
Targeted Mybpc3 Knock-Out Mice with Cardiac Hypertrophy Exhibit Structural Mitral Valve Abnormalities

 and
and
Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
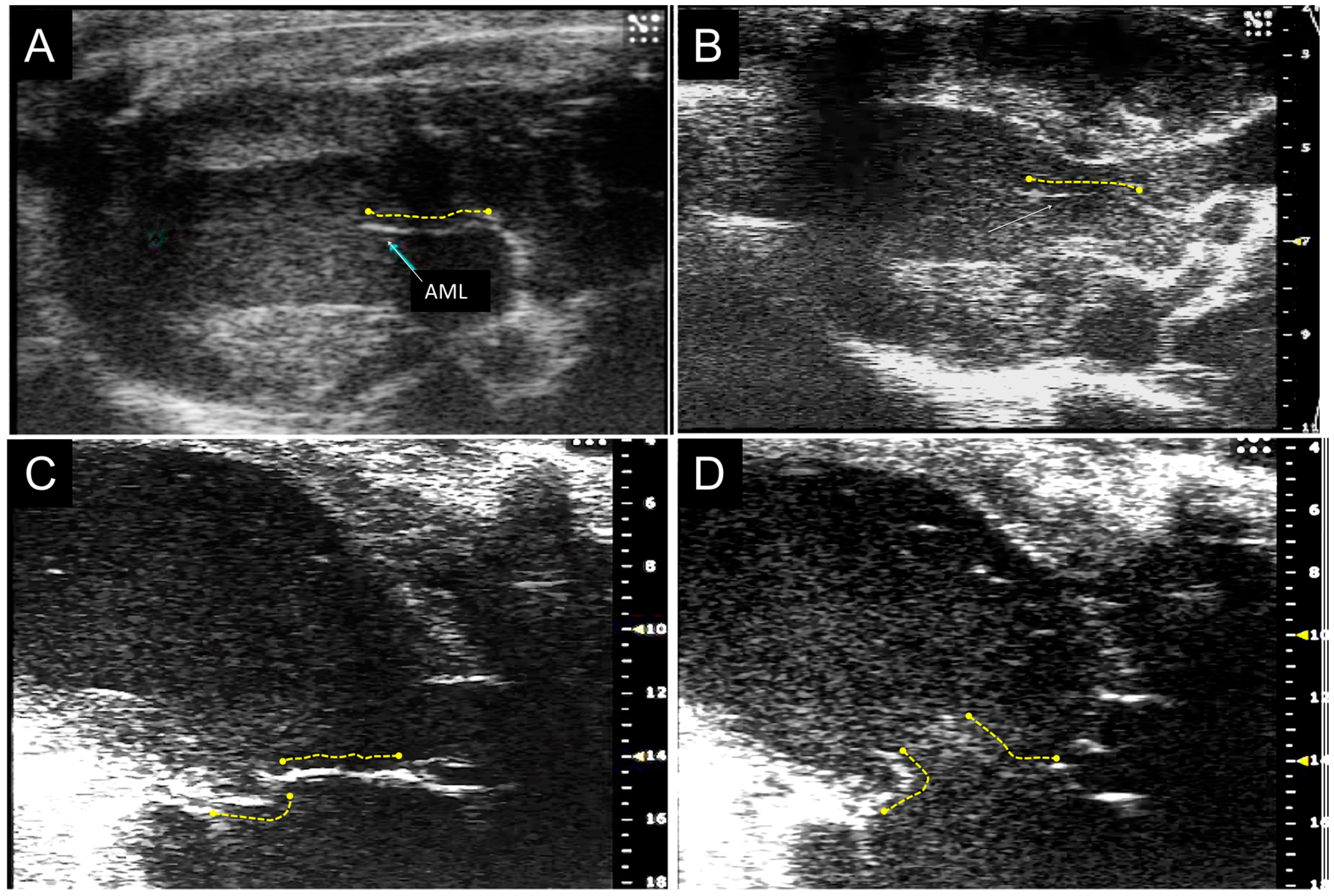
3.1. Mybpc3 (−/−) Anterior Mitral Leaflet is Longer and Thicker than Wild-Type Anterior Mitral Leaflet by Echocardiographic Analysis
3.2. MicroCT Imaging Confirms the Results of Echocardiographic Imaging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | +/+ | +/− | −/− | p |
|---|---|---|---|---|
| N = 22 | N = 33 | N = 29 | ||
| Length AML (mm) | 1.72 ± 0.08 | 1.73 ± 0.07 | 1.92 ± 0.08 | <0.001 * |
| (1.64–1.94) | (1.55–1.90) | (1.80–2.09) | ||
| Length PML (mm) | 1.36 ± 0.07 | 1.33 ± 0.07 | 1.35 ± 0.07 | NS * |
| (1.23–1.46) | (1.20–1.45) | (1.23–1.51) | ||
| Thickness AML + PML (mm) | 0.15 ± 0.02 | 0.18 ± 0.04 | 0.23 ± 0.04 | <0.001 * |
| (0.11–0.18) | (0.10–0.19) | (0.14–0.29) | ||
| LV Mass (mg) | 80 ± 16 | 88 ± 7 | 129 ± 26 | <0.0001 |
| (55–109) | (71–105) | (99–173) | ||
| EF (%) | 63 ± 5 | 62 ± 7 | 35 ± 7 | <0.0001 |
| (55–76) | (52–80) | (22–49) | ||
| ED Volume (mL) | 67 ± 11 | 63 ± 11 | 118 ± 40 | <0.0001 |
| (43–88) | (39–89) | (42–175) |
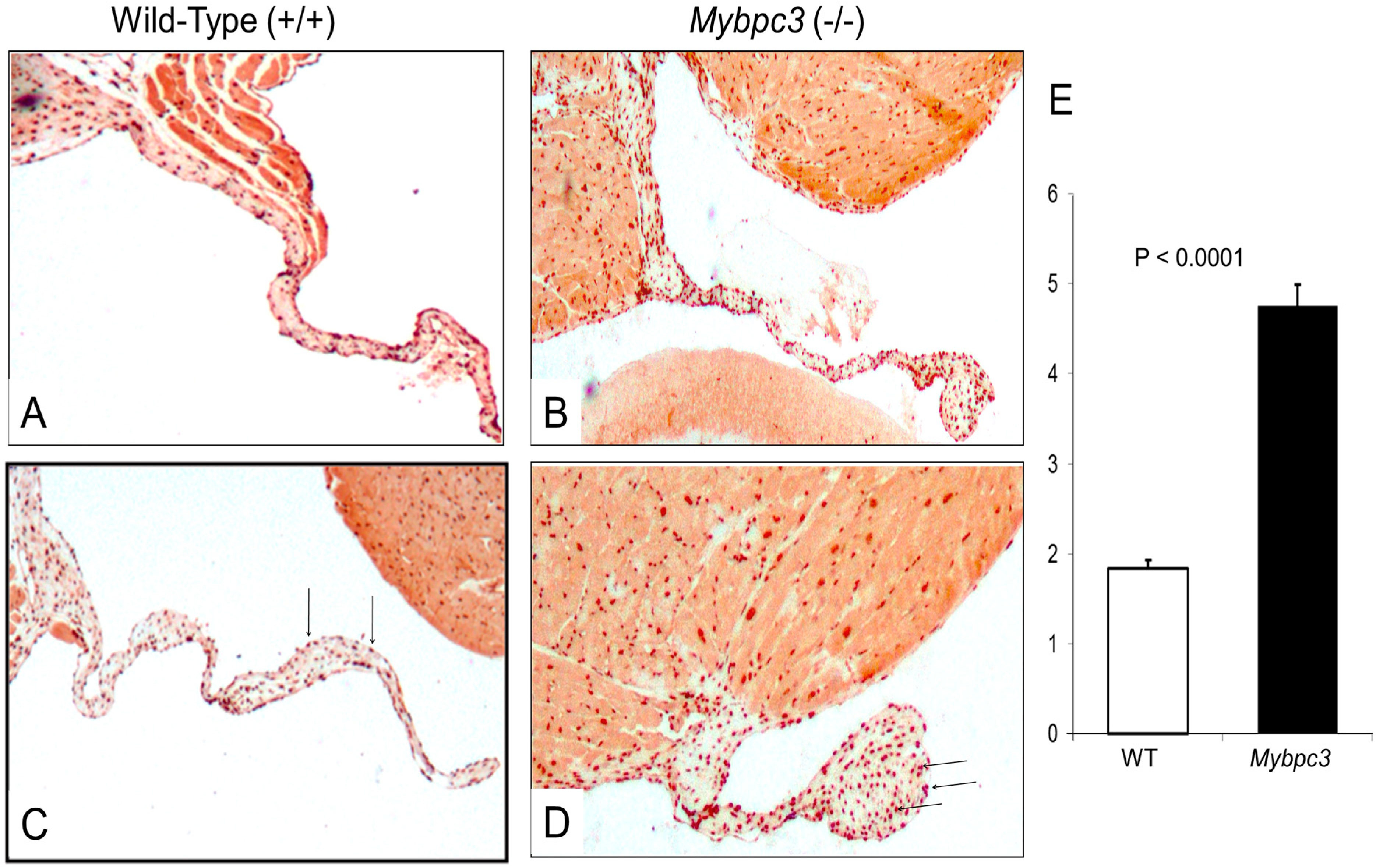
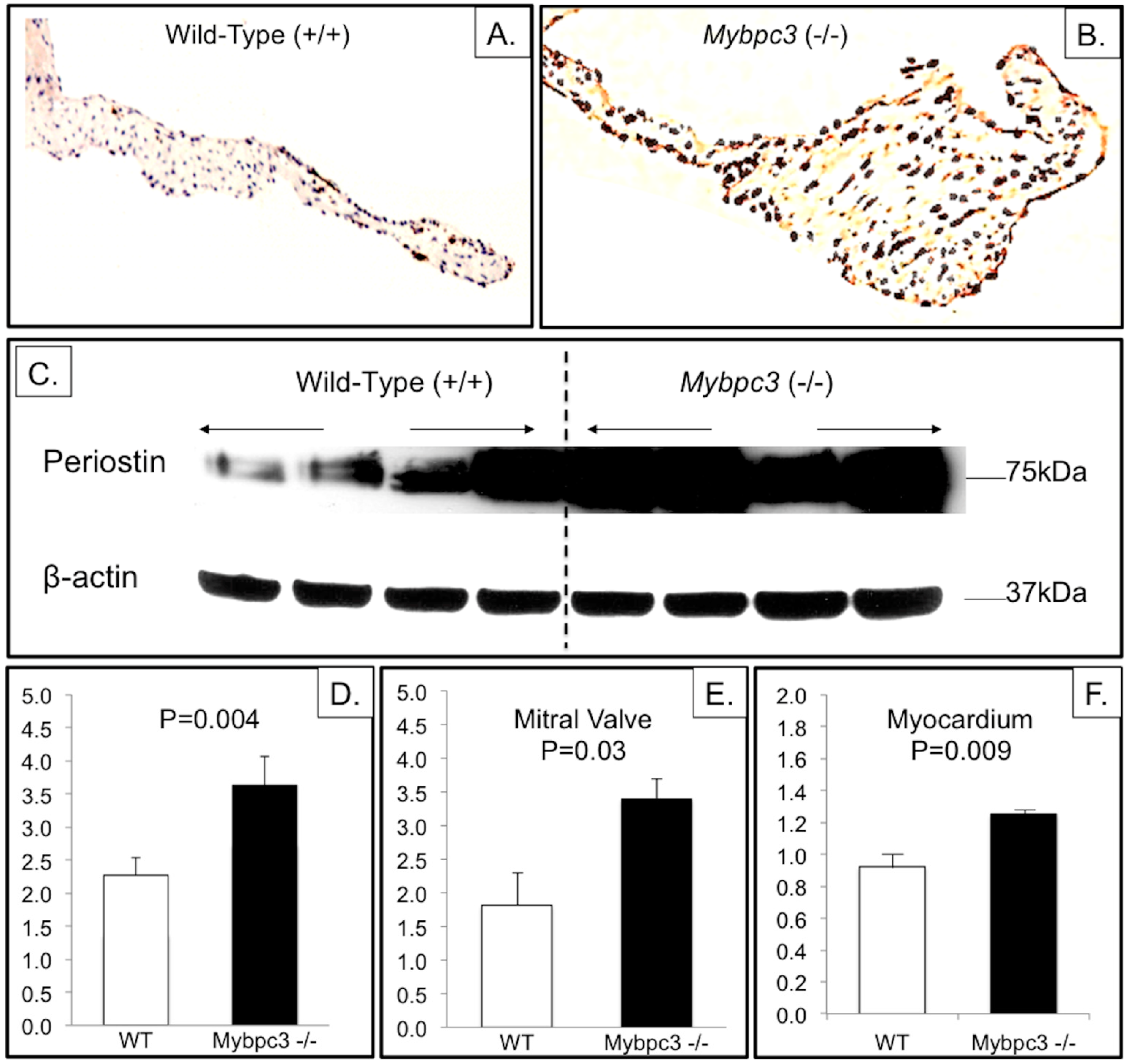
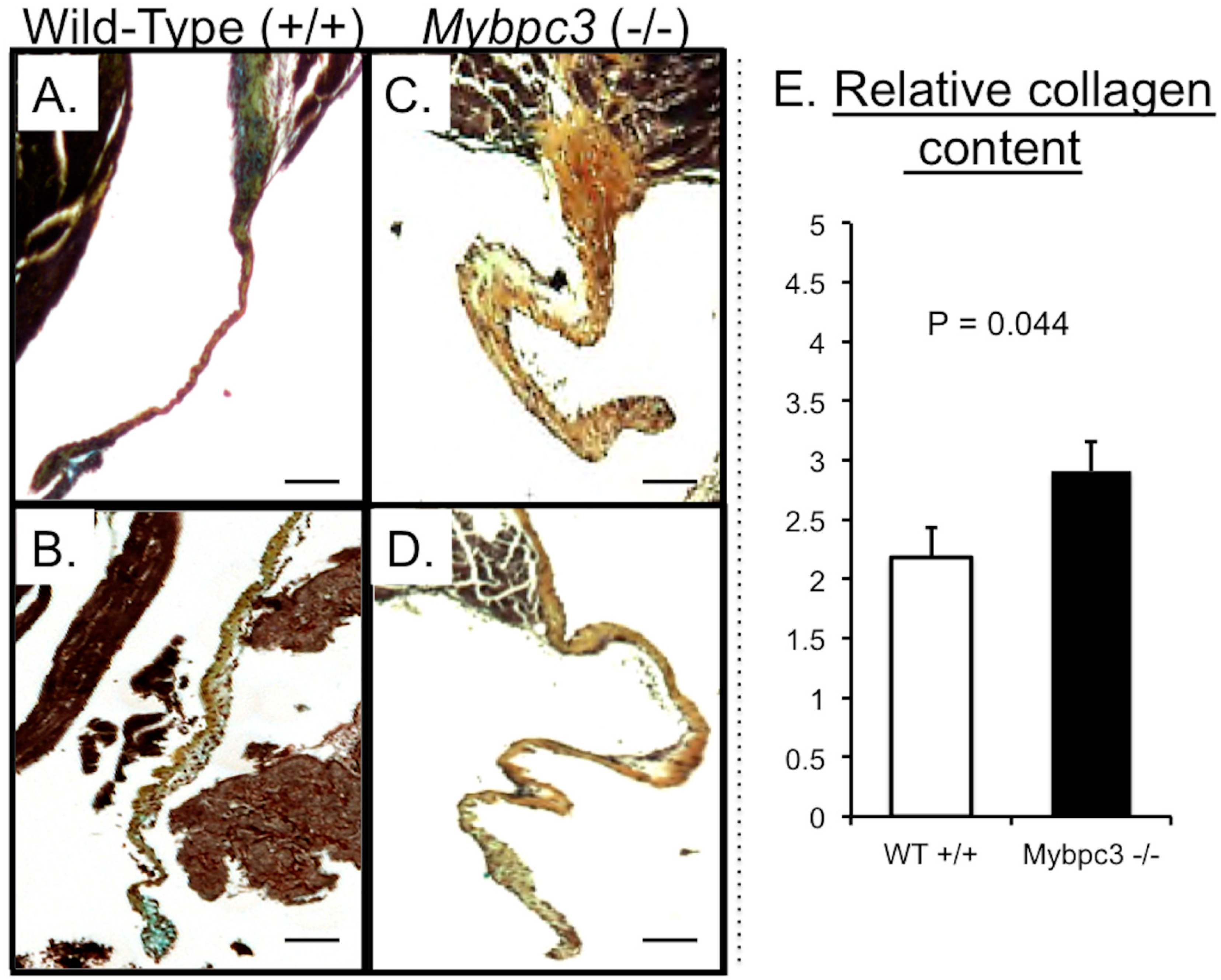
3.3. Mybpc3-(−/−) Mitral Valve Leaflets Have Evidence of Increased TGFβ Signaling


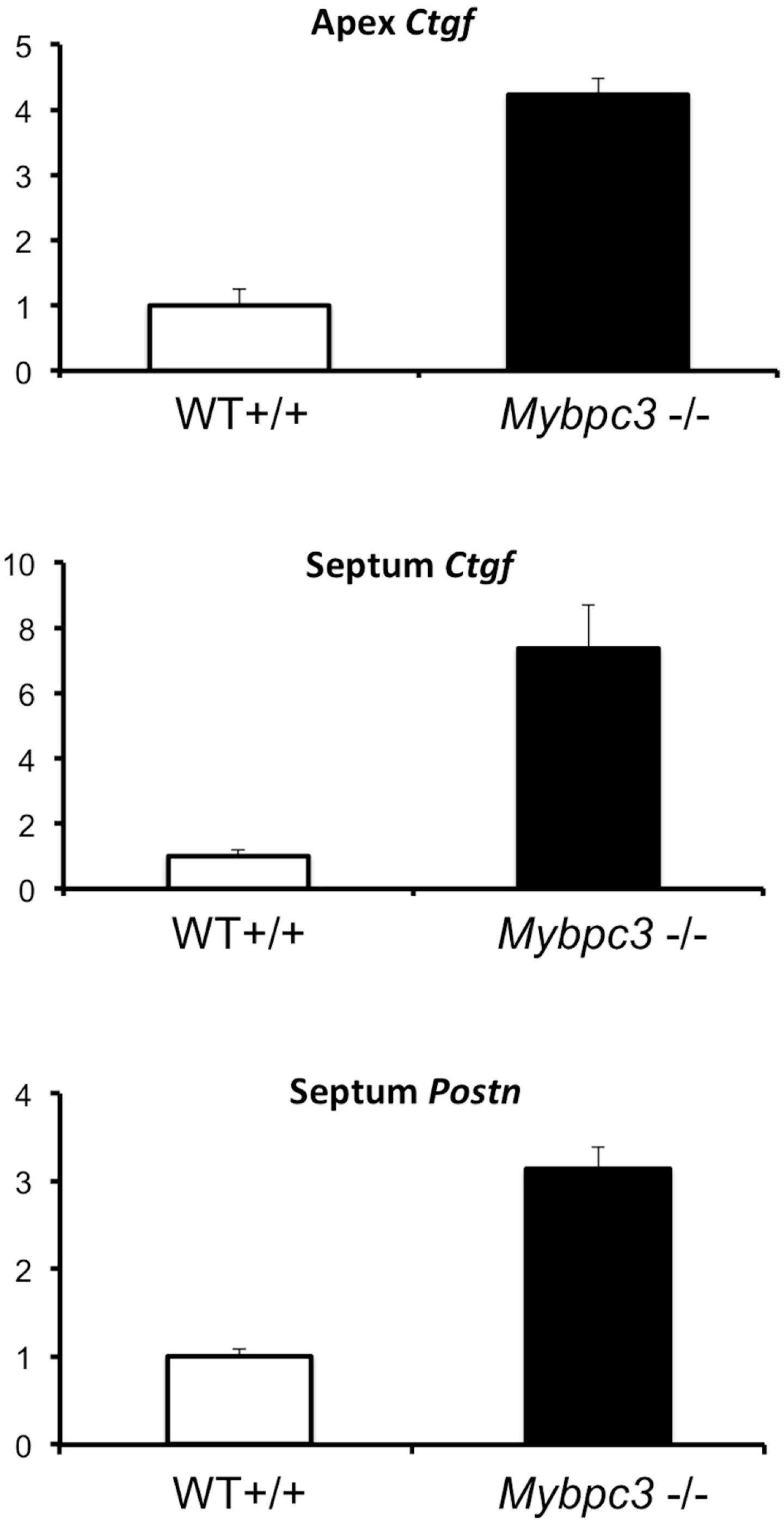
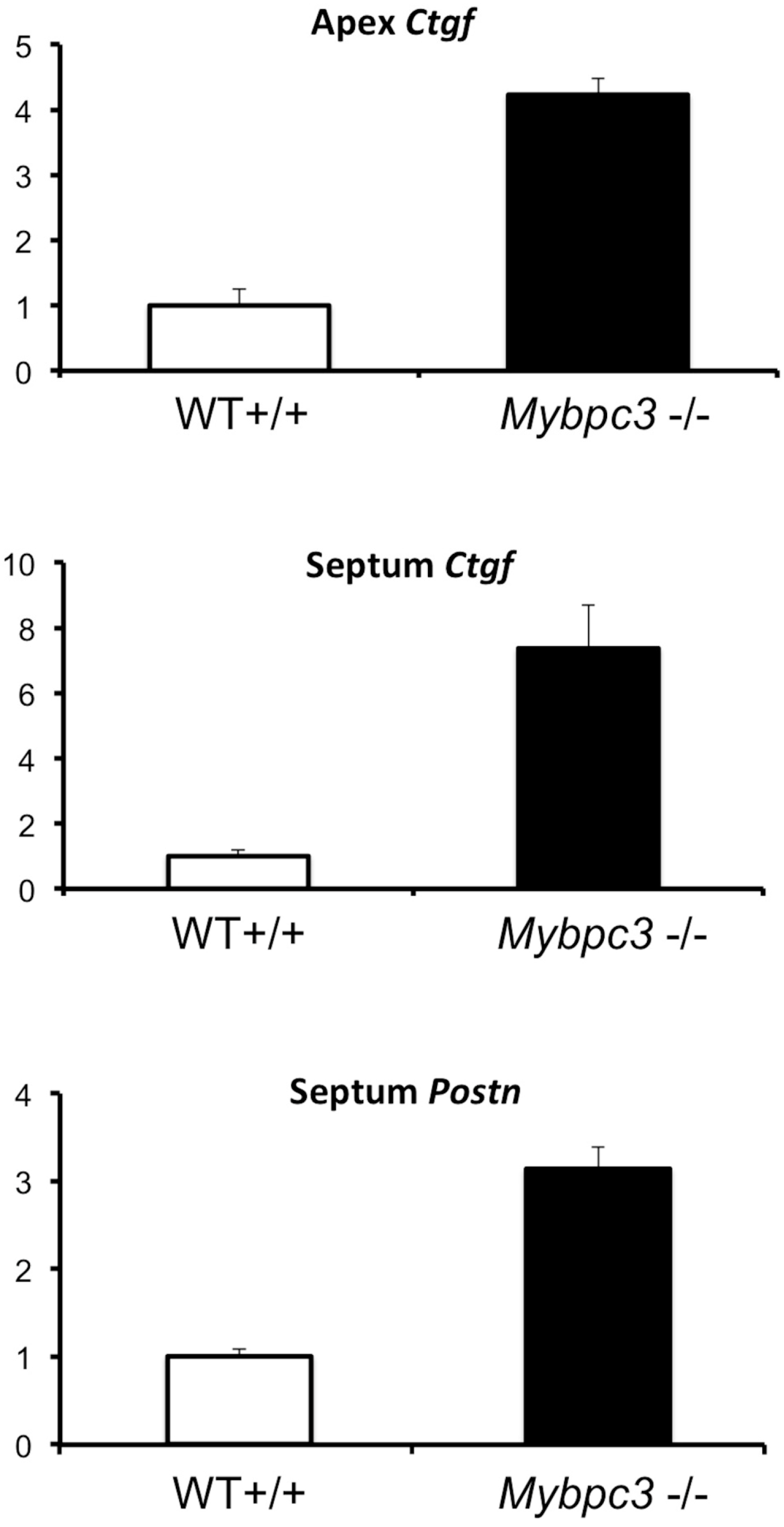
3.4. Quantitative PCR Analysis Confirms Increased Expression of TGFβ-Related Genes in Myocardium
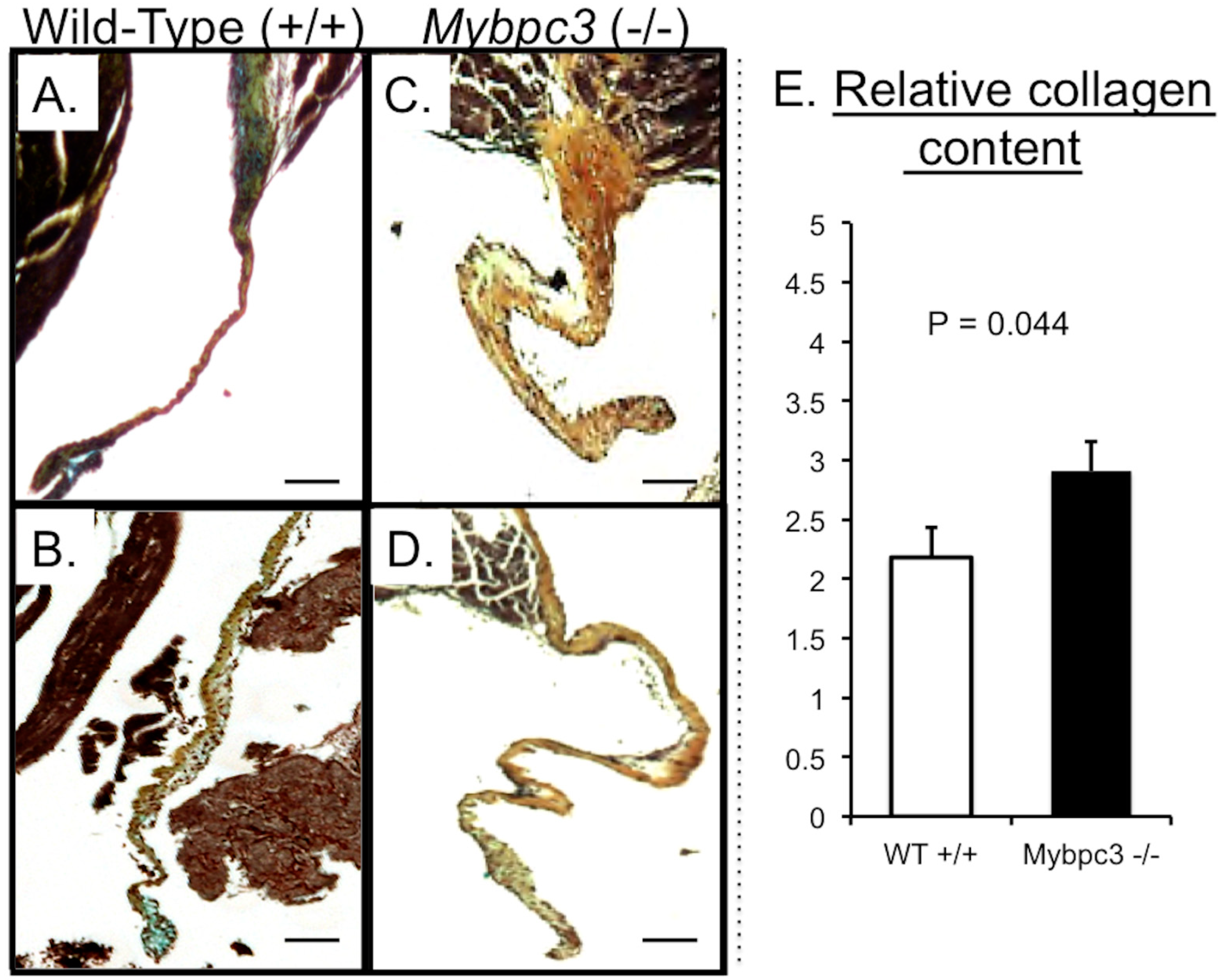
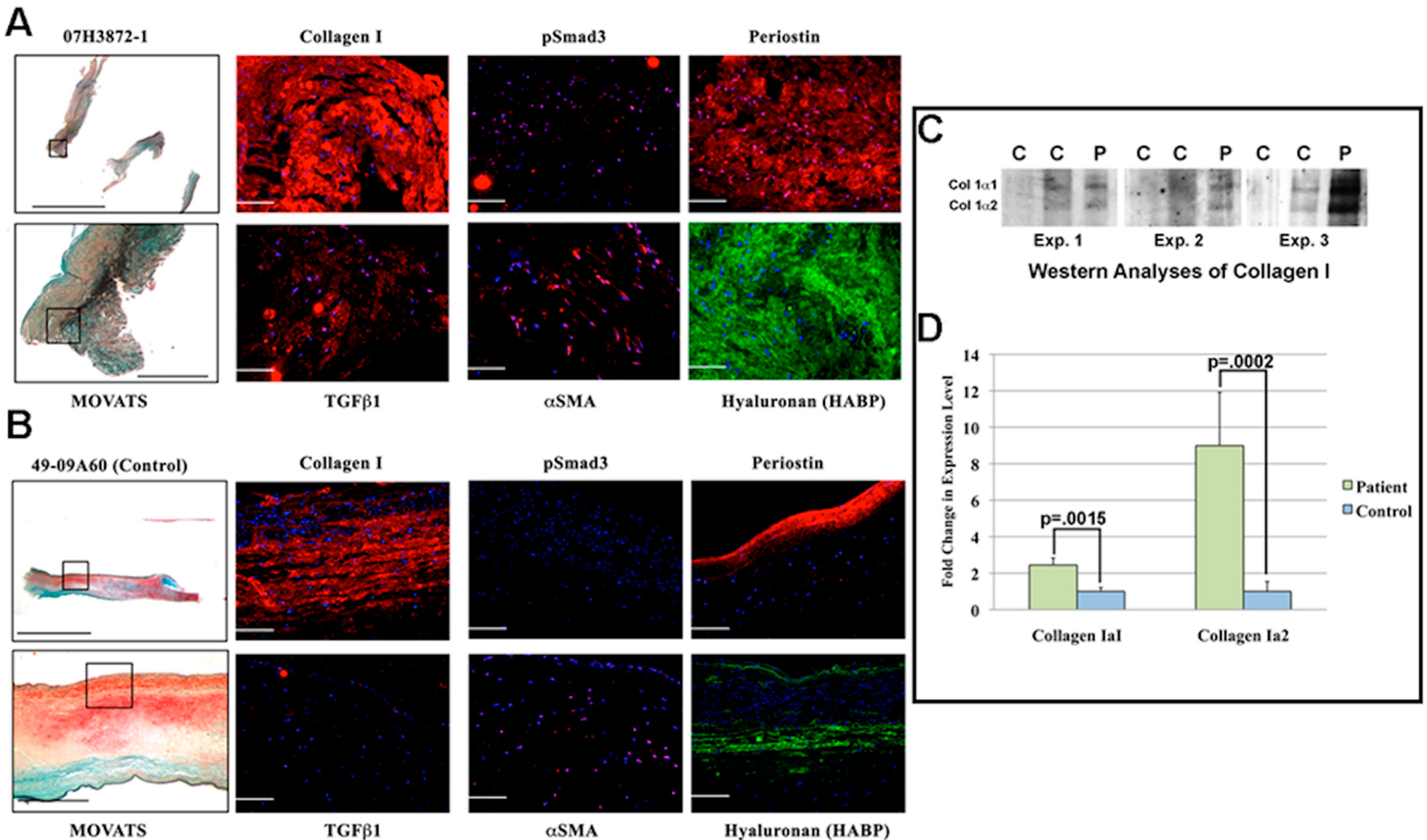
3.5. MV Leaflets in Human Non-Obstructive Hypertrophic Cardiomyopathy (HCM) Exhibit Increase in Collagen along with Molecular Changes in TGFβ Signaling
3.6. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jiang, L.; Levine, R.A.; King, M.E.; Weyman, A.E. An integrated mechanism for systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. Am. Heart J. 1987, 113, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.A.; Bruneval, P.; Levine, R.A.; Neamatalla, H.; Judge, D.P. The mitral valve in hypertrophic cardiomyopathy: Old versus new concepts. J. Cardiovasc. Transl. Res. 2011, 4, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.A.; Dubourg, O.; Desnos, M.; Mirochnik, R.; Isnard, G.; Bonne, G.; Carrier, L.; Guicheney, P.; Bouhour, J.B.; Schwartz, K.; Komajda, M. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur. Heart J. 1998, 19, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Harrigan, C.; Appelbaum, E.; Gibson, C.M.; Lesser, J.R.; Haas, T.S.; Udelson, J.E.; Manning, W.J.; Maron, B.J. Mitral Valve Abnormalities Identified by Cardiovascular Magnetic Resonance Represent a Primary Phenotypic Expression of Hypertrophic Cardiomyopathy. Circulation 2011, 124, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Schwammenthal, E.; Levine, R.A. Dynamic subaortic obstruction: A disease of the mitral valve suitable for surgical repair? J. Am. Coll. Cardiol. 1996, 28, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.A.; Vlahakes, G.J.; Lefebvre, X.; Guerrero, J.L.; Cape, E.G.; Yoganathan, A.P.; Weyman, A.E. Papillary muscle displacement causes systolic anterior motion of the mitral valve. Experimental validation and insights into the mechanism of subaortic obstruction. Circulation 1995, 91, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Wigle, E.D.; Adelman, A.G.; Auger, P.; Marquis, Y. Mitral regurgitation in muscular subaortic stenosis. Am. J. Cardiol. 1969, 24, 698–706. [Google Scholar] [CrossRef]
- Kim, D.H.; Handschumacher, M.D.; Levine, R.A.; Choi, Y.S.; Kim, Y.J.; Yun, S.C.; Song, J.M.; Kang, D.H.; Song, J.K. In vivo measurement of mitral leaflet surface area and subvalvular geometry in patients with asymmetrical septal hypertrophy: Insights into the mechanism of outflow tract obstruction. Circulation 2010, 122, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martinez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, F.; Gueffet, J.P.; Probst, V.; Jaafar, P.; Legendre, A.; le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the Gene Encoding Filamin A as a Cause for Familial Cardiac Valvular Dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Sauls, K.; de Vlaming, A.; Harris, B.S.; Williams, K.; Wessels, A.; Levine, R.A.; Slaugenhaupt, S.A.; Goodwin, R.L.; Pavone, L.; Merot, J.; et al. Developmental Basis for Filamin-A Associated Myxomatous Mitral Valve Disease. Cardiovasc. Res. 2012, 96, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Disse, S.; Abergel, E.; Berrebi, A.; Houot, A.M.; le Heuzey, J.Y.; Diebold, B.; Guize, L.; Carpentier, A.; Corvol, P.; Jeunemaitre, X. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2–p12.1. Am. J. Hum. Genet. 1999, 65, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Freed, L.A.; Acierno, J.S., Jr.; Dai, D.; Leyne, M.; Marshall, J.E.; Nesta, F.; Levine, R.A.; Slaugenhaupt, S.A. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am. J. Hum. Genet. 2003, 72, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Nesta, F.; Leyne, M.; Yosefy, C.; Simpson, C.; Dai, D.; Marshall, J.E.; Hung, J.; Slaugenhaupt, S.A.; Levine, R.A. New locus for autosomal dominant mitral valve prolapse on chromosome 13: Clinical insights from genetic studies. Circulation 2005, 112, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; da Costa, A.; Prieur, F.; Bresson, J.L.; Faivre, L.; Eicher, J.C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Conner, D.; Thierfelder, L.; Jarcho, J.A.; MacRae, C.; McKenna, W.J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 1995, 11, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Carrier, L.; Knoll, R.; Vignier, N.; Keller, D.I.; Bausero, P.; Prudhon, B.; Isnard, R.; Ambroisine, M.L.; Fiszman, M.; Ross, J., Jr.; et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc. Res. 2004, 63, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Lekanne Deprez, R.H.; Muurling-Vlietman, J.J.; Hruda, J.; Baars, M.J.H.; Wijnaendts, L.C.D.; Stolte-Dijkstra, I.; Alders, M.; van Hagen, J.M. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J. Med. Genet. 2006, 43, 829–832. [Google Scholar]
- Xin, B.; Puffenberger, E.; Tumbush, J.; Bockoven, J.R.; Wang, H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am. J. Med. Genet. A 2007, 143A, 2662–2667. [Google Scholar] [CrossRef]
- Bhan, A.; Sirker, A.; Zhang, J.; Protti, A.; Catibog, N.; Driver, W.; Botnar, R.; Monaghan, M.J.; Shah, A.M. High-frequency speckle tracking echocardiography in the assessment of left ventricular function and remodeling after murine myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1371–H1383. [Google Scholar] [CrossRef] [PubMed]
- Henning, A.L.; Jiang, M.X.; Yalcin, H.C.; Butcher, J.T. Quantitative three-dimensional imaging of live avian embryonic morphogenesis via micro-computed tomography. Dev. Dyn. 2011, 240, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Potts, J.D.; Yost, M.J.; Junor, L.; Brooks, T.; Tan, H.; Hoffman, S.; Hart, M.M.; Kern, M.J.; Damon, B.; et al. Periostin promotes a fibroblastic lineage pathway in atrioventricular valve progenitor cells. Dev. Dyn. 2009, 238, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires TGF-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 Antagonist, Prevents Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Morner, S.; Richard, P.; Kazzam, E.; Hellman, U.; Hainque, B.; Schwartz, K.; Waldenstrom, A. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J. Mol. Cell. Cardiol. 2003, 35, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Klues, H.G.; Maron, B.J.; Dollar, A.L.; Roberts, W.C. Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation 1992, 85, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Kaple, R.K.; Murphy, R.T.; DiPaola, L.M.; Houghtaling, P.L.; Lever, H.M.; Lytle, B.W.; Blackstone, E.H.; Smedira, N.G. Mitral valve abnormalities in hypertrophic cardiomyopathy: Echocardiographic features and surgical outcomes. Ann. Thorac. Surg. 2008, 85, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Mautner, S.L.; Klues, H.G.; Mautner, G.C.; Proschan, M.A.; Roberts, W.C.; Maron, B.J. Comparison of mitral valve dimensions in adults with valvular aortic stenosis, pure aortic regurgitation and hypertrophic cardiomyopathy. Am. J. Cardiol 1993, 71, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.M.; Taylor, R.D.; Wong, M. Abnormal mitral valve coaptation in hypertrophic obstructive cardiomyopathy: Proposed role in systolic anterior motion of mitral valve. Am. J. Cardiol. 1981, 48, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Schwammenthal, E.; Nakatani, S.; He, S.; Hopmeyer, J.; Sagie, A.; Weyman, A.E.; Lever, H.M.; Yoganathan, A.P.; Thomas, J.D.; Levine, R.A. Mechanism of Mitral Regurgitation in Hypertrophic Cardiomyopathy: Mismatch of Posterior to Anterior Leaflet Length and Mobility. Circulation 1998, 98, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Petrone, R.K.; Klues, H.G.; Panza, J.A.; Peterson, E.E.; Maron, B.J. Coexistence of mitral valve prolapse in a consecutive group of 528 patients with hypertrophic cardiomyopathy assessed with echocardiography. J. Am. Coll. Cardiol. 1992, 20, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Chaput, M.; Handschumacher, M.D.; Tournoux, F.; Hua, L.; Guerrero, J.L.; Vlahakes, G.J.; Levine, R.A. Mitral leaflet adaptation to ventricular remodeling: Occurrence and adequacy in patients with functional mitral regurgitation. Circulation 2008, 118, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Messas, E.; Bel, A.; Szymanski, C.; Cohen, I.; Touchot, B.; Handschumacher, M.D.; Desnos, M.; Carpentier, A.; Menasche, P.; Hagege, A.A.; et al. Relief of mitral leaflet tethering following chronic myocardial infarction by chordal cutting diminishes left ventricular remodeling. Circ. Cardiovasc. Imaging 2009, 3, 679–686. [Google Scholar] [CrossRef]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Rouf, R.; Habashi, J.; Dietz, H.C. Mitral valve disease in Marfan syndrome and related disorders. J. Cardiovasc. Transl. Res. 2011, 4, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.A.; Sinha, R.; Aziz, H.; Rouf, R.; Dietz, H.C., 3rd; Judge, D.P.; Butcher, J. Multi-scale biomechanical remodeling in aging and genetic mutant murine mitral valve leaflets: Insights into Marfan syndrome. PLoS One 2012, 7, e44639. [Google Scholar] [CrossRef]
- Seidman, J.G.; Seidman, C. The Genetic Basis for Cardiomyopathy: From Mutation Identification to Mechanistic Paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the Thick of It. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Shephard, R.; Semsarian, C. Role of animal models in HCM research. J. Cardiovasc. Transl. Res. 2009, 2, 471–482. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.K.; Jones, K.A.; Fatkin, D.; Arroyo, L.H.; Lee, R.T.; Aristizabal, O.; Turnbull, D.H.; Georgakopoulos, D.; Kass, D.; Bond, M.; et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Investig. 1999, 104, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Johnson, N.M. Genetic evaluation of familial cardiomyopathy. J. Cardiovasc. Transl. Res. 2008, 1, 144–154. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Judge, D.P.; Neamatalla, H.; Norris, R.A.; Levine, R.A.; Butcher, J.T.; Vignier, N.; Kang, K.H.; Nguyen, Q.; Bruneval, P.; Perier, M.-C.; et al. Targeted Mybpc3 Knock-Out Mice with Cardiac Hypertrophy Exhibit Structural Mitral Valve Abnormalities. J. Cardiovasc. Dev. Dis. 2015, 2, 48-65. https://doi.org/10.3390/jcdd2020048
Judge DP, Neamatalla H, Norris RA, Levine RA, Butcher JT, Vignier N, Kang KH, Nguyen Q, Bruneval P, Perier M-C, et al. Targeted Mybpc3 Knock-Out Mice with Cardiac Hypertrophy Exhibit Structural Mitral Valve Abnormalities. Journal of Cardiovascular Development and Disease. 2015; 2(2):48-65. https://doi.org/10.3390/jcdd2020048
Chicago/Turabian StyleJudge, Daniel P., Hany Neamatalla, Russell A. Norris, Robert A. Levine, Jonathan T. Butcher, Nicolas Vignier, Kevin H. Kang, Quangtung Nguyen, Patrick Bruneval, Marie-Cécile Perier, and et al. 2015. "Targeted Mybpc3 Knock-Out Mice with Cardiac Hypertrophy Exhibit Structural Mitral Valve Abnormalities" Journal of Cardiovascular Development and Disease 2, no. 2: 48-65. https://doi.org/10.3390/jcdd2020048