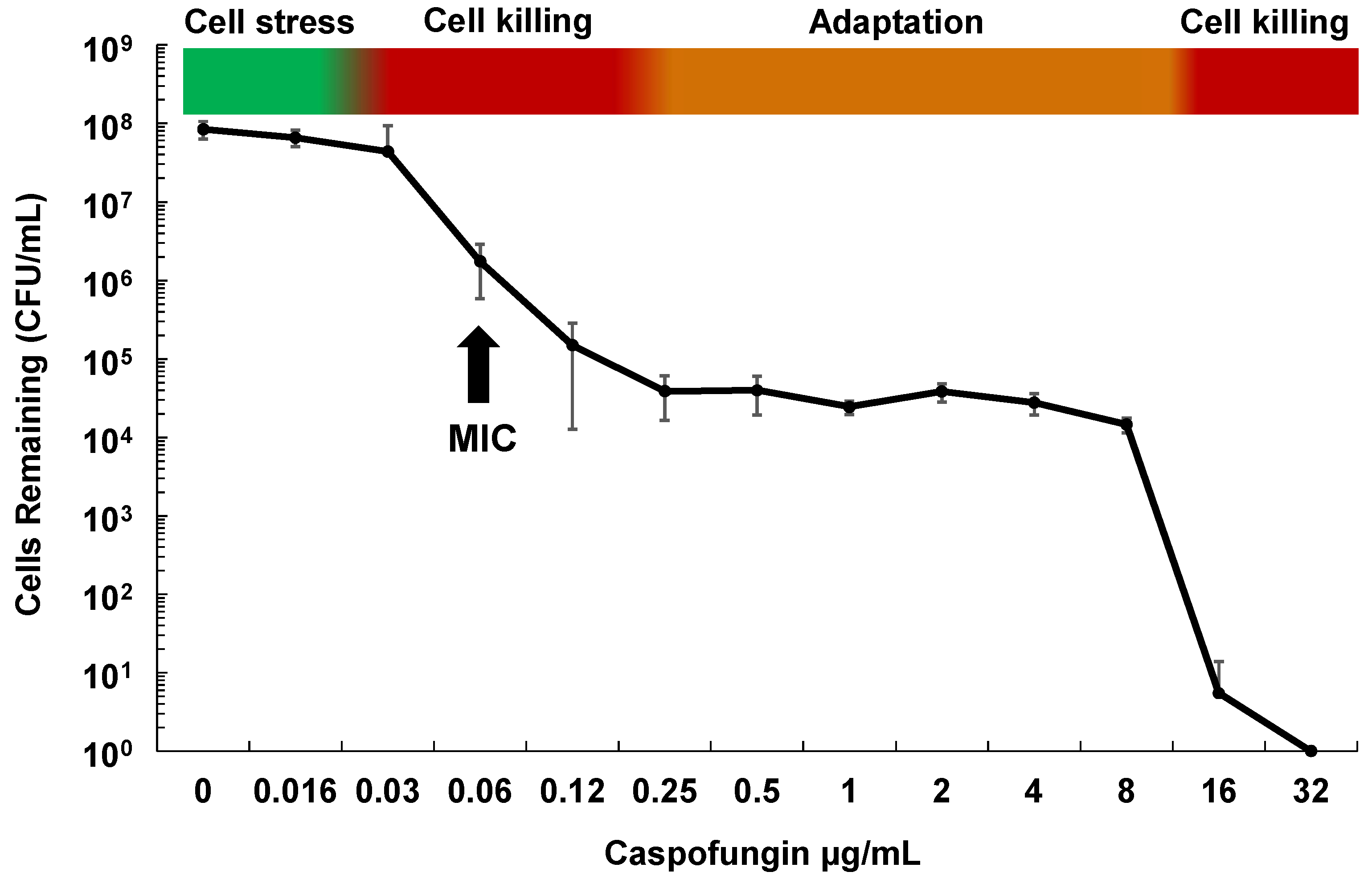
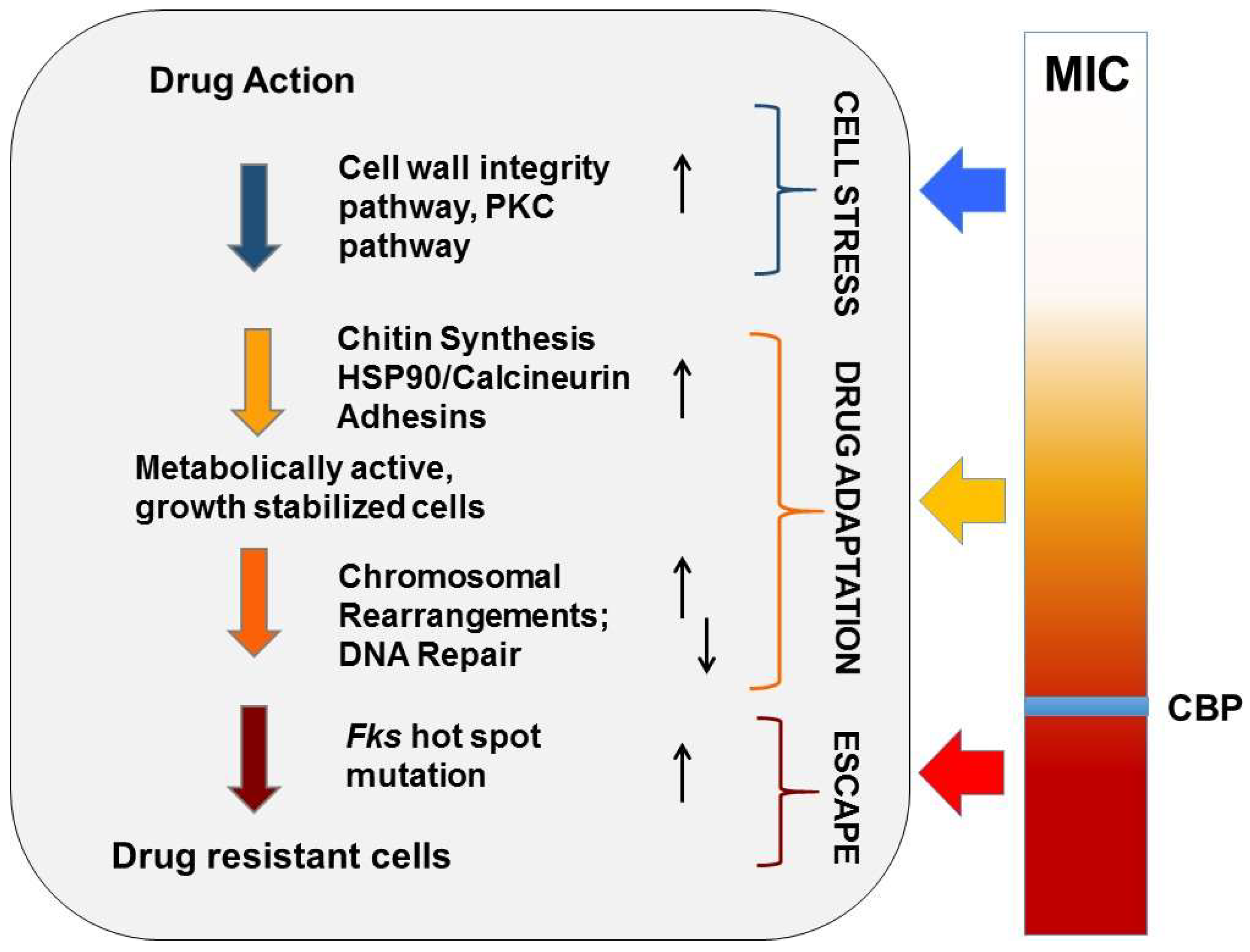
Figure 2.
Evolution of echinocandin resistance. Cellular factors that influence the ability of yeast to adapt to echinocandin drug pressure are represented in a multistep model of resistance. Steps include initial cellular stress, drug adaptation, and genetic escape (FKS mutation). The clinical breakpoint (CBP) of a species is the MIC measured prior to the formation of FKS escape mutants.
Figure 2.
Evolution of echinocandin resistance. Cellular factors that influence the ability of yeast to adapt to echinocandin drug pressure are represented in a multistep model of resistance. Steps include initial cellular stress, drug adaptation, and genetic escape (FKS mutation). The clinical breakpoint (CBP) of a species is the MIC measured prior to the formation of FKS escape mutants.
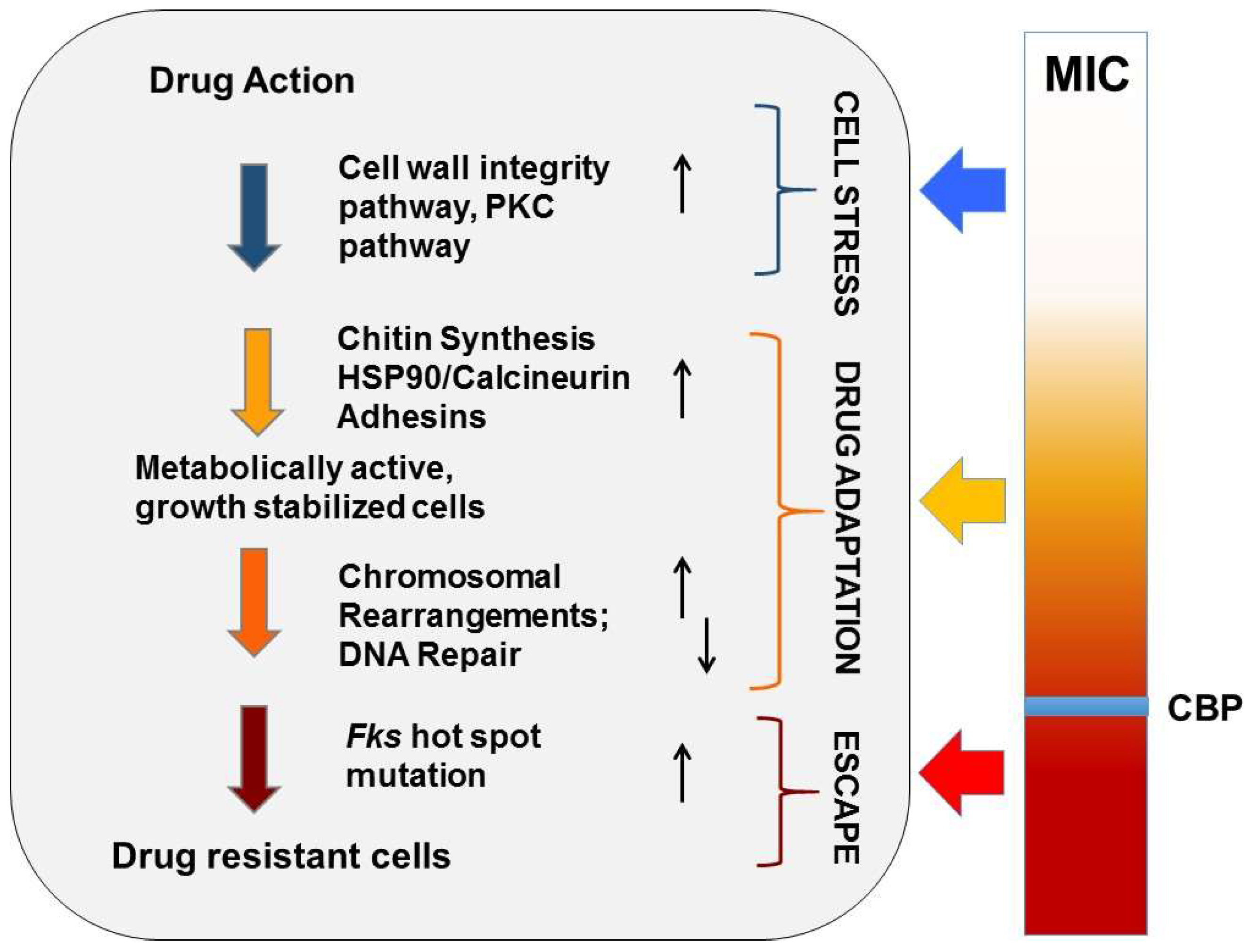
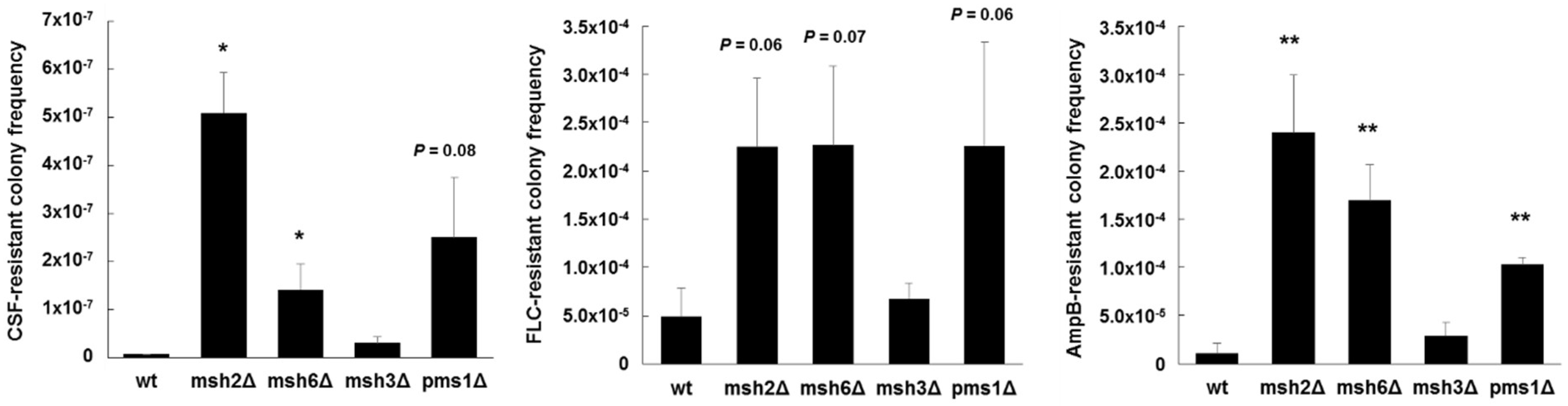
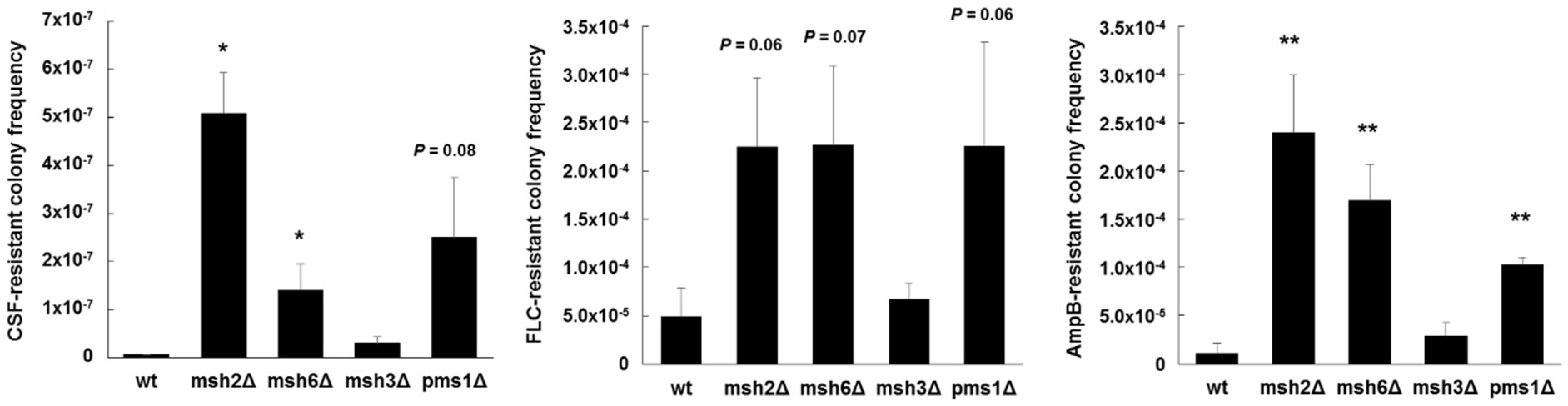
Figure 3.
Echinocandin, azole, and polyene resistant colony frequencies of C. glabrata mismatch repair deletion strains. Strains were selected agar plates containing 1 µg/mL of caspofungin (CSF), 256 µg/mL of fluconazole (FLC), or 2 µg/mL of amphotericin B (AmpB) (panels left to right) (all concentrations 8-16x the corresponding MIC). Dilutions were plated onto drug-free media to determine exact CFU counts. Frequencies were calculated as the number of colonies on the drug plate divided by the total CFU plated. Frequency averages were calculated from at least three independent selections. * p < 0.05 and ** p < 0.01 (student’s t-test; two-tailed).
Figure 3.
Echinocandin, azole, and polyene resistant colony frequencies of C. glabrata mismatch repair deletion strains. Strains were selected agar plates containing 1 µg/mL of caspofungin (CSF), 256 µg/mL of fluconazole (FLC), or 2 µg/mL of amphotericin B (AmpB) (panels left to right) (all concentrations 8-16x the corresponding MIC). Dilutions were plated onto drug-free media to determine exact CFU counts. Frequencies were calculated as the number of colonies on the drug plate divided by the total CFU plated. Frequency averages were calculated from at least three independent selections. * p < 0.05 and ** p < 0.01 (student’s t-test; two-tailed).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Healey, K.R.; Perlin, D.S. Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata. J. Fungi 2018, 4, 105. https://doi.org/10.3390/jof4030105
AMA Style
Healey KR, Perlin DS. Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata. Journal of Fungi. 2018; 4(3):105. https://doi.org/10.3390/jof4030105
Chicago/Turabian StyleHealey, Kelley R., and David S. Perlin. 2018. "Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata" Journal of Fungi 4, no. 3: 105. https://doi.org/10.3390/jof4030105
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.