Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease
by
 ,
,
Margherita Bertuzzi
1,†, 
Gemma E. Hayes
2,†,
Uju J. Icheoku
1,†,
Norman Van Rhijn
1, 
David W. Denning
3,
Nir Osherov
4 and
Elaine M. Bignell
1,* 1
Manchester Fungal Infection Group, Faculty of Biology, Medicine and Health, University of Manchester, Manchester M13 9NT, UK
2
Northern Devon Healthcare NHS Trust, North Devon District Hospital, Raleigh Park, Barnstaple EX31 4JB, UK
3
The National Aspergillosis Centre, Education and Research Centre, Wythenshawe Hospital, Manchester University NHS Foundation Trust, Manchester M23 9LT, UK
4
Department of Clinical Microbiology and Immunology, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Fungi 2018, 4(1), 8; https://doi.org/10.3390/jof4010008
Submission received: 18 December 2017
/
Revised: 3 January 2018
/
Accepted: 5 January 2018
/
Published: 8 January 2018
(This article belongs to the Special Issue Mucosal Fungal Infections)
{kind=link}
{kind=link}
Abstract
:Respiratory epithelia fulfil multiple roles beyond that of gaseous exchange, also acting as primary custodians of lung sterility and inflammatory homeostasis. Inhaled fungal spores pose a continual antigenic, and potentially pathogenic, challenge to lung integrity against which the human respiratory mucosa has developed various tolerance and defence strategies. However, respiratory disease and immune dysfunction frequently render the human lung susceptible to fungal diseases, the most common of which are the aspergilloses, a group of syndromes caused by inhaled spores of Aspergillus fumigatus. Inhaled Aspergillus spores enter into a multiplicity of interactions with respiratory epithelia, the mechanistic bases of which are only just becoming recognized as important drivers of disease, as well as possible therapeutic targets. In this mini-review we examine current understanding of Aspergillus-epithelial interactions and, based upon the very latest developments in the field, we explore two apparently opposing schools of thought which view epithelial uptake of Aspergillus spores as either a curative or disease-exacerbating event.
1. Introduction
Aspergillus-related diseases occur frequently in the human population and pathological outcomes are diverse [1]. Amongst more than 2.3 million episodes occurring annually in Europe alone, the vast majority of aspergilloses initiate in the respiratory tract via fungal spore inhalation resulting in up to 2 million cases of allergic disease in asthma and cystic fibrosis sufferers, and more than 300,000 cases of semi-invasive or invasive aspergillosis of which 70,000 prove fatal [2]. A unifying feature of all respiratory aspergilloses is the interaction of inhaled fungal particles with the respiratory epithelium, the outcome of which differs dramatically according to the respiratory niche(s) involved and the immunological status of the affected host.
A detailed understanding of the host and pathogen processes driving healthy clearance of fungal spores, as well as the mechanistic defects which drive adverse pathologies will better enable a more targeted therapeutic strategy in the future. To this end we review the most recent developments in the field, with a particular focus upon spore attachment to, and uptake by the respiratory epithelium, epithelial responses to A. fumigatus challenge, and their likely impacts upon disease outcomes. As authoritative reviews of the earlier literature have already dealt with experimental systems, founding concepts in the field, and relevant pathogenicity factors [3,4,5], we restrict our focus to aspects of the host-pathogen interaction having most pertinence to the outcome of Aspergillus-related lung diseases.
It is now widely accepted that epithelial uptake of A. fumigatus spores is an important component of the interaction with cultured human epithelia leading either to fungal killing or intraphagosomal occupation of airway epithelial cells (AECs). Despite multiple studies reporting spore internalisation by immortalised and/or primary AECs in in vitro infection systems and ex vivo organ culture models [6,7,8,9,10,11,12,13,14], no compelling published evidence of in vivo spore internalisation by AECs is available. On the contrary, using a novel bioimaging approach combined with transmission electron microscopy (TEM), Rammaert et al. recently demonstrated the absence, in vivo, of fungal spore internalisation in the bronchial epithelium of mice [15]. Due to the resolution limit of TEM, and the likely low frequency of spore uptake in vivo, these findings do not exclude the possibility that spore internalisation might occur during mammalian infection. Furthermore, the observation of Rammaert et al. derives from the study of bronchial, but not alveolar, epithelium [15]. The relevance of spore uptake to health likely encompasses several outcomes of the host-pathogen interaction, including those where uptake serves as a useful means of neutralising low-level spore exposure or as driving invasive growth and pathogenesis [6,8,14,16,17] where fungal killing is unachievable (such as during disease [14]). Here, we examine the latest advances in understanding of the host-A. fumigatus interaction and its role in driving or curtailing lung disease.
Due to the occurrence of an obligatory morphological shift during conidial germination of Aspergillus species, Aspergillus-epithelial interactions are most usefully regarded within a temporal framework commencing with spore-mediated host interactions, and outcomes thereof, and ending with either fungal death, or fungal invasion of the lung parenchyma (Figure 1 and Figure 2). Germination of inhaled Aspergillus spores involves a period of isotropic growth (swelling) which, if not apprehended, will be followed by the outgrowth of a tip-extending elongated cell called a primary hypha, which actively secretes fungal proteases and secondary metabolites whilst growing in a polarised fashion into surrounding tissues [8]. Since the cell surface of resting A. fumigatus conidia differs markedly from that of metabolically active, germinating spores and hyphae [18,19,20], and cytotoxic secreted factors derive predominantly from mature hyphal cells [8], it is likely that within the time frame of the host-pathogen interaction, individual epithelial cells will become iteratively exposed to one or multiple fungal morphotypes, as well as fungal cell surface-associated and secreted factors. Host responses to Aspergillus encounters, including the ability to contain the pathogenic threat and the nature of ensuing host damage, vary according to both the fungal and epithelial cell types involved (Figure 2), and are critically impacted by host immune status.
2. Attachment of A. fumigatus to the Respiratory Epithelium
A. fumigatus conidia bind in a concentration-dependent manner to A549 epithelia and extracellular matrix (ECM), and to purified basement membrane components, such as fibrinogen, fibronectin, laminin, type I collagen, and type IV collagen [21,22,23]. Basement membrane components are not normally accessible to pathogens in healthy lungs, but often become exposed in lungs of asthmatic patients, hence, adhesion to the basal lamina could facilitate fungal infection and persistence, exacerbating the risk of aspergillosis in this cohort of patients [21], as well as in other disease settings where integrity of the pulmonary epithelium is diminished.
Several fungal surface components, many of which are expressed in a morphotype-specific manner, play a role in A. fumigatus attachment to AECs [18,19,20], thereby reflecting the structural complexity and dynamism of the cell wall during germination and hyphal growth. For example, the conidial hydrophobin RodA, which renders A. fumigatus spores immunologically inert, but is shed from the spore surface during germination, is required for adherence to collagen, but not to A549 cells or other basement membrane components, such as laminin and fibronectin [24] (Figure 1). In the context of healthy defence against inhaled fungal spores, RodA is argued as being a facilitator of immune evasion at the earliest of time points following spore inhalation [46]. Since RodA extracted from A. fumigatus conidia is immunologically inert and fails to induce human dendritic cell maturation, it might have a role to play in inducing human tolerance to everyday spore exposure [46]. However, in diseased settings, and on the basis of current evidence, any role for RodA in the establishment of epithelial colonisation would necessarily involve (a) ungerminated A. fumigatus spores and (b) a pathological setting in which collagen is exposed.
Morphotype-specific adhesion factors are also expressed on swollen and germinating spores and hyphae, for example, CspA [18], which becomes unmasked during conidial germination [47], and is required for full adhesion to A549-derived ECM (Figure 1). Notably, a cspA null mutant exhibits only a 50% reduction in ECM binding relative to the progenitor clinical isolate, thereby supporting the hypothesis of multiple conidial adhesion factors expressed by A. fumigatus [47]. Furthermore, a recent study of 30 clinical and environmental isolates screened for conidial phenotypes and adherence to A549 cells, revealed four A. fumigatus strains with decreased adherence to A549 cells having a reduced conidial size, number, or hydrophobicity [20]. RNAseq analysis of the four most highly adherent isolates revealed a common cluster of 31 differentially-expressed genes during conidial formation, two of which (AFUA_4G01030 and AFUA_4G08805) were found by mutational analysis to be important for adhesion to AECs [20].
Due to the abundance of fucose on human cells, the recent discovery of an A. fumigatus soluble fucose-specific lectin, FleA [25,26], prompted speculation that FleA might be the conidial surface protein responsible for directing attachment of A. fumigatus to host cells, such as epithelial cells and macrophages, via membrane-tethered mucins [48]. Recombinant FleA prompts haemagglutination of human red blood cells in a fucose-dependent manner, directly binds fucose-containing oligosaccharides in glycan array studies and elicits IL-8 expression in cultured BEAS-2B bronchial epithelial cells. An anti-FleA rabbit polyclonal antibody revealed conidia-, but not hypha-, specific localisation of FleA in microscopy-mediated immunofluorescence assays [48]. Subsequent analyses of FleA-mediated glycoprotein binding revealed that FleA mediates binding of A. fumigatus conidia to airway mucin in a fucose-dependent manner (Figure 1) and is a necessary prerequisite for macrophage-mediated phagocytosis of A. fumigatus spores. Concordant with a role for FleA in driving host-mediated clearance of inhaled A. fumigatus spores, a fleA null mutant promoted invasive growth and heightened inflammatory cell recruitment in immunocompetent mice, however, the role of FleA in driving epithelial uptake of A. fumigatus spores remains untested [48].
Carbohydrate residues on the surface of A. fumigatus germinating conidia are ligands for ficolins (M-, L-, and H-ficolin in human serum), a family of lectin-like opsonins whose secretion is increased in bronchoalveolar lavage fluid and infected lungs of individuals with invasive aspergillosis [27,28,29] (Figure 1). Competitive binding assays using a range of carbohydrates and H-ficolin pre-incubation revealed that H-ficolin binds to A. fumigatus conidia via l-fucose, d-mannose, and N-acetylglucosamine (GalNAc) on the conidial surface and in a calcium- and pH-dependent manner, with maximal binding observed in acidic conditions (pH 5.7). At this pH, H-ficolin opsonised conidia are twice as adherent to A549 cells relative to their non-opsonised counterparts [29]. Recombinant M-ficolin was found via glycan arrays [49] to bind to acetylated compounds including GalNAc, and via pull-down assays [27] to bind to chitin and β-1,3 glucan on the A. fumigatus cell wall. Quantitation of C4 complement consumption and deposition and IL-8 ELISA demonstrated that opsonisation of the alkali-insoluble fraction of A. fumigatus cell wall with recombinant M-ficolin mediates complement activation and potentiates IL-8 secretion by A549 cells in a dose-dependent manner. Although H-ficolin has been demonstrated to positively influence binding of A. fumigatus spores to A549 cells [29], the role of M-ficolin adherence to host cells remains unexplored. In support of a protective role for ficolin-mediated activities during A. fumigatus infection, a recent study showed that mice lacking both ficolin A and B (murine orthologues of the human L- and M-ficolins [50]) suffer significantly higher pulmonary fungal burden compared to their wild-type counterparts [51]. Cytokine ELISA of broncoalveolar lavage fluids (BALF) revealed a trend towards decreased production of IL-1β, IL-6, and TNFα in the lungs of A. fumigatus-infected ficolin-deficient mice [51]. However, defective fungal clearance did not correlate with muted recruitment of inflammatory cells or aberrant complement activation during A. fumigatus infection as measured via quantification of myeloperoxidase in murine lung homogenates and complement component C3a in BALFs.
E-cadherin has also been suggested to play a role in A. fumigatus spore adhesion to, and uptake by, A549 cells given the reduction in E-cadherin expression via application of a blocking antibody or siRNA expression results in reduced cell-conidial association and conidial phagocytosis [31]. Accordingly, co-immunoprecipitation and mass spectrometry analysis following co-incubation of A. fumigatus spore lysates with purified recombinant human E-cadherin recently identified the metalloprotease designated as MEP (or AFUA_8G07080), and another two uncharacterised proteins, AFUA_1G11480 and AFUA_6G02870, as putative mediators of A. fumigatus spore binding to the respiratory epithelium via the adhesion molecule E-cadherin although this hypothesis remains to be substantiated [30,31,32].
The secreted and hyphal cell wall-associated exopolysaccharide galactosaminogalactan (GAG) has been demonstrated to mediate a number of virulence-associated traits, including adherence of A. fumigatus hyphae to fibronectin and A549 epithelial cells [19] (Figure 1). GAG is a heteroglycan composed of galactose and N-acetyl-galactosamine (GalNAc), produced by hyphae but not conidia [52], via the activity of the fungal epimerase Uge3. Uge3 mediates the synthesis of the GalNAc component of GAG [19,53] and a uge3-deficient mutant shows a 10-fold reduction in adhesion to A549 monolayers compared to the respective parental isolate [19]. Uge3 is also required for the induction of epithelial cell damage by A. fumigatus, as a uge3-deficient mutant is 10-fold less able to cause lysis of A549 cells after 16 h of infection compared to its wild-type counterpart [19]. Furthermore, survival analyses and fungal burden analyses performed in cortisone acetate-treated or leukopenic mice indicate that the Δuge3 mutant is less efficient at colonising the murine lung [19], suggesting an important role for adherence to host cells during infection. Recently, the deacetylation of GalNAc residues within GAG has been identified as an essential requirement for the adherence of GAG to hyphae. The Agd3 deacetylase is required for the deacetylation of GAG [54], and a Δagd3 isolate does not display detectable cell wall-associated GAG as measured by direct immunofluorescence with the GalNAc-specific soybean agglutinin lectin [54]. More importantly, a Δagd3 mutant is unable to adhere to negatively charged surfaces because de-N-acetylated GAG confers a positive charge on the hyphal surface [54]. Deletion of agd3 attenuates A. fumigatus virulence in a leukopenic mouse model of invasive aspergillosis, demonstrating that deacetylation of GAG is required for full virulence of A. fumigatus [54]. The epithelial receptor for GAG is still unknown.
3. Epithelial Uptake of A. fumigatus Spores
Cultured bronchial cells and type II alveolar cells are able to internalise approximately 30–50% of adherent A. fumigatus conidia in a concentration- and time-dependent manner [6,7,8,9,12,13,14,32,33,55]. The fungal ligands required for uptake of A. fumigatus by AECs remain largely unknown but, recently, a thaumatin-like protein, namely CalA, was identified as the first invasin of A. fumigatus [17]. Analogously to a previously identified A. nidulans calA gene homologue, which is highly expressed during and required for, spore germination and hyphal formation [56], an A. fumigatus ΔcalA mutant exhibits delayed germination and curved hyphal tips when grown in tissue-culture medium. A. fumigatus CalA was predicted as a candidate adhesin via bioinformatics analysis, supported by the finding that a recombinant version of CalA produced in Escherichia coli is able to bind laminin and AECs in vitro [57]. However, an A. fumigatus strain lacking calA was not deficient in binding immobilized laminin or A549 cells, indicating that any role CalA plays in adhesion can be served by other adhesion factors [17]. A differential fluorescence approach demonstrated that, compared to the respective parental isolate, a calA null mutant shows a significant reduction (50%) in uptake by A549 cells after 2.5 h of infection. CalA therefore mediates, at least in part, fungal internalisation by A549 and primary human alveolar epithelial cells. Both whole-cell affinity purification, and subsequent immunoblotting with anti-integrin antibodies, indicated that CalA-dependent internalisation is mediated by direct interaction with the host cell receptor α5β1 integrin [17] (Figure 1). Accordingly, in differential fluorescence assays, both β1 and α5 integrin antibody- and siRNA-mediated inhibition reduce internalisation of A. fumigatus by ~50% when infecting A549 monolayers for 2.5 h [17]. Importantly, CalA is also required for normal virulence and lung invasion as demonstrated via survival and fungal burden analyses in cortisone- acetate treated mice following aerosol infection with A. fumigatus conidia. In the same infection model an anti-CalA antibody reduced A. fumigatus uptake by A549 cells by 50% and, when injected intraperitoneally prior to A. fumigatus infection, increased the survival of the infected mice by 20%, suggesting CalA to be a plausible target for novel therapeutic interventions.
Redundancy of function apparently also extends to the host cell repertoire of internalisation factors. Spore internalisation is dependent on two crucial regulators of host actin dynamics, phospholipase D (PLD) and cofilin-1, whose activation and phosphorylation state, respectively, are modulated by exposure to β-1,3-glucan on the surface of germinating conidia [9,33] (Figure 1). Accordingly, chemical or siRNA-mediated downregulation of host PLD or aberrant phosphorylation of cofilin-1 via the RhoA-ROCK-LIM kinase pathway causes a decrease in A. fumigatus internalisation [9,33]. In A549 cells, PLD co-localises with internalised conidia [9], which also quickly become co-localised with late endosomal/lysosomal markers, such as LAMP-1, and CD63 and cathepsin D, indicating rapid trafficking through the endosomal system to the phagolysosomes [6]. Following phagolysosomal acidification most of the internalised conidia are killed, but after 24 h 3% of the internalised conidia remain viable and 34% of these can eventually germinate to escape the phagolysosome by 36 h, without lysis of the host cell [6]. Two additional host proteins (ARC and EGR1) upregulated at 8 h in response to A. fumigatus challenge, have been implicated as influencing uptake of conidia by A549 cells, as siRNA mediated knockdown of the respective gene functions diminished conidial uptake by approximately 20% and 40%, respectively [58].
In AEC, induction of PLD following exposure to β-1,3-glucan on the surface of germinating conidia has been demonstrated to occur in a Dectin-1 dependent manner [9]. Accordingly, anti-Dectin-1 antibodies significantly reduce A. fumigatus conidia internalisation by A549 cells [8,9]. Supporting a role for Dectin-1 in mediating curative spore clearance in vivo a high risk of invasive pulmonary aspergillosis has been reported in hematopoietic stem cell transplantees carrying a non-functional truncated version of Dectin-1 [59]. This suggests that Dectin-1 mediated activities of non-hematopoietic cells, such as AECs, play a crucial role in defence against aspergillosis. Studies in A549 and immortalised human bronchial epithelial cells (HBECs) have revealed conflicting reports [9,36,60,61] of Dectin-1 expression levels whereby immunohistochemical staining of human lung sections reveals predominantly apical expression of Dectin-1 on bronchial and alveolar human epithelium [60], while flow cytometric analyses of primary small airway epithelial cells (SAECs) and primary HBECs confirmed Dectin-1 expression [60], but failed to detect the receptor in the A549 cell line unless induced by challenge with Mycobacterium tuberculosis or A. fumigatus [61]. Given that Han et al. (2011) reported constitutive Dectin-1 expression in A549 cells [9], Sun et al., reported Dectin-1 expression in a papilloma virus-immortalised human bronchial epithelial cell line [47] and anti-Dectin-1 antibodies significantly ameliorate conidial internalisation and epithelial damage following A. fumigatus infection [8], it is feasible that Dectin-1 is specifically responsive to A. fumigatus challenge in both cultured and primary respiratory epithelia. A targeted comparative study will be necessary in order to resolve this important question.
Conidial dihydroxynaphthalane (DHN) melanin increases internalisation of A. fumigatus spores by A549 cells [34], as demonstrated by an analysis of epithelial uptake of non-pigmented ΔpksP mutants versus that of the respective parental isolate. In professional phagocytes DHN melanin on the surface of A. fumigatus spores prevents the acidification of phagolysosomes and, consequently, spore killing by professional phagocytes [62,63,64]. Concordant with intraphagosomal behaviour in macrophages, melanised wild-type A. fumigatus spores inhibit the acidification of A549 phagolysosomes thereby achieving higher intracellular survival rates compared to those of the strain ΔpksP [34]. In human monocyte-derived macrophages, intracellular swelling of A. fumigatus conidia and, in particular, cell wall melanin removal during germination, is required for the activation of a specialized autophagy pathway called LC3-associated phagocytosis (LAP) which ultimately results in fungal killing [65,66]. Accordingly, a non-pigmented ΔpksP mutant lacking DHN-melanin is avirulent in wild-type mice [17,56,57], but not in mice with conditional inactivation of Atg5 in hematopoietic cells. The role of autophagy in fungal killing within AECs remains untested, as does the mechanistic basis of AEC-mediated A. fumigatus killing.
4. Epithelial Responses to A. fumigatus
In common with the direct physical interactions between host and pathogen described above, it is now clear that epithelial responses to A. fumigatus also vary dramatically according to the fungal morphotype and host cell origin. Interpretation of findings in this field is therefore significantly confounded by immense experimental heterogeneity, including multiplicity of infection, fungal isolate, culture medium, etc. For example, in cultured HBECs (BEAS-2B), recognition of germinating (but not resting) conidia and hyphae leads to activation of phosphatidylinositol3-kinase (PI3K), mitogen-activated protein kinase (MAPK) p38 and ERK1/2 after 10 h of co-incubation with A. fumigatus conidia [37]. Similarly, expression and production of β-defensin 2 (HBD-2) and HBD-9 is significantly increased in response to live, swollen conidia, but not in response to resting conidia or heat dried hyphal fragments [38]. Further, when challenged with A. fumigatus conidia a papilloma virus-immortalised bronchial epithelial cell line upregulates Dectin-1 expression at the cell membrane at 6 and 18 h post challenge with resting conidia compared to challenge with heat-killed conidia, latex beads, or laminarin, thereby suggesting that fungal viability, rather than mere particulate contact with epithelial cells is a critical factor in driving Dectin-1 mediated epithelial activities [36]. Other groups have reported that physical contact with conidia, irrespective of fungal viability, is also important [67], as only inactivated resting, but not swollen or germinated conidia, induced IFN-β signalling through the RIP-1/TBK1 pathway in human primary bronchial epithelial cells. It is well documented that mere contact with A. fumigatus spores is sufficient to prompt actin cytoskeletal reorganisation and loss of focal adherence [35], however, the mechanistic basis of such cytotoxicity remains unknown. Recently, Gauthier et al., identified five main spore-associated toxins: tryptoquivaline F, fumiquinazoline C, questin, monomethylsulochrin, and trypacidin, amongst which trypacidin was shown to be the most toxic, as evidenced by MT-mediated cytotoxicity assays and LDH quantitation in A549 cells and primary HBECs [68]. In A549 epithelial cells, lysis was detectable as early as 1 h post exposure and was directly proportional to the dose of trypacidin used [68]. Trypacidin triggers cell death by initiating intracellular formation of nitric oxide, leading to death of the cells after about 24 h by necrosis. It is as yet unclear whether these toxins are retained in conidia killed by various methods.
Concordant with a role for mature hyphal morphotypes in prompting host responses, fluorescence activated cell sorting (FACS) analysis and confocal immunofluorescence microscopy, applied to papilloma virus-immortalised HBECs, revealed a time-dependent increase in Dectin-1 cell surface expression peaking at 24 h post-infection and correlating with transcriptional upregulation of TNFα, IL-8, HBD-2, and HBD-9, an effect which could be abrogated by pre-incubation with an anti-Dectin-1 antibody [36]. Accordingly, in a leukopenic murine model of invasive pulmonary aspergillosis, expression of TNFα, GM-CSF, IL-1β, and IL-10, and secretion of TNFα and GM-CSF, was significantly increased following adenovirus mediated Dectin-1 upregulation in lung tissue resulting in increased neutrophil recruitment into the lungs, lower fungal burden, and a higher survival rate in response to intratracheal infection with 3 × 105 A. fumigatus conidia [69].
Proteomic analysis of the secretome of BEAS-2B cells infected with A. fumigatus via difference in-gel electrophoresis (DIGE) and immunoblotting showed a time-dependent induction of the release of lysosomal enzymes (namely NAGase, cathepsin B, and cathepsin D) and proteins of the thioredoxin system in response to fungal challenge [70]. Cell degranulation required live fungus, whereby heat-killed spores or hyphae were unable to induce NAGase release [70]. Furthermore, PI3K and the p38 MAPK inhibitors diminished NAGase release by approximately 60% and 70%, respectively, thereby demonstrating that lysosomal enzyme release is partly mediated by PI3K and the p38 MAPK pathways [70]. The demonstration that A. fumigatus induces the release and activation of lysosomal enzymes by bronchial cells and proteins of the redox system, suggests that degranulation of lysosomal enzymes may participate in controlling fungal growth and human cell self-damage [70].
Differential responses to A. fumigatus conidia, germ tubes, and secreted fungal products are also mounted by cultured alveolar A549 cells. Exposure of A549 cells to A. fumigatus conidia for 3 h resulted in only a slight increase in ERK1/2 phosphorylation compared to polystyrene beads and no significant MAPK p38 activation, as monitored by Western blot using anti-p-ERK and anti-p-p38 specific antibodies [39]. In contrast, challenge with culture filtrate from A. fumigatus at the same time point led to MAPK ERK1/2, p38, and JNK phosphorylation. ERK1/2 and JNK, but not p38 phosphorylation, was abolished during exposure to culture filtrate from an A. fumigatus mutant (ΔprtT) lacking the PrtT transcription factor suggesting a role for proteolytic activities of secreted fungal proteases in prompting host responses [39]. Recent RNAseq analyses of A549 gene expression at 8 h post-infection with viable A. fumigatus conidia revealed upregulation of as many as 302 genes compared to uninfected cells most of which are involved in immune response, chemotaxis, cell activation, and regulation of phosphorylation [58]. Analysis of the mRNA levels of inflammatory cytokines by targeted qRT-PCR showed significant increase in expression of IL-6, IL-8, and TNFα from 6–8 h post interaction with conidia [58].
Given the wide range of host cell types, fungal challenges, and time-points employed for studying A. fumigatus-respiratory epithelial interactions it has not yet been possible to assemble a coherent account of the host-pathogen interaction [3,4,37,38,39,58,67,71]. The iterative nature of host contact with different fungal morphotypes as fungal growth progresses suggests that localised manipulation of host responses and host damage elicited by A. fumigatus challenge occurs in a stepwise and cumulative fashion. In support of this hypothesis, the recent discovery of a tissue non-invasive A. fumigatus mutant lacking the pH-responsive transcription factor PacC [8] revealed stage-specific features of the A. fumigatus-A549 interaction to comprise of a minimum of two mechanistically distinct phases of damage, the first (prior to 16 h of co-incubation) occurring in a contact-mediated manner and independently of secreted fungal proteins and the second occurring via the action of secreted fungal products. PacC null mutants were found to exhibit a non-invasive phenotype in A549 alveolar epithelia, where contact-mediated detachment and epithelial decay elicited by fungal culture filtrate were highly attenuated relative to infection with non-mutated clinical isolates. This phenotype was replicated in leukopenic mice where ΔpacC conidia germinate but fail to invade the respiratory epithelium [8].
For cultured A549 epithelia Icheoku et al. recently constructed the first comprehensive temporal map of host responses to A. fumigatus revealing discrete responses to resting, swollen, and germinating spores, hyphae, and secreted fungal products [40]. Modest (two-fold relative to PBS challenge), but sustained, phosphorylation of IκBα was observed from as early as 4 h post-infection and was maintained throughout a 48 h time-course with viable A. fumigatus. In stark contrast, however, hyphal maturation or direct exposure to culture filtrates from mature (48 h) fungal cultures reproducibly elicited heightened phosphorylation of JNK (seven-fold), p38 (six-fold), and ERK1/2 (two-fold) relative to PBS challenge. Epithelial cell lysis, quantified via a lactate dehydrogenase enzyme assay, was first measurable at 12 h post-infection and coincided with initiation of hyphal growth indicating significant toxicity of secreted fungal products. The application of JNK or p38 inhibitors prior to fungal challenge reduced lytic cell death by 70% and 40%, respectively, indicating that the host response to secreted fungal products is an important driver of host damage. The latter finding is concordant with that of Sharon et al. [39], whereby protease inhibitors were found to ameliorate epithelial damage caused by fungal culture filtrate [39]. The same authors also demonstrated that inhibition of JNK or ERK1/2 kinase activity substantially decreased CF-induced cell damage, including cell peeling, actin-cytoskeleton damage, and reduction in metabolic activity and necrotic death. In NCI-H292 bronchial epithelial cells, increased production of IL-6, IL-8, MCP-1, and mucin in response to A. fumigatus culture filtrate for 48 h is also abrogated by treating with serine protease inhibitors [71] (Figure 2).
Due to the difficulties in isolating primary alveolar epithelial cells (AECs) from human patients, animal models remain, notwithstanding the significant influence of host cell isolation and handling on cellular physiology, the closest alternative for the study of host-A. fumigatus interactions. Seddigh et al. recently developed a novel negative immunomagnetic protocol for isolating untouched primary AECs from murine lung for quantitative-label free in vivo proteomics [72]. By circumventing flow-cytometry-based cell sorting, which, by itself, massively influences cellular physiology, this approach permitted the targeted comparative ex vivo analysis, in nine biological replicates, of type II AECs from A. fumigatus-infected and sham-infected C57BL/6 mice at 24 h post-challenge. The study demonstrated a remarkable increase in abundance of L-amino acid oxidase (also known as interleukin 4-induced gene-1, IL4I1) mRNA and protein in primary alveolar epithelial cells from immunocompetent mice infected intratracheally with A. fumigatus spores for 24 h [72]. Via immunofluorescence-mediated histopathology it was determined that IL4I1 was often co-localized with the presence, and in some cases even germination of, A. fumigatus conidia an observation which did not apply to tissues harvested from sham-infected mice. To assess the relevance of IL4I1 expression in human disease AEC II, in the vicinity of human aspergilloma lesions, were analysed revealing clear positivity for IL4I1 in ten patient samples. In contrast, tissue from the same lung but more distant from mycetoma was negative for IL4I1 signal. IL4I1 belongs to the family of L-amino acid oxidases (LAAO), the major enzymatic function of which is catalysis of LAAO deamination, predominantly phenylalanine. Reported as also being abundant in B-cells through STAT6 activation and in antigen presenting cells, such as dendritic cells and macrophages, this work by Seddigh et al. yields the first published data of primary AEC II responses to A. fumigatus challenge, demonstrates for the first time that IL4I1 is present in primary murine AEC II during A. fumigatus infection, and adds weight to an intriguing theory that AEC II function as an antigen presenting cell type. In phagocytes IL4I1 abundance has been shown to have a direct bactericidal activity suggesting a plausible role in host protection against A. fumigatus infection [73]. Considering these facts, and coupled with the role of IL4I1 in driving M2 macrophage polarization [73], expression of IL4I1 by alveolar epithelia cells in response to A. fumigatus could be an important driver of airway hypersensitivity, allergic T helper 2 responses, and fibrosis.
5. Secreted Effectors of Epithelial Damage
In humans, exposure to A. fumigatus can lead to the development of allergic lung disease, including allergic bronchopulmonary aspergillosis (ABPA), and severe asthma with fungal sensitization (SAFS). The secreted products of cultured A. fumigatus have been recently demonstrated to provoke remodelling of the respiratory epithelium in immunocompetent BALB/c mice [41]. Repeated intranasal dosing with culture filtrate, conducted over the course of five weeks led to airway hyperreactivity as measured by whole-body plethysmography, ELISA, and cell counts. Significant increases were seen in the expression of classical Th2 cytokines (IL-4, IL-5, IL-13) and serum IgE in lung homogenate compared to PBS-treated control and significant airway remodelling revealed by histopathological analysis [41]. Th2 cytokine and IgE levels in mice challenged with culture filtrates harvested from A. fumigatus mutants lacking the Aspf5 or Aspf13 proteases were indistinguishable from those from mice challenged with wild type culture filtrates. However, total inflammatory and white blood cell counts revealed a significant reduction in total BALF cell and blood neutrophil content following challenge with culture filtrate from Δaspf5 and Δaspf13 mutants compared to challenge with wild-type culture filtrate. Further, immunohistochemistry of serial wax lung sections from mice challenged with Δaspf5 and Δaspf13 filtrate showed significantly reduced peribronchiole eosinophil counts and bronchiole goblet cells, as well as reduced sub-epithelial collagen deposition in the lung suggesting a role for Aspf5 and 13 in airway remodelling during A. fumigatus infection [41]. The presence of Aspf13 (also known as Alp1, a 33 kDa serine protease) in sputum has recently been reported to correlate with asthma severity in atopic patients, having been repeatedly detected in sputum from asthmatic but not healthy patients [74].
Bhushan et al. recently showed in HBECs that A. fumigatus extracts suppress JAK-STAT signalling, potentially promoting Th2 bias [75]. In a follow-up study, using CXCL10 transcription and protein expression, and NF-κB and tyrosine-protein phosphatase non-receptor type II (PTPNII) protein expression as markers of Th1 bias, Homma et al. showed that A. fumigatus extracts suppressed Th1 bias in primary normal human bronchial epithelial cells [76]. siRNA-mediated knockdown of the protease-activated receptor-2 (PAR-2) or PTPNII suppressed CXCL10 expression, thereby implicating PAR-2 activation in suppression of the endogenous epithelial IFN response, a pathway which ordinarily functions to keep Th2 and Th17 responses in check. A. fumigatus extract and culture filtrate also suppressed the activation of epithelial signal transducer and activator of transcription 1 (STAT 1) which is a Th1 cytokine signature transcription factor [75].
Gliotoxin (Gt) is a major secondary metabolite and virulence factor produced by A. fumigatus. It is an epipolythiodioxopiperazine with a reactive disulfide bridge. At moderate levels, Gt is a powerful immunosuppressant in T cells, macrophages and neutrophils. At higher levels (>250 ng/mL) it induces apoptotic cell death (reviewed in [77]). Gt also exhibits strong anti-angiogenic activity, disrupting the formation of capillaries [78].
Gt exposure in vitro, of lung epithelial cells of all types, induces cytoskeletal collapse, cell peeling, and apoptosis [42,43,44,45] (Figure 2). The signalling pathways mediating the response of lung epithelial cells to Gt have been analysed in A549 alveolar cells, primary type II alveolar cells, and BEAS-2B bronchial epithelial cells in culture [42,44]. In A549 cells, sub-lethal Gt concentrations increased the internalization of A. fumigatus conidia approximately five-fold by stimulating PLD activity and actin reorganization. This might protect the fungus from immune cells and enable it to germinate into the surrounding tissue and capillaries. In primary type II alveolar and BEAS-2B cells, Gt activates the JNK pathway leading to apoptosis. JNK triple-phosphorylates the pro-apoptotic protein Bim, leading to pro-apoptotic Bak and Bax activation, mitochondrial membrane permeabilization and apoptosis.
Several studies have analysed the importance of Gt during in vivo infection, however, its effect on the integrity of the lung epithelium remains unclear. Gt is secreted by A. fumigatus during murine and human infection and can be detected in the lungs at bioactive levels [79,80]. Abolition of Gt biosynthesis by deletion of GliP, encoding the Gt peptide synthase, strongly reduces the cytotoxic activity of culture filtrates and results in attenuated virulence in infected non-neutropenic mice [81,82]. Attenuated virulence in this model was attributed to reduced apoptosis of neutrophils entering the lungs of mice infected by the Gt null mutant, resulting in improved host survival. Interestingly, in neutropenic mice, the A. fumigatus Gt null mutant was as virulent as the wild-type strain. This suggests that, in the absence of immune cells, when the fungus interacts only with lung epithelial cells, Gt apparently plays no active role in exacerbating the infection. This is surprising, as we know that in vitro Gt kills epithelial cells. Clearly, these results suggest that a more detailed microscopic analysis of the lung epithelial cell layer needs to be performed in neutropenic mice infected with wild-type and Gt null A. fumigatus.
6. Conclusions
Recent studies have delineated morphotype-specific epithelial responses to conidial and germinated forms of A. fumigatus. Given that germinated forms of A. fumigatus pose a major threat, both in vivo and in vitro, to epithelial integrity, and that epithelial cells can potently neutralise the vast majority of internalised spores [6], we, and others [72], hypothesise that epithelial cells, in collaboration with professional phagoctyes, provide an important, directly microbicidal, defence against A. fumigatus which can become dysfunctional in settings of respiratory disease or immune compromise. Within this context, epithelial entry would serve, in the healthy host, as a means to control everyday A. fumigatus exposure, and, in the diseased setting, as a critical gateway through which to elicit host entry and damage. Opportunities to study airway infection have been severely limited by the difficulties associated with isolating and culturing primary Type I and II cells. However, a stably transformed Type I cell line has been generated recently [83] and a move towards new proteomic and single-cell technologies will move the field on further still [84]. The experimental heterogeneity and intrinsic limitations of population-scale in vitro analysis leave several unanswered questions with regards to the mode of uptake of A. fumigatus by AECs and the relevance of this process in the context of mammalian disease. Multiple studies report spore internalisation by immortalised and/or primary AECs in in vitro infection systems and ex vivo organ culture models [6,7,8,9,10,11,12,13,14], but no compelling published evidence of in vivo spore internalisation by AECs is available. Several, currently insurmountable, technical challenges might be usefully circumvented by the use of computational approaches. For example, in support of a protective role for AEC-mediated activities, agent-based modelling of the human alveolus predicts that chemotactic signalling by AECs is required for the efficient recruitment of alveolar macrophages during early A. fumigatus infection [85] and low-dose exposure to A. fumigatus spores prompts fungal persistence in both in silico and murine models of pulmonary aspergillosis [85].
Studies of Aspergillus-epithelial interactions remain in their infancy and have not yet progressed well enough to meaningfully consider the important implications of the resident microbiota and/or biofilm formation. Co-infection studies, both in vitro and in vivo, are lacking and fungal hyphae are too noxious to in vitro-cultured epithelial cells to promote longevity of co-cultured AECs and fungal hyphae. Although the propensity to form biofilms is not hindered by co-culture with human bronchial epithelial cells [86], too few intact epithelial cells remain after 2–3 days of co-culture to promote the study of the host-pathogen interaction over a sustained period of time. Similarly, whilst it is known that co-habiting microbes, such as Pseudomonas aeruginosa can manipulate redox and iron homeostasis of A. fumigatus in in vitro co-culture [87], it is unknown how this might impact upon the outcome of host-pathogen interactions at the epithelial interface neither in healthy or diseased settings. Since physiologically-relevant studies of such aspects of disease demand whole animal studies performed in real time, it is likely that advances in imaging technology will provide the opportunity to surmount the technical hurdles still obscuring a full understanding of Aspergillus-host interactions with relevant inoculum sizes, host cell context, and resident microbiota.
Acknowledgments
This work was supported in part by grants to Elaine M. Bignell from the Medical Research Council (G0501164, MR/L000822/1 and MR/M02010X/1) the Biotechnology and Biological Sciences Research Council (BB/G009619/1) and the Wellcome Trust (WT093596MA); to Margherita Bertuzzi from Imperial College London (Division of Investigative Sciences Ph.D. Studentships); to Elaine M. Bignell and Margherita Bertuzzi from a University of Manchester MRC Discovery Award (MC_PC_15072); to Uju J. Icheoku from a Wellcome Trust Strategic Award (097377/Z/11/Z); and to Norman van Rhijn from the BBSRC Doctoral Training Programme (BB/M011208/1). Nir Osherov is funded by the Israel Science Foundation (ISF no. 1347/14).
Author Contributions
Margherita Bertuzzi, Gemma E. Hayes, Uju J. Icheoku, David W. Denning, Nir Osherov and Elaine M. Bignell critically reviewed the literature and wrote the paper. Norman van Rhijn designed and drew the figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- Kleinkauf, N.; Verweij, P.; Arendrup, M.; Donnelly, P.; Cuenca-Estrella, M.; Fraaije, B.; Melchers, W.; Adriaenssens, N.; Kema, G.; Ullmann, A. Risk Assessment on the Impact of Environmental Usage of Triazoles on the Development and Spread of Resistance to Medical Triazoles in Aspergillus Species; European Centre for Disease Prevention and Control (ECDC): Stockholm, Sweden, 2013; ISBN 978-92-9193-444-7. [Google Scholar]
- Osherov, N. Interaction of the pathogenic mold Aspergillus fumigatus with lung epithelial cells. Front. Microbiol. 2012, 3, 346. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.A.; Culibrk, L.; Moore, M.M.; Tebbutt, S.J. Interactions of Aspergillus fumigatus conidia with airway epithelial cells: A critical review. Front. Microbiol. 2016, 7, 472. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.C.; Filler, S.G. Host cell invasion by medically important fungi. Cold Spring Harb. Perspect. Med. 2015, 5, a019687. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J. Cell Sci. 2003, 116, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Uptake of Aspergillus fumigatus conidia by phagocytic and nonphagocytic cells in vitro: Quantitation using strains expressing green fluorescent protein. Infect. Immun. 2002, 70, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Munoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D.; et al. The pH-responsive pacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Yu, R.; Zhen, D.; Tao, S.; Schmidt, M.; Han, L. β-1,3-glucan-induced host phospholipase D activation is involved in Aspergillus fumigatus internalization into type II human pneumocyte A549 cells. PLoS ONE 2011, 6, e21468. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Boisvieux-Ulrich, E.; Crestani, B.; Houcine, O.; Taramelli, D.; Lombardi, L.; Latgé, J.P. Internalization of Aspergillus fumigatus conidia by epithelial and endothelial cells. Infect. Immun. 1997, 65, 1510–1514. [Google Scholar] [PubMed]
- Zhang, Z.; Liu, R.; Noordhoek, J.A.; Kauffman, H.F. Interaction of airway epithelial cells (A549) with spores and mycelium of Aspergillus fumigatus. J. Infect. 2005, 51, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Gomez, P.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Functional genomics of human bronchial epithelial cells directly interacting with conidia of Aspergillus fumigatus. BMC Genom. 2010, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, J.L.; Gomez, P.; Ruan, J.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS ONE 2011, 6, e20527. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Datta, K.; Askin, F.B.; Staab, J.F.; Marr, K.A. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am. J. Respir. Crit. Care Med. 2012, 185, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Rammaert, B.; Jouvion, G.; de Chaumont, F.; Garcia-Hermoso, D.; Szczepaniak, C.; Renaudat, C.; Olivo-Marin, J.C.; Chretien, F.; Dromer, F.; Bretagne, S. Absence of fungal spore internalization by bronchial epithelium in mouse models evidenced by a new bioimaging approach and transmission electronic microscopy. Am. J. Pathol. 2015, 185, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Botterel, F.; Gross, K.; Ibrahim-Granet, O.; Khoufache, K.; Escabasse, V.; Coste, A.; Cordonnier, C.; Escudier, E.; Bretagne, S. Phagocytosis of Aspergillus fumigatus conidia by primary nasal epithelial cells in vitro. BMC Microbiol. 2008, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lee, M.J.; Solis, N.V.; Phan, Q.T.; Swidergall, M.; Ralph, B.; Ibrahim, A.S.; Sheppard, D.C.; Filler, S.G. Aspergillus fumigatus calA binds to integrin α5β1 and mediates host cell invasion. Nat. Microbiol. 2016, 2, 16211. [Google Scholar] [CrossRef] [PubMed]
- Levdansky, E.; Romano, J.; Shadkchan, Y.; Sharon, H.; Verstrepen, K.J.; Fink, G.R.; Osherov, N. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot. Cell 2007, 6, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Gravelat, F.N.; Beauvais, A.; Liu, H.; Lee, M.J.; Snarr, B.D.; Chen, D.; Xu, W.; Kravtsov, I.; Hoareau, C.M.; Vanier, G.; et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog. 2013, 9, e1003575. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Nakaguchi, A.; Sakai, K.; Takahashi, H.; Hagiwara, D.; Toyotome, T.; Chibana, H.; Watanabe, A.; Yaguchi, T.; Yamaguchi, M.; Kamei, K.; et al. Aspergillus fumigatus adhesion factors in dormant conidia revealed through comparative phenotypic and transcriptomic analyses. Cell. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bromley, I.M.; Donaldson, K. Binding of Aspergillus fumigatus spores to lung epithelial cells and basement membrane proteins: Relevance to the asthmatic lung. Thorax 1996, 51, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- DeHart, D.J.; Agwu, D.E.; Julian, N.C.; Washburn, R.G. Binding and germination of Aspergillus fumigatus conidia on cultured A549 pneumocytes. J. Infect. Dis. 1997, 175, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Adhesion of Aspergillus species to extracellular matrix proteins: Evidence for involvement of negatively charged carbohydrates on the conidial surface. Infect. Immun. 2000, 68, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Debeaupuis, J.P.; Crameri, R.; Carey, M.; Charles, F.; Prevost, M.C.; Schmitt, C.; Philippe, B.; Latge, J.P. Conidial hydrophobins of Aspergillus fumigatus. Appl. Environ. Microbiol. 2003, 69, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Kuboi, S.; Ishimaru, T.; Tamada, S.; Bernard, E.M.; Perlin, D.S.; Armstrong, D. Molecular characterization of AfuFleA, an L-fucose-specific lectin from Aspergillus fumigatus. J. Infect. Chemother. 2013, 19, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Houser, J.; Komarek, J.; Kostlanova, N.; Cioci, G.; Varrot, A.; Kerr, S.C.; Lahmann, M.; Balloy, V.; Fahy, J.V.; Chignard, M.; et al. A soluble fucose-specific lectin from Aspergillus fumigatus conidia—Structure, specificity and possible role in fungal pathogenicity. PLoS ONE 2013, 8, e83077. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Lund, K.P.; Christensen, K.B.; Holm, A.T.; Dubey, L.K.; Moeller, J.B.; Jepsen, C.S.; Schlosser, A.; Galgoczy, L.; Thiel, S.; et al. M-ficolin is present in Aspergillus fumigatus infected lung and modulates epithelial cell immune responses elicited by fungal cell wall polysaccharides. Virulence 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Abdolrasouli, A.; Shah, A.; Reed, A.; Armstrong-James, D.; Schelenz, S. The serum opsonin L-ficolin is detected in lungs of human transplant recipients following fungal infections and modulates inflammation and killing of Aspergillus fumigatus. J. Infect. Dis. 2015, 212, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Yates, M.; Abdolrasouli, A.; Shah, A.; Wallis, R.; Reed, A.; Armstrong-James, D.; Schelenz, S. H-ficolin binds Aspergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology 2015, 146, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Han, J.; Yu, X. E-cadherin mediates adhesion of Aspergillus fumigatus to non-small cell lung cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Chen, F.; Sun, H.; Chen, C.; Zhao, B.L. Important factors mediates the adhesion of Aspergillus fumigatus to alveolar epithelial cells with E-cadherin. Am. J. Transl. Res. 2016, 8, 2419–2425. [Google Scholar] [PubMed]
- Bidula, S.; Kenawy, H.; Ali, Y.M.; Sexton, D.; Schwaeble, W.J.; Schelenz, S. Role of ficolin-A and lectin complement pathway in the innate defense against pathogenic Aspergillus species. Infect. Immun. 2013, 81, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Han, X.; Chen, F.; Jia, X.; Zhao, J.; Zhang, C.; Yong, C.; Tian, S.; Zhou, X.; Han, L. Evidence for the involvement of cofilin in Aspergillus fumigatus internalization into type II alveolar epithelial cells. BMC Microbiol. 2015, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Thywissen, A.; Heinekamp, T.; Saluz, H.P.; Brakhage, A.A. Melanin dependent survival of Aspergillus fumigatus conidia in lung epithelial cells. Int. J. Med. Microbiol. IJMM 2014, 304, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kogan, T.V.; Jadoun, J.; Mittelman, L.; Hirschberg, K.; Osherov, N. Involvement of secreted Aspergillus fumigatus proteases in disruption of the actin fiber cytoskeleton and loss of focal adhesion sites in infected A549 lung pneumocytes. J. Infect. Dis. 2004, 189, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.K.; Lu, X.; Li, X.; Sun, Q.Y.; Su, X.; Song, Y.; Sun, H.M.; Shi, Y. Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune responses in human bronchial epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Sallenave, J.M.; Wu, Y.; Touqui, L.; Latge, J.P.; Si-Tahar, M.; Chignard, M. Aspergillus fumigatus-induced interleukin-8 synthesis by respiratory epithelial cells is controlled by the phosphatidylinositol 3-kinase, p38 MAPK, and ERK1/2 pathways and not by the Toll-like receptor-myd88 pathway. J. Biol. Chem. 2008, 283, 30513–30521. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, L.; Huet, D.; Femenia, F.; Mouyna, I.; Abdelouahab, M.; Cagna, A.; Guerrier, D.; Tichanne-Seltzer, V.; Baeza-Squiban, A.; Chermette, R.; et al. Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol. 2009, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, H.; Amar, D.; Levdansky, E.; Mircus, G.; Shadkchan, Y.; Shamir, R.; Osherov, N. PrtT-regulated proteins secreted by Aspergillus fumigatus activate MAPK signaling in exposed A549 lung cells leading to necrotic cell death. PLoS ONE 2011, 6, e17509. [Google Scholar] [CrossRef] [PubMed]
- Icheoku, U.; Bertuzzi, M.; Thomson, D.; Moyes, D.L.; Bignell, E.M.; Naglik, J.R. Temporal analysis of the Aspergillus fumigatus host-pathogen interaction reveals morphotype-specific host activation and a dominant role for secreted fungal products in epithelial damage. J. Infect. Dis. 2018, submitted. [Google Scholar]
- Namvar, S.; Warn, P.; Farnell, E.; Bromley, M.; Fraczek, M.; Bowyer, P.; Herrick, S. Aspergillus fumigatus proteases, Asp f5 and Asp f13, are essential for airway inflammation and remodelling in a murine inhalation model. Clin. Exp. Allergy 2015, 45, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chen, F.; Pan, W.; Yu, R.; Tian, S.; Han, G.; Fang, H.; Wang, S.; Zhao, J.; Li, X.; et al. Gliotoxin promotes Aspergillus fumigatus internalization into type II human pneumocyte A549 cells by inducing host phospholipase d activation. Microbes Infect. 2014, 16, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Amitani, R.; Taylor, G.; Elezis, E.N.; Llewellyn-Jones, C.; Mitchell, J.; Kuze, F.; Cole, P.J.; Wilson, R. Purification and characterization of factors produced by Aspergillus fumigatus which affect human ciliated respiratory epithelium. Infect. Immun. 1995, 63, 3266–3271. [Google Scholar] [PubMed]
- Geissler, A.; Haun, F.; Frank, D.O.; Wieland, K.; Simon, M.M.; Idzko, M.; Davis, R.J.; Maurer, U.; Borner, C. Apoptosis induced by the fungal pathogen gliotoxin requires a triple phosphorylation of Bim by JNK. Cell Death Differ. 2013, 20, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zuo, J.; Han, Y.; Zhang, L.; Yan, L.; Shen, L.; Jiang, Y.; Cao, Y.; Zhao, J. Gliotoxin produced by Aspergillus fumigatus induces apoptosis of human bronchial epithelial cells via the Bcl-2 pathway. Int. J. Clin. Exp. Med. 2017, 10, 12. [Google Scholar]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Levdansky, E.; Kashi, O.; Sharon, H.; Shadkchan, Y.; Osherov, N. The Aspergillus fumigatus cspA gene encoding a repeat-rich cell wall protein is important for normal conidial cell wall architecture and interaction with host cells. Eukaryot. Cell 2010, 9, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.C.; Fischer, G.J.; Sinha, M.; McCabe, O.; Palmer, J.M.; Choera, T.; Yun Lim, F.; Wimmerova, M.; Carrington, S.D.; Yuan, S.; et al. FleA expression in Aspergillus fumigatus is recognized by fucosylated structures on mucins and macrophages to prevent lung infection. PLoS Pathog. 2016, 12, e1005555. [Google Scholar] [CrossRef] [PubMed]
- Gout, E.; Garlatti, V.; Smith, D.F.; Lacroix, M.; Dumestre-Pérard, C.; Lunardi, T.; Martin, L.; Cesbron, J.-Y.; Arlaud, G.J.; Gaboriaud, C.; et al. Carbohydrate recognition properties of human ficolins: Glycan array screening reveals the sialic acid binding specificity of M-ficolin. J. Biol. Chem. 2010, 285, 6612–6622. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Liu, Y.; Kanno, K.; Takahashi, M.; Matsushita, M.; Fujita, T. Identification of the mouse H-ficolin gene as a pseudogene and orthology between mouse ficolins a/b and human L-/M-ficolins. Genomics 2004, 84, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Genster, N.; Praestekjaer Cramer, E.; Rosbjerg, A.; Pilely, K.; Cowland, J.B.; Garred, P. Ficolins promote fungal clearance in vivo and modulate the inflammatory cytokine response in host defense against Aspergillus fumigatus. J. Innate Immun. 2016, 8, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Delangle, A.; Simenel, C.; Coddeville, B.; van Vliet, S.J.; van Kooyk, Y.; Bozza, S.; Moretti, S.; Schwarz, F.; Trichot, C.; et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Gravelat, F.N.; Cerone, R.P.; Baptista, S.D.; Campoli, P.V.; Choe, S.I.; Kravtsov, I.; Vinogradov, E.; Creuzenet, C.; Liu, H.; et al. Overlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J. Biol. Chem. 2014, 289, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Geller, A.M.; Bamford, N.C.; Liu, H.; Gravelat, F.N.; Snarr, B.D.; Le Mauff, F.; Chabot, J.; Ralph, B.; Ostapska, H.; et al. Deacetylation of fungal exopolysaccharide mediates adhesion and biofilm formation. mBio 2016, 7, e00252-16. [Google Scholar] [CrossRef] [PubMed]
- Escobar, N.; Ordonez, S.R.; Wösten, H.A.B.; Haas, P.-J.A.; de Cock, H.; Haagsman, H.P. Hide, keep quiet, and keep low: Properties that make Aspergillus fumigatus a successful lung pathogen. Front. Microbiol. 2016, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Belaish, R.; Sharon, H.; Levdansky, E.; Greenstein, S.; Shadkchan, Y.; Osherov, N. The Aspergillus nidulans cetA and calA genes are involved in conidial germination and cell wall morphogenesis. Fungal Genet. Biol. 2008, 45, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Mahajan, L.; Ramjee, S.; Singh, Y.; Basir, S.F.; Madan, T. Identification and characterization of a laminin-binding protein of Aspergillus fumigatus: Extracellular thaumatin domain protein (AfCalAp). J. Med. Microbiol. 2009, 58, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, C.; Jia, X.; Wang, S.; Wang, J.; Chen, Y.; Zhao, J.; Tian, S.; Han, X.; Han, L. Transcriptome profiles of human lung epithelial cells A549 interacting with Aspergillus fumigatus by RNA-seq. PLoS ONE 2015, 10, e0135720. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Di Ianni, M.; Bozza, S.; Giovannini, G.; Zagarella, S.; Zelante, T.; D’Angelo, C.; Pierini, A.; Pitzurra, L.; Falzetti, F.; et al. Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 2010, 116, 5394–5402. [Google Scholar] [CrossRef] [PubMed]
- Heyl, K.A.; Klassert, T.E.; Heinrich, A.; Muller, M.M.; Klaile, E.; Dienemann, H.; Grunewald, C.; Bals, R.; Singer, B.B.; Slevogt, H. Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable Haemophilus influenzae. mBio 2014, 5, e01492-14. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Yuk, J.M.; Shin, D.M.; Jo, E.K. Dectin-1 is inducible and plays an essential role for mycobacteria-induced innate immune responses in airway epithelial cells. J. Clin. Immunol. 2009, 29, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Jahn, B.; Boukhallouk, F.; Lotz, J.; Langfelder, K.; Wanner, G.; Brakhage, A.A. Interaction of human phagocytes with pigmentless Aspergillus conidia. Infect. Immun. 2000, 68, 3736–3739. [Google Scholar] [CrossRef] [PubMed]
- Volling, K.; Thywissen, A.; Brakhage, A.A.; Saluz, H.P. Phagocytosis of melanized aspergillus conidia by macrophages exerts cytoprotective effects by sustained PI3K/AKT signalling. Cell. Microbiol. 2011, 13, 1130–1148. [Google Scholar] [CrossRef] [PubMed]
- Thywißen, A.; Heinekamp, T.; Dahse, H.-M.; Schmaler-Ripcke, J.; Nietsche, S.; Zipfel, P.; Brakhage, A. Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front. Microbiol. 2011, 2, 96. [Google Scholar] [CrossRef] [PubMed]
- Chamilos, G.; Akoumianaki, T.; Kyrmizi, I.; Brakhage, A.; Beauvais, A.; Latge, J.-P. Melanin targets LC3-associated phagocytosis (LAP): A novel pathogenetic mechanism in fungal disease. Autophagy 2016, 12, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Akoumianaki, T.; Kyrmizi, I.; Valsecchi, I.; Gresnigt Mark, S.; Samonis, G.; Drakos, E.; Boumpas, D.; Muszkieta, L.; Prevost, M.-C.; Kontoyiannis Dimitrios, P.; et al. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe 2016, 19, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, C.; Hess, C.; Bals, R. Aspergillus fumigatus conidia induce interferon-beta signalling in respiratory epithelial cells. Eur. Respir. J. 2012, 39, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.; Wang, X.; Sifuentes Dos Santos, J.; Fysikopoulos, A.; Tadrist, S.; Canlet, C.; Artigot, M.P.; Loiseau, N.; Oswald, I.P.; Puel, O. Trypacidin, a spore-borne toxin from Aspergillus fumigatus, is cytotoxic to lung cells. PLoS ONE 2012, 7, e29906. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Wang, M.; Sun, W.K.; Xia, D.; Tan, M.M.; Ding, Y.; Qian, Q.; Su, X.; Shi, Y. Up-regulation of Dectin-1 in airway epithelial cells promotes mice defense against invasive pulmonary aspergillosis. Int. J. Clin. Exp. Med. 2015, 8, 17489–17497. [Google Scholar] [PubMed]
- Fekkar, A.; Balloy, V.; Pionneau, C.; Marinach-Patrice, C.; Chignard, M.; Mazier, D. Secretome of human bronchial epithelial cells in response to the fungal pathogen Aspergillus fumigatus analyzed by differential in-gel electrophoresis. J. Infect. Dis. 2012, 205, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Tomee, J.F.; Wierenga, A.T.; Hiemstra, P.S.; Kauffman, H.K. Proteases from Aspergillus fumigatus induce release of proinflammatory cytokines and cell detachment in airway epithelial cell lines. J. Infect. Dis. 1997, 176, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Seddigh, P.; Bracht, T.; Molinier-Frenkel, V.; Castellano, F.; Kniemeyer, O.; Schuster, M.; Weski, J.; Hasenberg, A.; Kraus, A.; Poschet, G.; et al. Quantitative analysis of proteome modulations in alveolar epithelial type II cells in response to pulmonary Aspergillus fumigatus infection. Mol. Cell. Proteom. 2017, 16, 2184–2198. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Huang, W.; Liang, J.; Guo, J.; Ji, J.; Yao, Y.; Zheng, M.; Cai, Z.; Lu, L.; Wang, J. IL4I1 is a novel regulator of M2 macrophage polarization that can inhibit T cell activation via L-tryptophan and arginine depletion and IL-10 production. PLoS ONE 2015, 10, e0142979. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.; Seyedmousavi, S.; Sugui, J.A.; Balenga, N.; Zhao, M.; Kwon Chung, K.J.; Biardel, S.; Laviolette, M.; Druey, K.M. Aspergillus fumigatus alkaline protease 1 (Alp1/Asp f13) in the airways correlates with asthma severity. J. Allergy Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Homma, T.; Norton, J.E.; Sha, Q.; Siebert, J.; Gupta, D.S.; Schroeder, J.W.; Schleimer, R.P. Suppression of epithelial signal transducer and activator of transcription 1 activation by extracts of Aspergillus fumigatus. Am. J. Respir. Cell Mol. Biol. 2015, 53, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kato, A.; Bhushan, B.; Norton, J.E.; Suh, L.A.; Carter, R.G.; Gupta, D.S.; Schleimer, R.P. Role of Aspergillus fumigatus in triggering protease-activated receptor-2 in airway epithelial cells and skewing the cells toward a T-helper 2 bias. Am. J. Respir. Cell Mol. Biol. 2016, 54, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Dolan, S.K.; O’Keeffe, G.; Jones, G.W.; Doyle, S. Resistance is not futile: Gliotoxin biosynthesis, functionality and utility. Trends Microbiol. 2015, 23, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, R.; Lewis, R.E.; Leventakos, K.; Kontoyiannis, D.P. Aspergillus fumigatus inhibits angiogenesis through the production of gliotoxin and other secondary metabolites. Blood 2009, 114, 5393. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E.; Wiederhold, N.P.; Chi, J.; Han, X.Y.; Komanduri, K.V.; Kontoyiannis, D.P.; Prince, R.A. Detection of gliotoxin in experimental and human aspergillosis. Infect. Immun. 2005, 73, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Domingo, M.P.; Colmenarejo, C.; Martinez-Lostao, L.; Mullbacher, A.; Jarne, C.; Revillo, M.J.; Delgado, P.; Roc, L.; Meis, J.F.; Rezusta, A.; et al. Bis(methyl)gliotoxin proves to be a more stable and reliable marker for invasive aspergillosis than gliotoxin and suitable for use in diagnosis. Diagn. Microbiol. Infect. Dis. 2012, 73, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sugui, J.A.; Pardo, J.; Chang, Y.C.; Zarember, K.A.; Nardone, G.; Galvez, E.M.; Mullbacher, A.; Gallin, J.I.; Simon, M.M.; Kwon-Chung, K.J. Gliotoxin is a virulence factor of Aspergillus fumigatus: GliP deletion attenuates virulence in mice immunosuppressed with hydrocortisone. Eukaryot. Cell 2007, 6, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Spikes, S.; Xu, R.; Nguyen, C.K.; Chamilos, G.; Kontoyiannis, D.P.; Jacobson, R.H.; Ejzykowicz, D.E.; Chiang, L.Y.; Filler, S.G.; May, G.S. Gliotoxin production in Aspergillus fumigatus contributes to host-specific differences in virulence. J. Infect. Dis. 2008, 197, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Thorley, A.J.; Grandolfo, D.; Lim, E.; Goldstraw, P.; Young, A.; Tetley, T.D. Innate immune responses to bacterial ligands in the peripheral human lung--role of alveolar epithelial TLR expression and signalling. PLoS ONE 2011, 6, e21827. [Google Scholar] [CrossRef] [PubMed]
- Pollmächer, J.; Figge, M.T. Agent-based model of human alveoli predicts chemotactic signaling by epithelial cells during early Aspergillus fumigatus infection. PLoS ONE 2014, 9, e111630. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.J.; Boon, N.J.; Vrcelj, K.; Nguyen, A.; Vinci, C.; Armstrong-James, D.; Bignell, E. In silico modeling of spore inhalation reveals fungal persistence following low dose exposure. Sci. Rep. 2015, 5, 13958. [Google Scholar] [CrossRef] [PubMed]
- Seidler, M.J.; Salvenmoser, S.; Müller, F.-M.C. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob. Agents Chemother. 2008, 52, 4130–4136. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Bomme, P.; Lechner, B.E.; Mislin, G.L.A.; Lair, V.; Prévost, M.-C.; Latgé, J.-P.; Haas, H.; Beauvais, A. Pseudomonas aeruginosa manipulates redox and iron homeostasis of its microbiota partner Aspergillus fumigatus via phenazines. Sci. Rep. 2015, 5, 8220. [Google Scholar] [CrossRef] [PubMed]
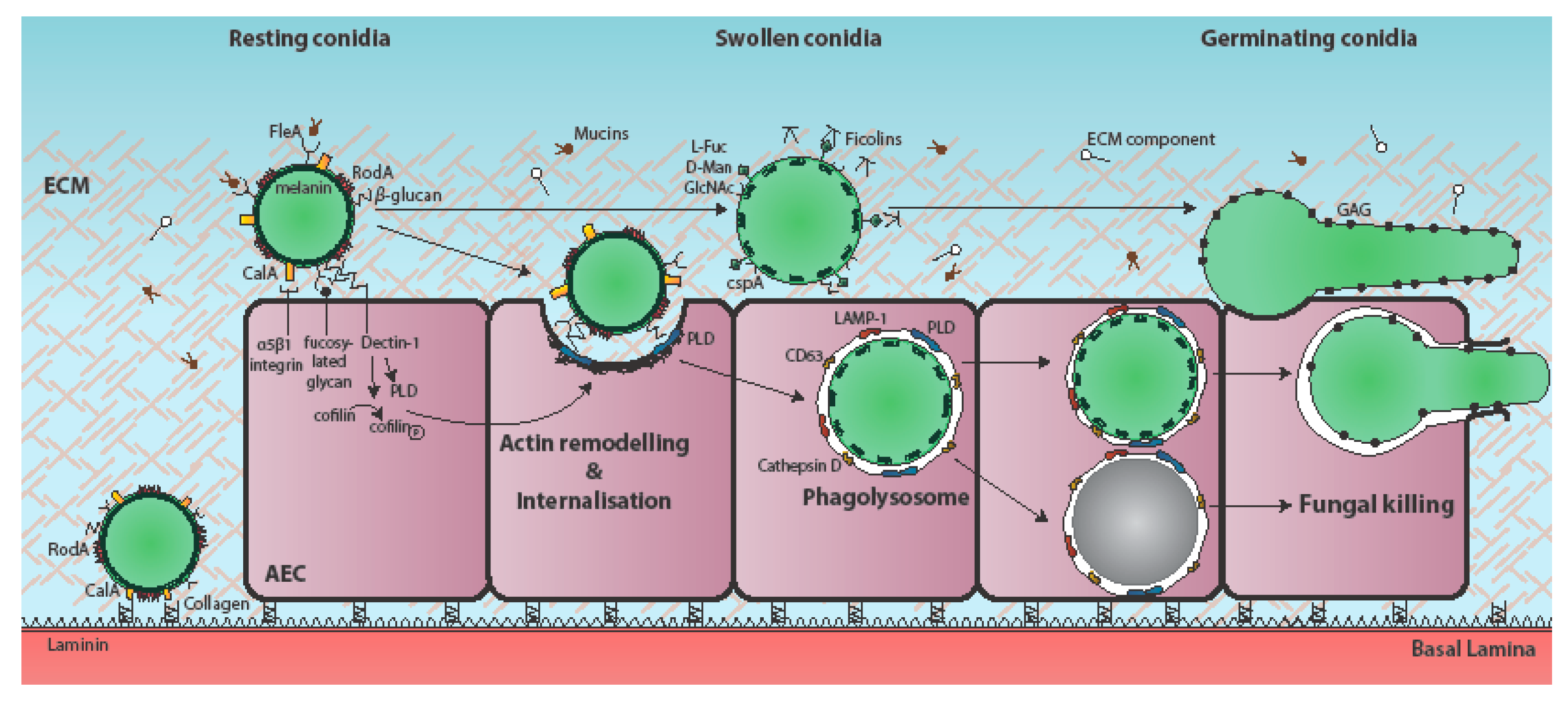
Figure 1.
Temporal and mechanistic basis of A. fumigatus attachment to, and uptake by, the respiratory epithelium. Respiratory aspergilloses commence with interaction of inhaled fungal particles with the respiratory epithelium. During vegetative growth of the pathogen, individual airway epithelial cells (AECs) may become iteratively exposed to one or multiple fungal morphotypes, as well as cell surface-associated and secreted fungal factors [8]. A. fumigatus conidia bind to AECs, extracellular matrix (ECM) components, and basement membrane components [21,22,23]. The immunoprotective conidial hydrophobin RodA is required for adherence to collagen [10,18,19,20,24] and the fucose-specific conidial lectin FleA mediates fucose-dependent binding of A. fumigatus conidia to airway mucin [25,26]. The cell surface protein CspA, which becomes unmasked during conidial germination, is also required for adhesion to A549-derived ECM [18]. The H-ficolin opsonin binds to A. fumigatus conidia via l-fucose, d-mannose and N-acetylglucosamine (GalNAc) on the conidial surface and moderates adhesion to A549 cells [27,28,29]. The secreted and hyphal cell wall-associated exopolysaccharide galactosaminogalactan (GAG) mediates adherence of A. fumigatus hyphae to fibronectin and epithelial cells [19]. CalA is the first identified invasin of A. fumigatus and is required for epithelial entry in a α5β1 integrin-dependent manner [17]. Spore internalisation is thought to be dependent on E-cadherin [30,31,32], and the actin regulators phospholipase D (PLD) [9] and cofilin-1 [33]. PLD co-localises with internalised conidia, as do the late endosomal/lysosomal markers LAMP-1, CD63, and cathepsin D [6,9]. Most internalised conidia are killed, but a few remain viable and eventually germinate to escape the phagolysosome without lysis of the host cell [6]. Induction of PLD following exposure to β-1,3-glucan on the surface of germinating conidia has been demonstrated to occur in a Dectin-1-dependent manner [9]. Conidial dihydroxynaphthalane (DHN) melanin increases internalisation of A. fumigatus spores by A549 cells and by preventing the acidification of AEC phagolysosomes, promotes spore viability [34].
Figure 1.
Temporal and mechanistic basis of A. fumigatus attachment to, and uptake by, the respiratory epithelium. Respiratory aspergilloses commence with interaction of inhaled fungal particles with the respiratory epithelium. During vegetative growth of the pathogen, individual airway epithelial cells (AECs) may become iteratively exposed to one or multiple fungal morphotypes, as well as cell surface-associated and secreted fungal factors [8]. A. fumigatus conidia bind to AECs, extracellular matrix (ECM) components, and basement membrane components [21,22,23]. The immunoprotective conidial hydrophobin RodA is required for adherence to collagen [10,18,19,20,24] and the fucose-specific conidial lectin FleA mediates fucose-dependent binding of A. fumigatus conidia to airway mucin [25,26]. The cell surface protein CspA, which becomes unmasked during conidial germination, is also required for adhesion to A549-derived ECM [18]. The H-ficolin opsonin binds to A. fumigatus conidia via l-fucose, d-mannose and N-acetylglucosamine (GalNAc) on the conidial surface and moderates adhesion to A549 cells [27,28,29]. The secreted and hyphal cell wall-associated exopolysaccharide galactosaminogalactan (GAG) mediates adherence of A. fumigatus hyphae to fibronectin and epithelial cells [19]. CalA is the first identified invasin of A. fumigatus and is required for epithelial entry in a α5β1 integrin-dependent manner [17]. Spore internalisation is thought to be dependent on E-cadherin [30,31,32], and the actin regulators phospholipase D (PLD) [9] and cofilin-1 [33]. PLD co-localises with internalised conidia, as do the late endosomal/lysosomal markers LAMP-1, CD63, and cathepsin D [6,9]. Most internalised conidia are killed, but a few remain viable and eventually germinate to escape the phagolysosome without lysis of the host cell [6]. Induction of PLD following exposure to β-1,3-glucan on the surface of germinating conidia has been demonstrated to occur in a Dectin-1-dependent manner [9]. Conidial dihydroxynaphthalane (DHN) melanin increases internalisation of A. fumigatus spores by A549 cells and by preventing the acidification of AEC phagolysosomes, promotes spore viability [34].
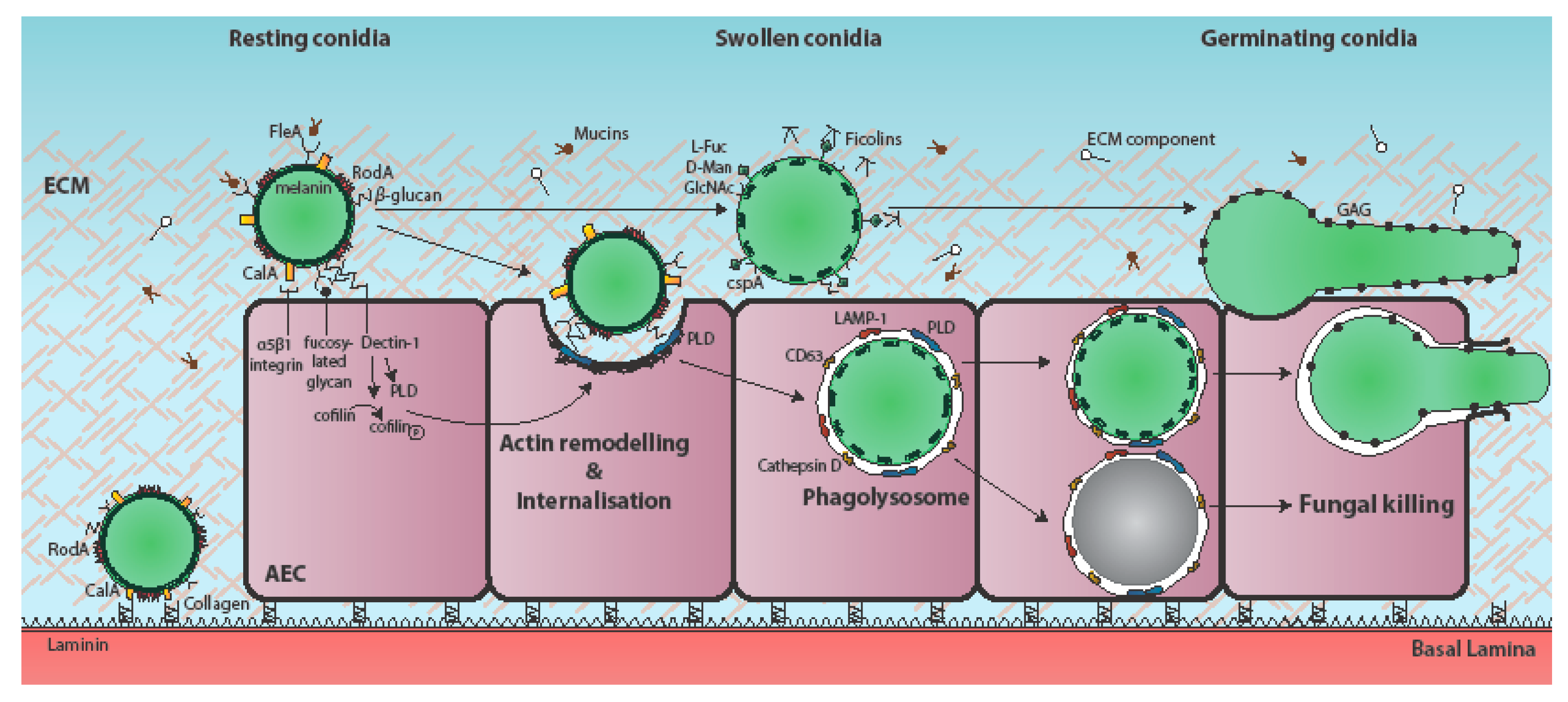
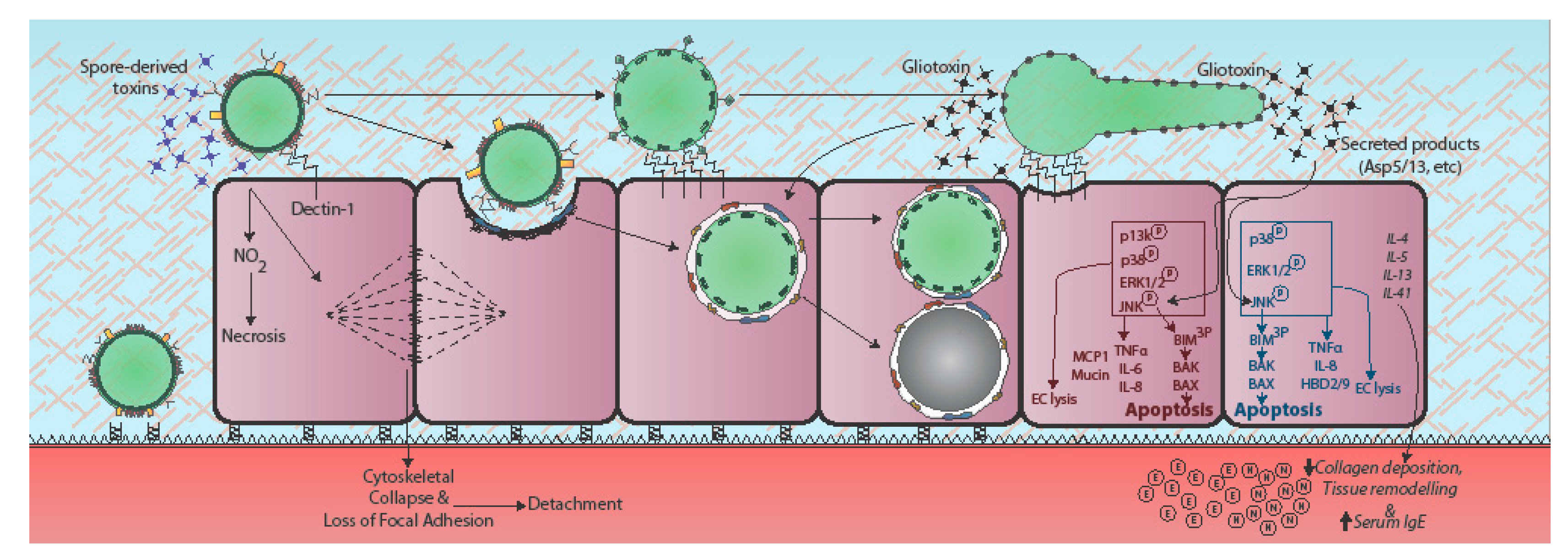
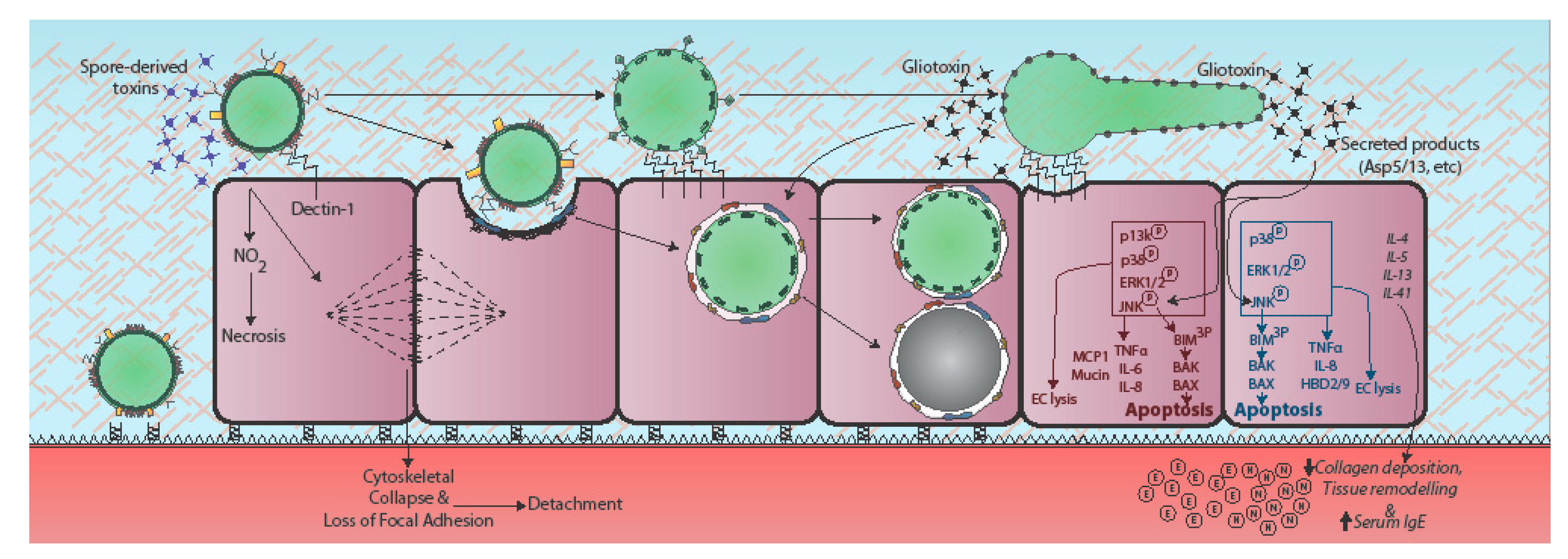
Figure 2.
Epithelial responses to A. fumigatus. Epithelial responses to A. fumigatus vary dramatically according to fungal morphotype and host cell origin, the latter denoted in red for alveolar epithelial cells and blue for bronchial epithelial cells. Contact with A. fumigatus spores prompts rapid cytoskeletal reorganisation and loss of focal adherence, possibly via spore-associated toxins [35]. In bronchial epithelial cells recognition of germinating conidia and hyphae leads to upregulation of Dectin-1 expression at the host cell membrane and phosphorylation-mediated activation of phosphatidylinositol3-kinase (PI3K), mitogen activated protein kinase (MAPK) p38 and ERK1/2 signalling, leading to TNFα, IL-8, and β-defensin expression (HBD-2 and -9) [36,37,38]. In AECs, challenge with A. fumigatus culture filtrate prompts MAPK ERK1/2, p38, and c-Jun N-terminal protein kinases (JNK) phosphorylation, in a partially fungal protease-dependent manner, leading to lytic death of the host cell [39,40]. JNK or p38 inhibitors protect against host damage [39]. The secreted products of cultured A. fumigatus also elicit remodelling of the respiratory epithelium involving heightened expression of classical Th2 cytokines (IL-4, IL-5, IL-13), serum IgE, collagen deposition, and neutrophil (N) and eosinophil (E) recruitment [41]. Gliotoxin exposure induces cytoskeletal collapse, cell peeling, and apoptosis. Gt activates the JNK pathway leading to apoptosis. JNK triple-phosphorylates the pro-apoptotic protein Bim, leading to pro-apoptotic Bak and Bax activation, mitochondrial membrane permeabilization, and apoptosis [42,43,44,45]. Italicised text indicates responses observed in whole animals or humans.
Figure 2.
Epithelial responses to A. fumigatus. Epithelial responses to A. fumigatus vary dramatically according to fungal morphotype and host cell origin, the latter denoted in red for alveolar epithelial cells and blue for bronchial epithelial cells. Contact with A. fumigatus spores prompts rapid cytoskeletal reorganisation and loss of focal adherence, possibly via spore-associated toxins [35]. In bronchial epithelial cells recognition of germinating conidia and hyphae leads to upregulation of Dectin-1 expression at the host cell membrane and phosphorylation-mediated activation of phosphatidylinositol3-kinase (PI3K), mitogen activated protein kinase (MAPK) p38 and ERK1/2 signalling, leading to TNFα, IL-8, and β-defensin expression (HBD-2 and -9) [36,37,38]. In AECs, challenge with A. fumigatus culture filtrate prompts MAPK ERK1/2, p38, and c-Jun N-terminal protein kinases (JNK) phosphorylation, in a partially fungal protease-dependent manner, leading to lytic death of the host cell [39,40]. JNK or p38 inhibitors protect against host damage [39]. The secreted products of cultured A. fumigatus also elicit remodelling of the respiratory epithelium involving heightened expression of classical Th2 cytokines (IL-4, IL-5, IL-13), serum IgE, collagen deposition, and neutrophil (N) and eosinophil (E) recruitment [41]. Gliotoxin exposure induces cytoskeletal collapse, cell peeling, and apoptosis. Gt activates the JNK pathway leading to apoptosis. JNK triple-phosphorylates the pro-apoptotic protein Bim, leading to pro-apoptotic Bak and Bax activation, mitochondrial membrane permeabilization, and apoptosis [42,43,44,45]. Italicised text indicates responses observed in whole animals or humans.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bertuzzi, M.; Hayes, G.E.; Icheoku, U.J.; Van Rhijn, N.; Denning, D.W.; Osherov, N.; Bignell, E.M. Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. J. Fungi 2018, 4, 8. https://doi.org/10.3390/jof4010008
AMA Style
Bertuzzi M, Hayes GE, Icheoku UJ, Van Rhijn N, Denning DW, Osherov N, Bignell EM. Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. Journal of Fungi. 2018; 4(1):8. https://doi.org/10.3390/jof4010008
Chicago/Turabian StyleBertuzzi, Margherita, Gemma E. Hayes, Uju J. Icheoku, Norman Van Rhijn, David W. Denning, Nir Osherov, and Elaine M. Bignell. 2018. "Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease" Journal of Fungi 4, no. 1: 8. https://doi.org/10.3390/jof4010008
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.