Cross Talk between NOTCH Signaling and Biomechanics in Human Aortic Valve Disease Pathogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
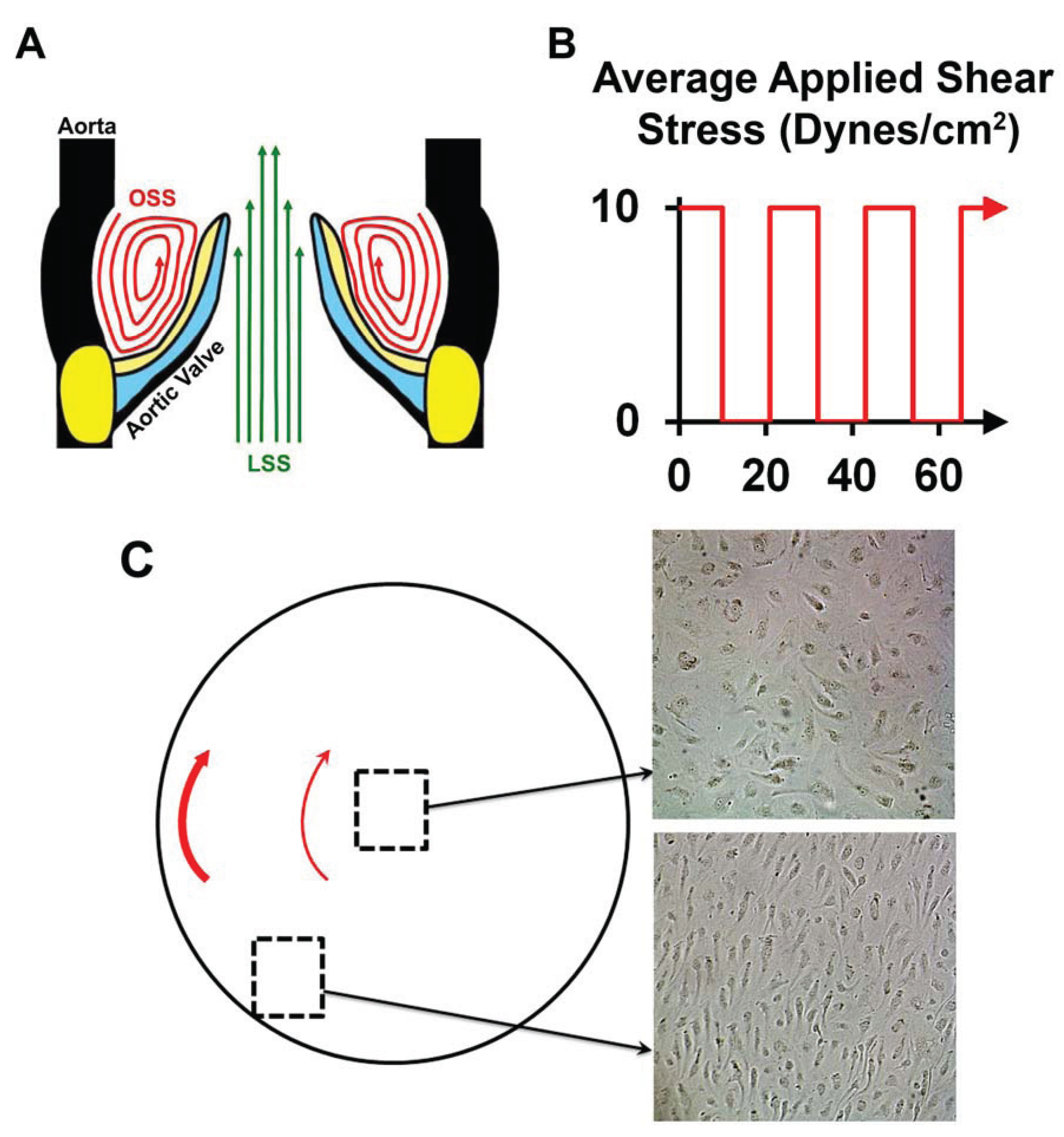
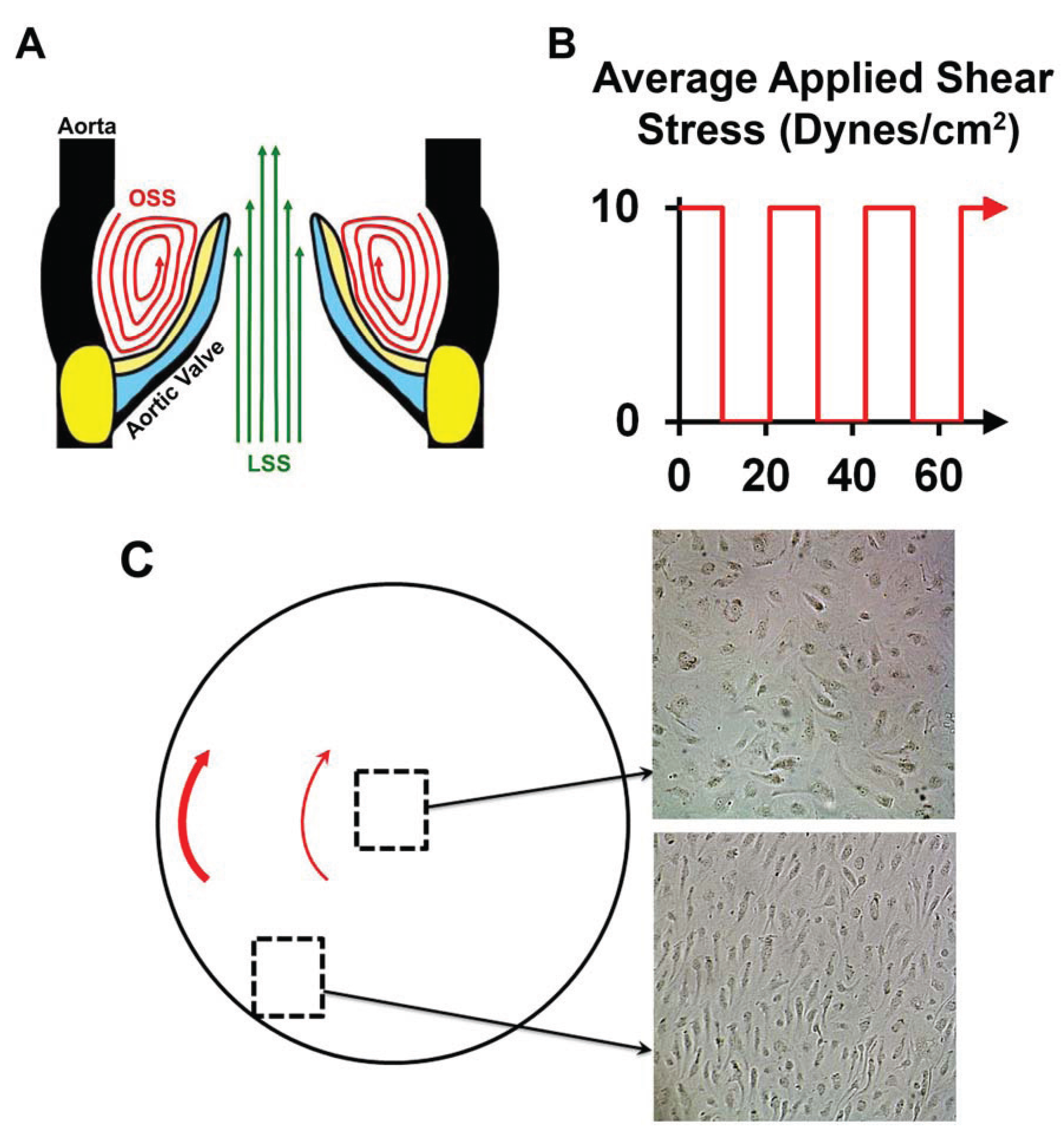
2.2. Oscillatory Shear Stress
2.3. Alizarin Red Staining
2.4. RNA Isolation and Real-Time Quantitative RT-PCR
2.5. VEGF-A ELISA
2.6. Statistics
3. Results
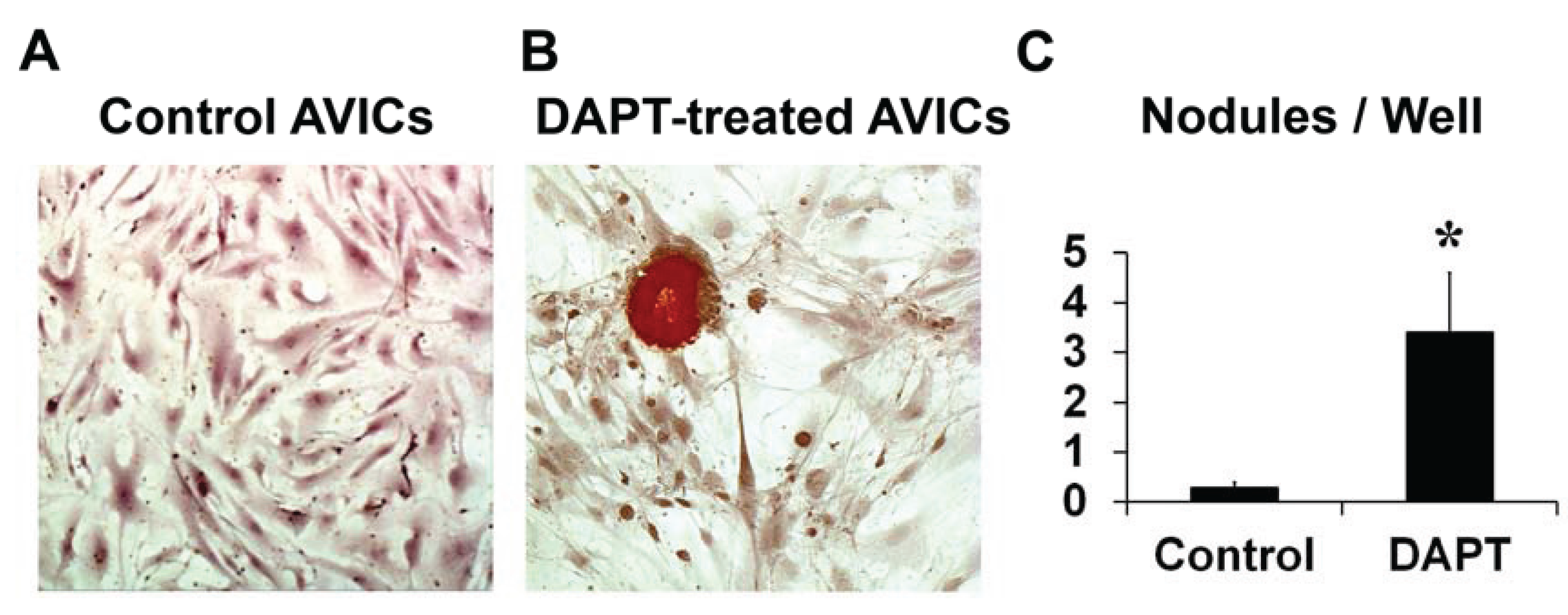
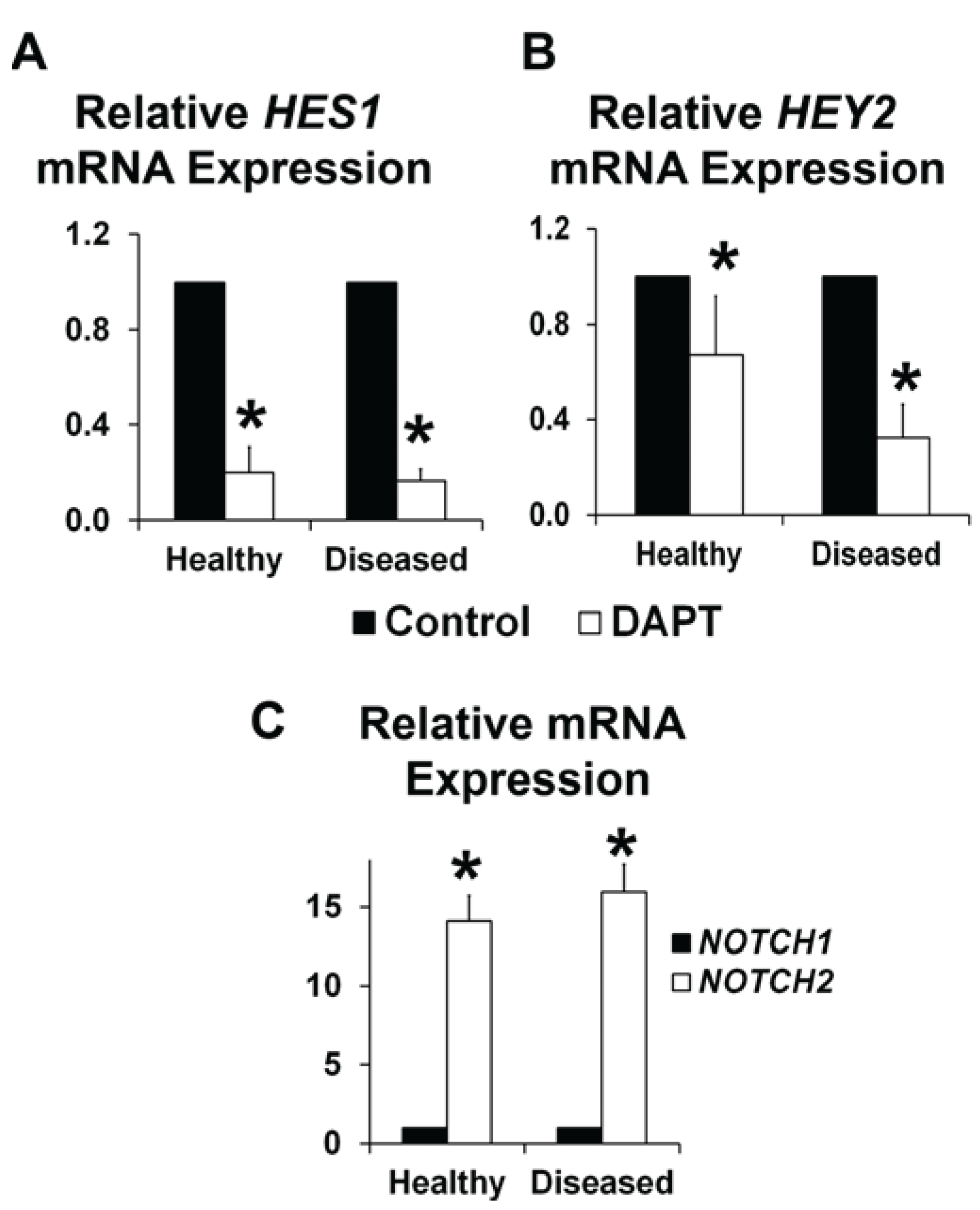
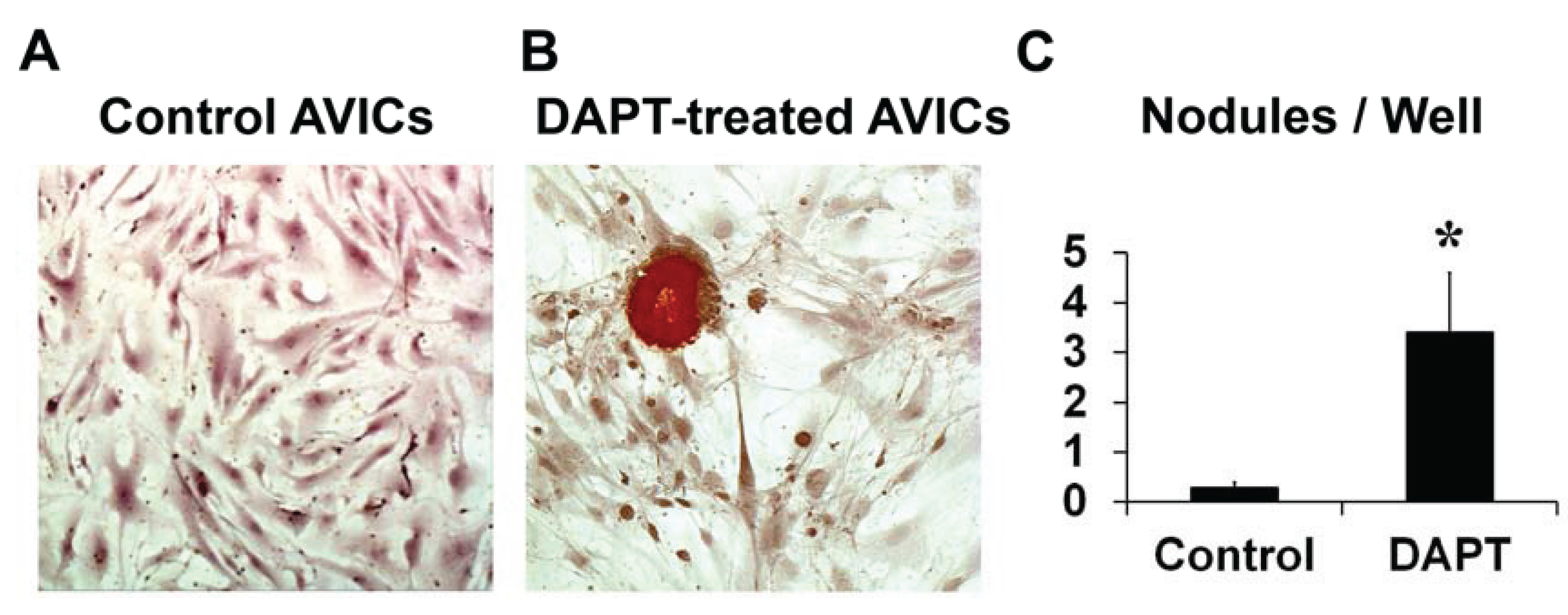
3.1. NOTCH Loss of Function in Human AVICs Leads to Calcifications in Vitro
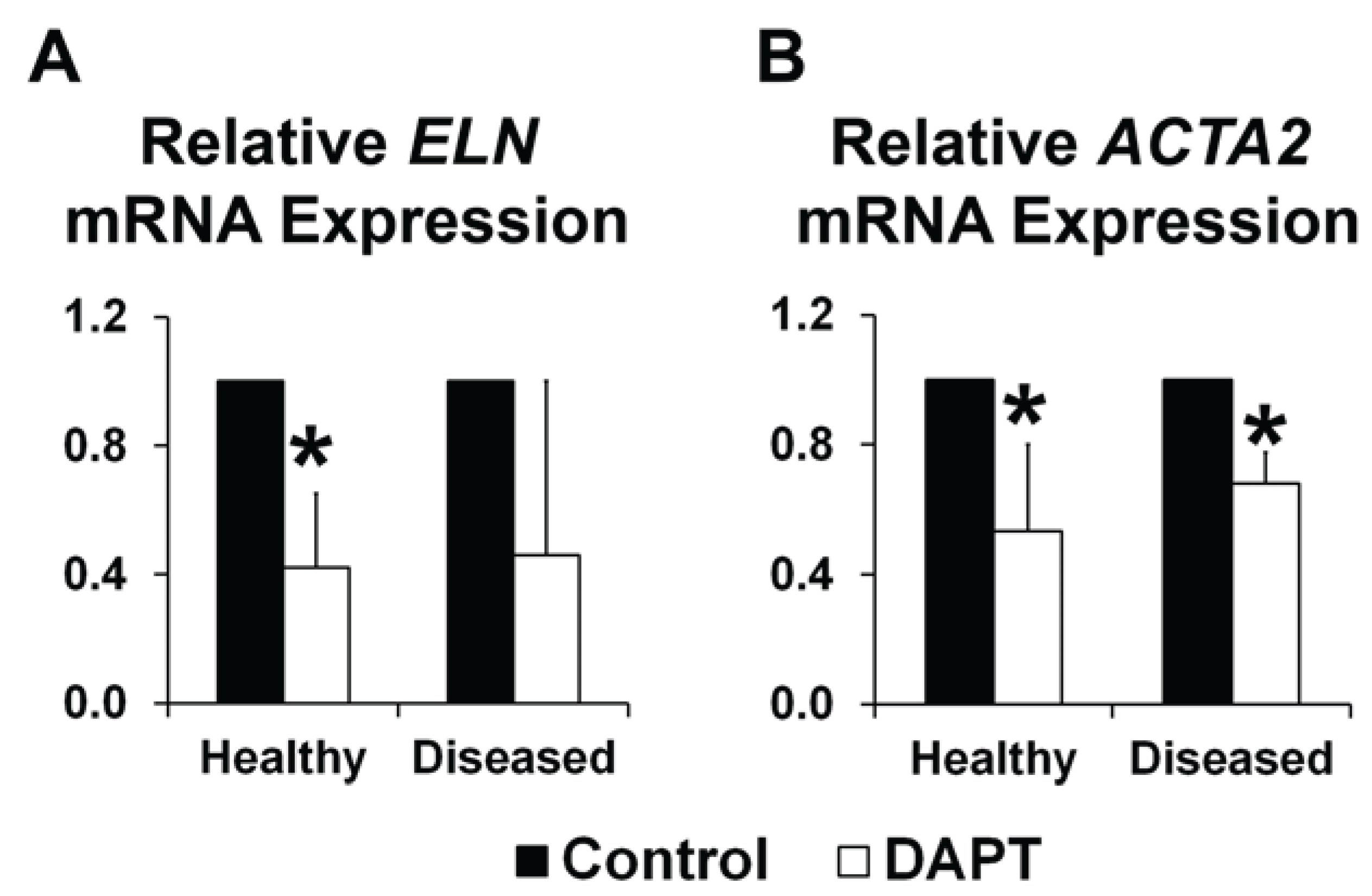
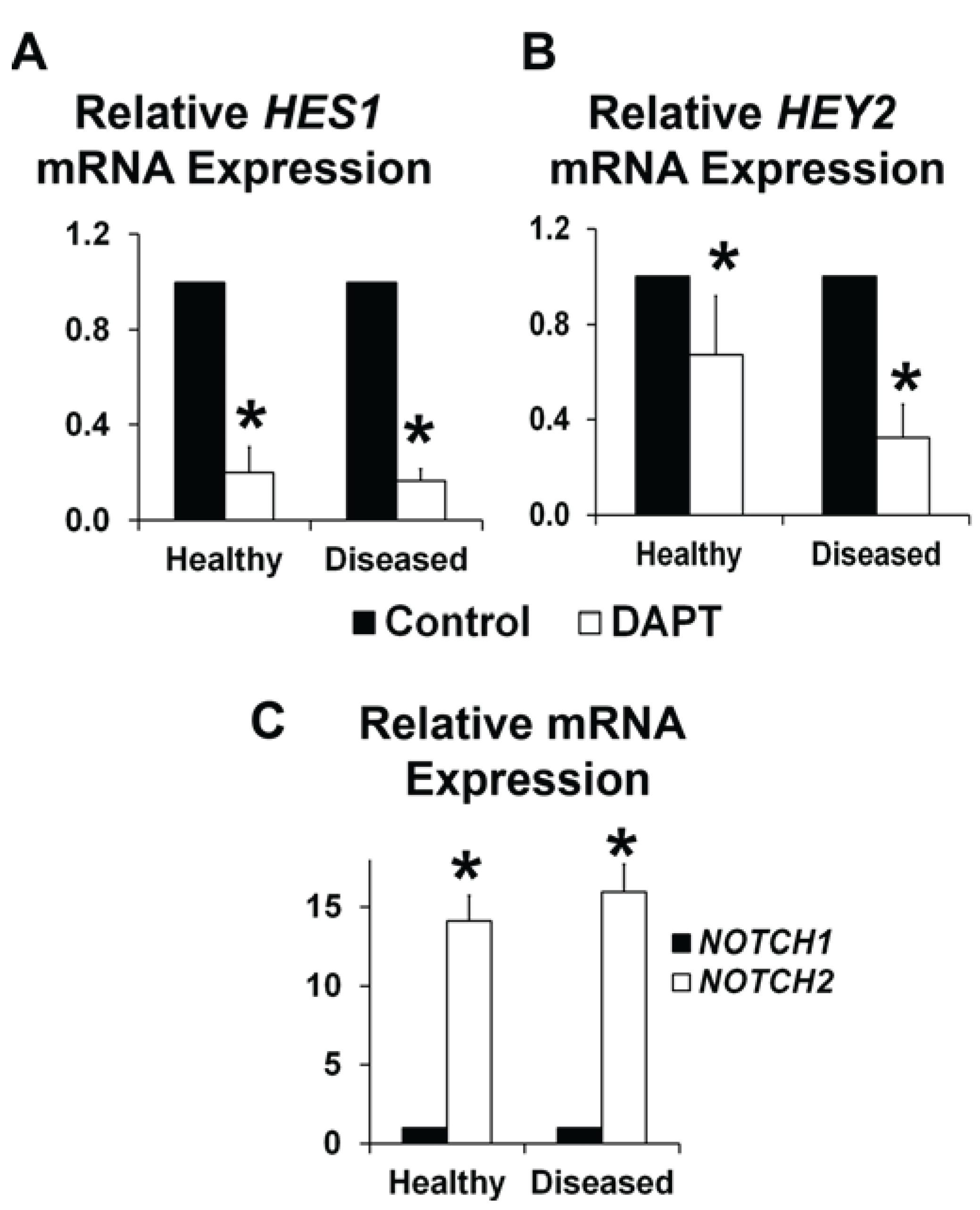
3.2. NOTCH Loss of Function Leads to Decreased ELN and α-SMA Expression in Human AVICs

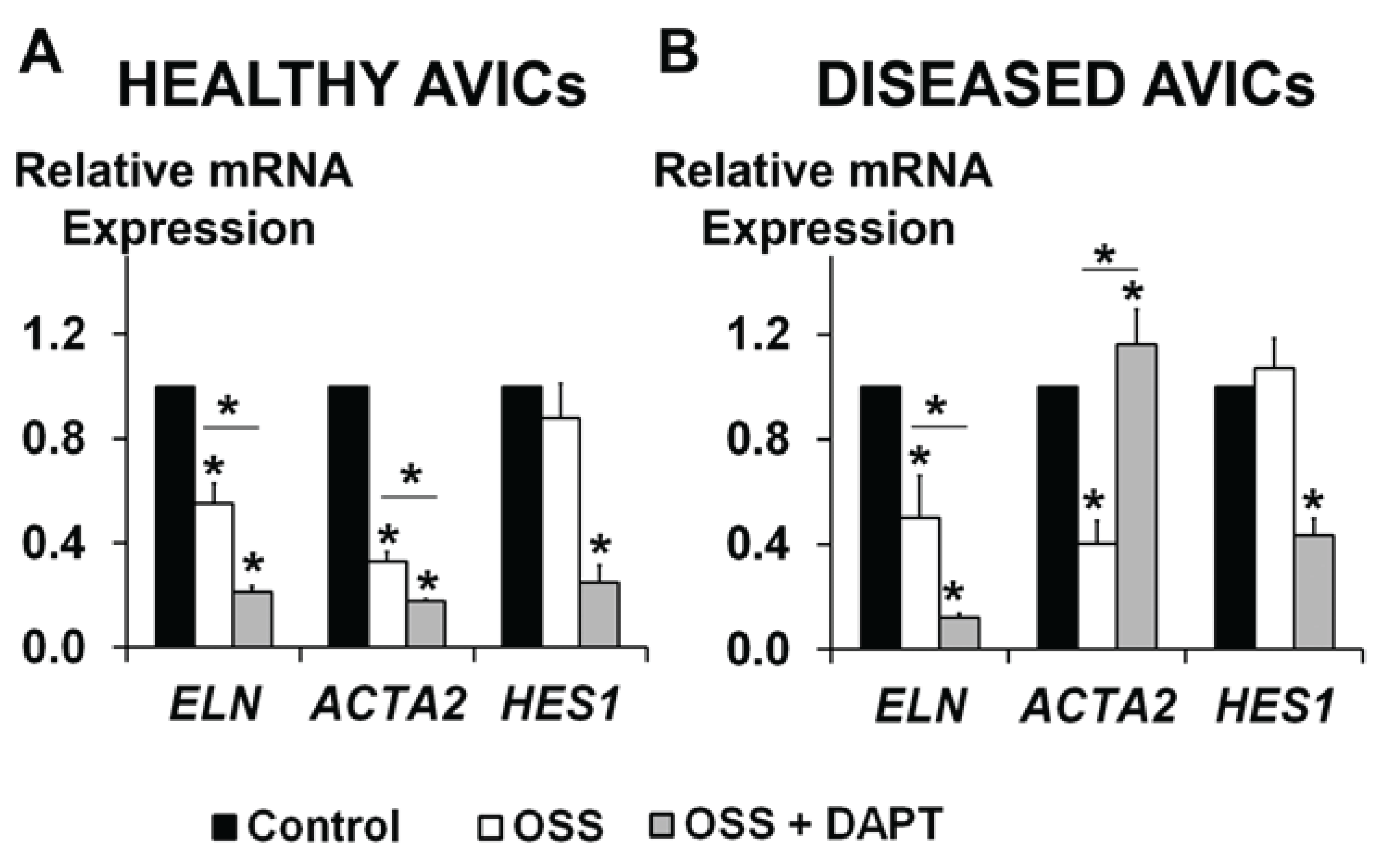
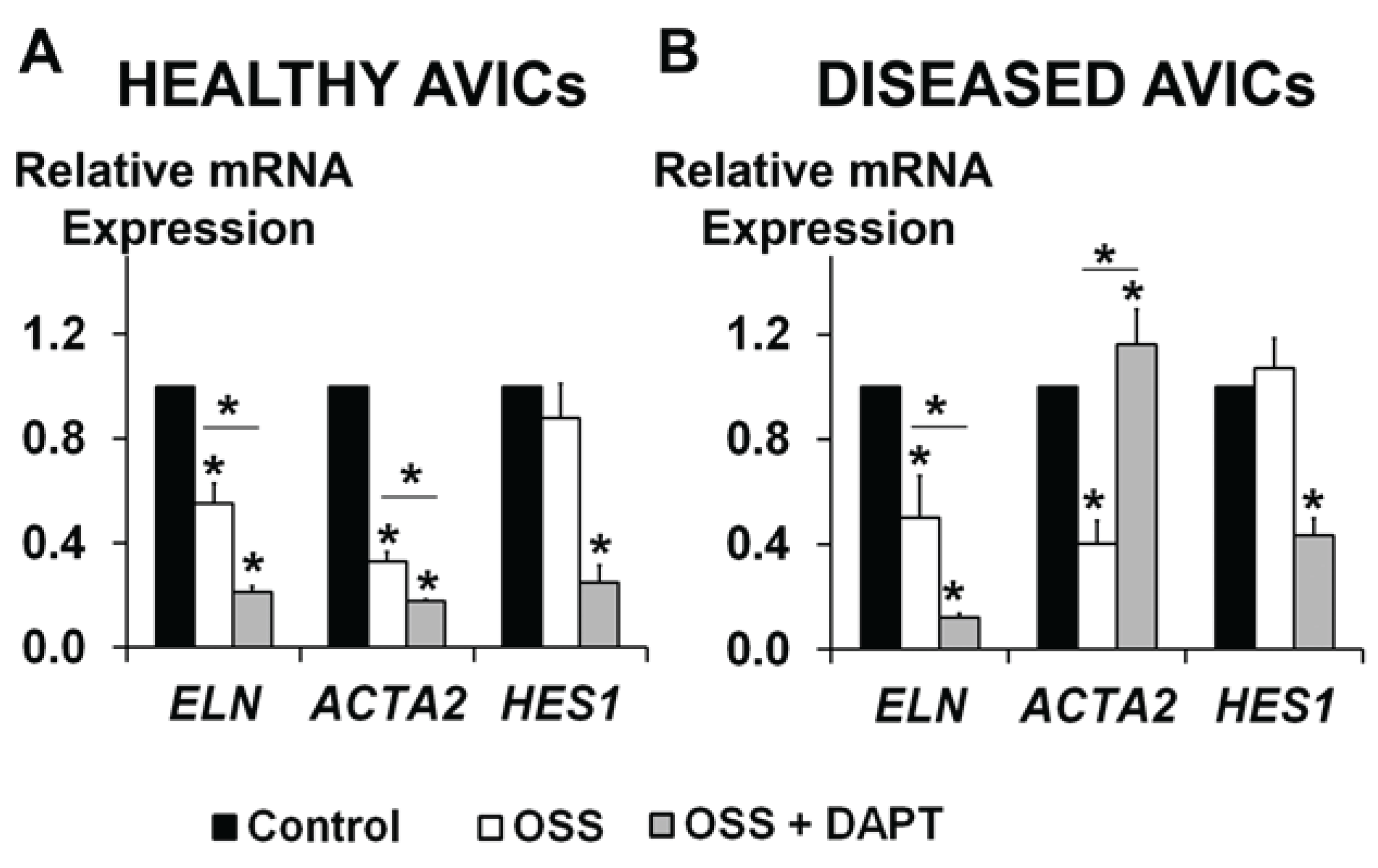
3.3. OSS and NOTCH Provide a Complex Regulation of ELN and ACTA2 Expression in Human AVICs
3.4. Disease Differentially Affects VEGFA Gene and Protein Expression Levels

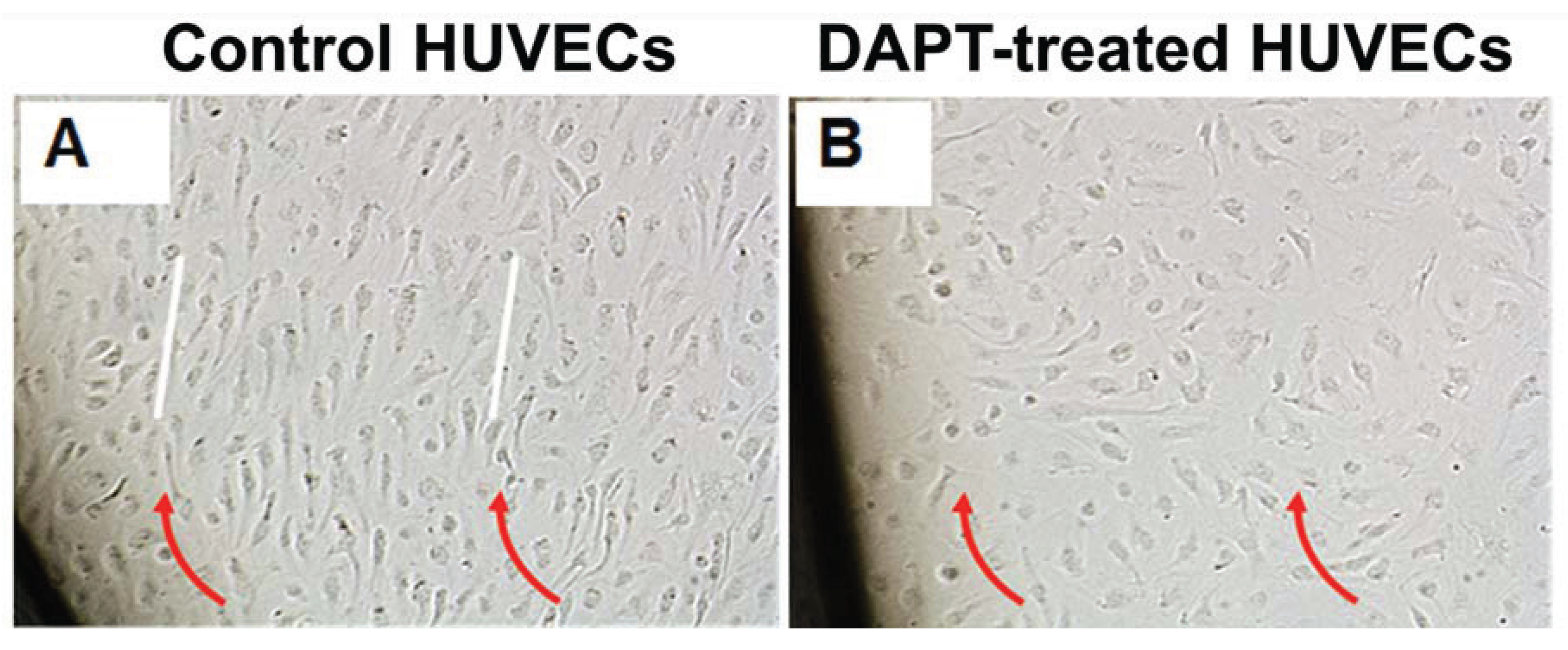
3.5. NOTCH Loss of Function Leads to Cellular Misalignment in HUVECs in Response to OSS

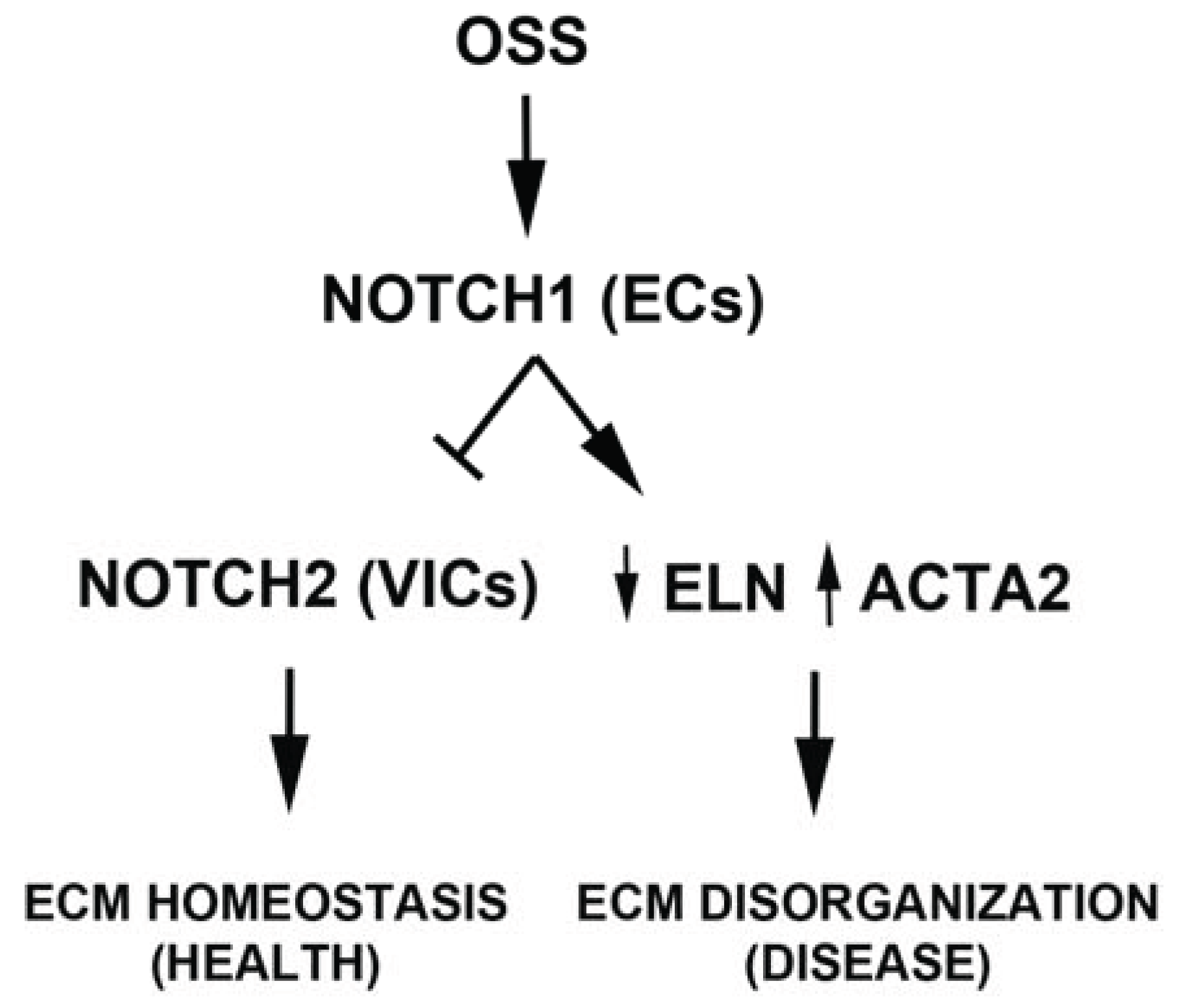
4. Discussion
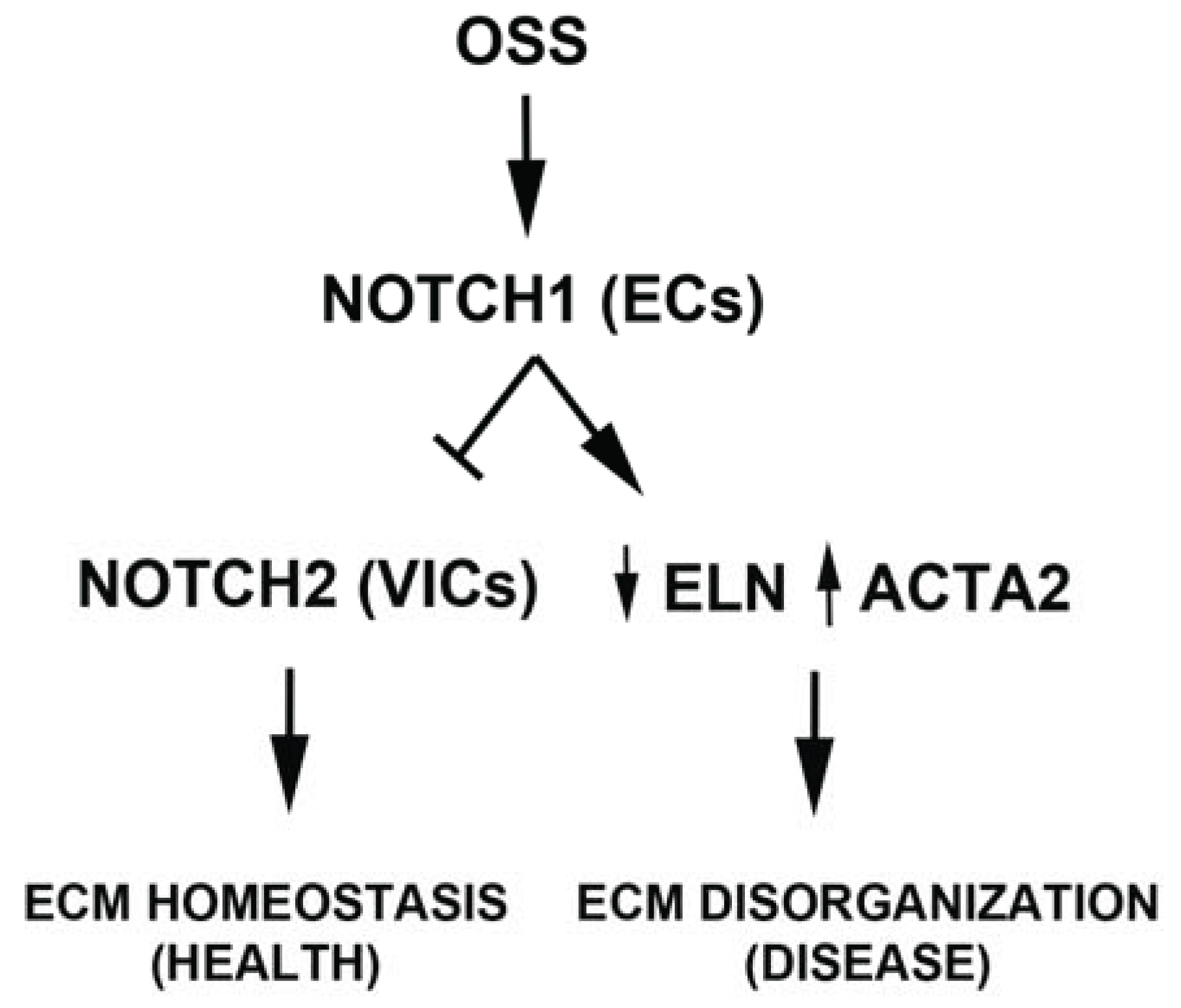
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yacoub, M.H.; Takkenberg, J.J.M. Will heart valve tissue engineering change the world? Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Gersh, B.; Bonow, R.O. Calcific aortic stenosis: From bench to the bedside—Emerging clinical and cellular concepts. Heart 2003, 89, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.A.; Otto, C.M.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P.; Guyton, R.A.; O’Gara, P.T.; Ruiz, C.E.; Skubas, N.J.; Sorajja, P.; et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: A report of the american college of cardiology/american heart association task force on practice guidelines. Circulation 2014, 129, e521–e643. [Google Scholar]
- Howell, E.J.; Butcher, J.T. Valvular heart diseases in the developing world: Developmental biology takes center stage. J. Heart Valve Dis. 2012, 21, 234–240. [Google Scholar] [PubMed]
- Hinton, R.B.; Yutzey, K.E. Heart valve structure and function in development and disease. Annu. Rev. Physiol. 2011, 73, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Vesely, I. The role of elastin in aortic valve mechanics. J. Biomech. 1998, 31, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Latif, N.; Sarathchandra, P.; Taylor, P.M.; Antoniw, J.; Yacoub, M.H. Localization and pattern of expression of extracellular matrix components in human heart valves. J. Heart Valve Dis. 2005, 14, 218–227. [Google Scholar] [PubMed]
- Bellhouse, B.J.; Talbot, L. The fluid mechanics of the aortic valve. J. Fluid Mechanics 1969, 35, 721–735. [Google Scholar] [CrossRef]
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of “degenerative” valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef]
- Wirrig, E.E.; Hinton, R.B.; Yutzey, K.E. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J. Mol. Cell. Cardiol. 2011, 50, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in notch1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- De la Pompa, J.L.; Epstein, J.A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev. Cell 2012, 22, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Fortini, M.E. Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Nigam, V.; Srivastava, D. Notch1 represses osteogenic pathways in aortic valve cells. J. Mol. Cell. Cardiol. 2009, 47, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Hans, C.P.; Koenig, S.N.; Nichols, H.A.; Galindo, C.L.; Garner, H.R.; Merrill, W.H.; Hinton, R.B.; Garg, V. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One 2011, 6, e27743. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; MacGrogan, D.; Martínez-Poveda, B.; Benito, Y.; Casanova, J.C.; Fernández-Avilés, F.; Bermejo, J.; de la Pompa, J.L. Diet-induced aortic valve disease in mice haploinsufficient for the notch pathway effector RBPJK/CSL. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Hans, C.P.; Koenig, S.N.; Huang, N.; Cheng, J.; Beceiro, S.; Guggilam, A.; Kuivaniemi, H.; Partida-Sánchez, S.; Garg, V. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.S.; Sainson, R.C.A.; Williams, C.K.; Taylor, J.M.; Shi, W.; Li, J.-L.; Harris, A.L. Regulation of multiple angiogenic pathways by Dll4 and notch in human umbilical vein endothelial cells. Microvasc. Res. 2008, 75, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Guha, S.; Sweeney, C.; Birney, Y.; Walshe, T.; O’Brien, C.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch and vascular smooth muscle cell phenotype. Circ. Res. 2008, 103, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-H.; Li, F.-D.; Tian, C.; Ren, H.-L.; Du, J.; Li, H.-H. Notch γ-secretase inhibitor dibenzazepine attenuates angiotensin ii-induced abdominal aortic aneurysm in apoe knockout mice by multiple mechanisms. PLoS One 2013, 8, e83310. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, G.; Grego-Bessa, J.; González-Rajal, A.; Bolós, V.; de La Pompa, J.L. Monitoring notch1 activity in development: Evidence for a feedback regulatory loop. Dev. Dyn. 2007, 236, 2594–2614. [Google Scholar] [CrossRef] [PubMed]
- Braddock, M.; Schwachtgen, J.-L.; Houston, P.; Dickson, M.C.; Lee, M.J.; Campbell, C.J. Fluid shear stress modulation of gene expression in endothelial cells. News Physiol. Sci. 1998, 13, 241–246. [Google Scholar] [PubMed]
- Butcher, J.T.; Simmons, C.A.; Warnock, J.N. Review—Mechanobiology of the aortic heart valve. J. Heart Valve Dis. 2008, 17, 62–73. [Google Scholar] [PubMed]
- Butcher, J.T.; Tressel, S.; Johnson, T.; Turner, D.; Sorescu, G.; Jo, H.; Nerem, R.M. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: Influence of shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Peterson, T.E.; Kovach, N.L.; Berk, B.C. Map kinase activation by flow in endothelial cells. Role of beta 1 integrins and tyrosine kinases. Circ. Res. 1996, 79, 310–316. [Google Scholar]
- Yap, C.H.; Saikrishnan, N.; Tamilselvan, G.; Vasilyev, N.; Yoganathan, A.P. The congenital bicuspid aortic valve can experience high-frequency unsteady shear stresses on its leaflet surface. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H721–H731. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Simmons, C.A. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 2011, 108, 1510–1524. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Likhitpanichkul, M.; Lam, C.J.; Beca, B.M.; Sun, Y.; Simmons, C.A. Microdevice array-based identification of distinct mechanobiological response profiles in layer-specific valve interstitial cells. Integr. Biol. (Camb) 2013, 5, 673–680. [Google Scholar] [CrossRef]
- Yip, C.Y.Y.; Chen, J.-H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Cujec, B.; Pollick, C. Isolated thickening of one aortic cusp: Preferential thickening of the noncoronary cusp. J. Am. Soc. Echocardiogr. 1988, 1, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Thubrikar, M.J.; Aouad, J.; Nolan, S.P. Patterns of calcific deposits in operatively excised stenotic or purely regurgitant aortic valves and their relation to mechanical stress. Am. J. Cardiol. 1986, 58, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, C.A.; Alfieri, C.M.; Joyce, E.M.; Watkins, S.C.; Yutzey, K.E.; Sacks, M.S. Gene expression and collagen fiber micromechanical interactions of the semilunar heart valve interstitial cell. Cell Mol. Bioeng. 2012, 5, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Arjunon, S.; Rathan, S.; Jo, H.; Yoganathan, A.P. Aortic valve: Mechanical environment and mechanobiology. Ann. Biomed. Eng. 2013, 41, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- David Merryman, W. Mechano-potential etiologies of aortic valve disease. J. Biomech. 2010, 43, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, K.; Sucosky, P.; Yoganathan, A.P. Hemodynamics and mechanobiology of aortic valve inflammation and calcification. Int. J. Inflam. 2011, 2011, 263870. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, K.; Alford, P.W.; Wylie-Sears, J.; Goss, J.A.; Grosberg, A.; Bischoff, J.; Aikawa, E.; Levine, R.A.; Parker, K.K. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc. Natl. Acad. Sci. USA 2011, 108, 19943–19948. [Google Scholar] [CrossRef] [PubMed]
- Levesque, M.J.; Nerem, R.M. The elongation and orientation of cultured endothelial cells in response to shear stress. J. Biomech. Eng. 1985, 107, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, S.; Yang, J.-H.; Aikawa, E.; Melero-Martin, J.M.; Khan, Z.A.; Loukogeorgakis, S.; Schoen, F.J.; Bischoff, J. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-a and transforming growth factor-beta2. Circ. Res. 2006, 99, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Nealis, T.B.; Subramaniam, M.; Pandya, S.; Stock, S.R.; Ignatiev, C.I.; Sebo, T.J.; Rosengart, T.K.; Edwards, W.D.; McCarthy, P.M.; et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation 2005, 111, 3296–3301. [Google Scholar]
- Yang, J.-H.; Wylie-Sears, J.; Bischoff, J. Opposing actions of notch1 and vegf in post-natal cardiac valve endothelial cells. Biochem. Biophys. Res. Commun. 2008, 374, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [PubMed]
- Santhanakrishnan, A.; Miller, L.A. Fluid dynamics of heart development. Cell Biochem. Biophys. 2011, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Goenezen, S.; Rennie, M.Y.; Rugonyi, S. Biomechanics of early cardiac development. Biomech. Model Mechanobiol. 2012, 11, 1187–1204. [Google Scholar] [CrossRef] [PubMed]
- Votteler, M.; Berrio, D.A.C.; Horke, A.; Sabatier, L.; Reinhardt, D.P.; Nsair, A.; Aikawa, E.; Schenke-Layland, K. Elastogenesis at the onset of human cardiac valve development. Development 2013, 140, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar]
- Zeng, Q.; Song, R.; Ao, L.; Weyant, M.J.; Lee, J.; Xu, D.; Fullerton, D.A.; Meng, X. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-κB activation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Osman, L.; Yacoub, M.H.; Latif, N.; Amrani, M.; Chester, A.H. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 2006, 114, I547–1552. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, A.; Simionescu, D.T.; Vyavahare, N.R. Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-beta1: Role of myofibroblasts in vascular calcification. Am. J. Pathol. 2007, 171, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Silverman, N.H. The systolic to diastolic duration ratio in children with heart failure secondary to restrictive cardiomyopathy. J. Am. Soc. Echocardiogr. 2006, 19, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.D.; Chakraborty, A.; Sharp, M.K.; Berson, R.E. Spatial and temporal resolution of shear in an orbiting petri dish. Biotechnol. Prog. 2011, 27, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.T.; Hexiu, J.; Kim, J.; Seonwoo, H.; Choung, P.H.; Chung, J.H. Synergistic effects of orbital shear stress on in vitro growth and osteogenic differentiation of human alveolar bone-derived mesenchymal stem cells. BioMed. Res. Int. 2014, 2014, 316803. [Google Scholar] [PubMed]
- Dardik, A.; Chen, L.; Frattini, J.; Asada, H.; Aziz, F.; Kudo, F.A.; Sumpio, B.E. Differential effects of orbital and laminar shear stress on endothelial cells. J. Vasc. Surg. 2005, 41, 869–880. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Frangos, J.A. The shear stress of it all: The cell membrane and mechanochemical transduction. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1459–1467. [Google Scholar] [CrossRef]
- Potter, C.M.F.; Lundberg, M.H.; Harrington, L.S.; Warboys, C.M.; Warner, T.D.; Berson, R.E.; Moshkov, A.V.; Gorelik, J.; Weinberg, P.D.; Mitchell, J.A. Role of shear stress in endothelial cell morphology and expression of cyclooxygenase isoforms. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.F.; Bussolari, S.R.; Gimbrone, M.A.; Davies, P.F. The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 1981, 103, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Kedes, L.; Hamamori, Y. Hes and herp families: Multiple effectors of the notch signaling pathway. J. Cell. Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, K.; Konduri, S.; Sucosky, P.; Jo, H.; Yoganathan, A.P. An ex vivo study of the biological properties of porcine aortic valves in response to circumferential cyclic stretch. Ann. Biomed. Eng. 2006, 34, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, A.; Philips, K.; Vyavahare, N. Elastin-derived peptides and tgf-beta1 induce osteogenic responses in smooth muscle cells. Biochem. Biophys. Res. Commun. 2005, 334, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Russo, E.; Camastra, C.; Filice, G.; di Mizio, G.; Colosimo, F.; Ricci, P.; Tripepi, S.; Amorosi, A.; Triumbari, F.; et al. New evidence for a critical role of elastin in calcification of native heart valves: Immunohistochemical and ultrastructural study with literature review. Histopathology 2011, 59, 504–513. [Google Scholar]
- Munjal, C.; Opoka, A.M.; Osinska, H.; James, J.F.; Bressan, G.M.; Hinton, R.B. Tgf-beta mediates early angiogenesis and latent fibrosis in an emilin1-deficient mouse model of aortic valve disease. Dis. Models Mech. 2014, 7, 987–996. [Google Scholar] [CrossRef]
- Hinton, R.B.; Adelman-Brown, J.; Witt, S.; Krishnamurthy, V.K.; Osinska, H.; Sakthivel, B.; James, J.F.; Li, D.Y.; Narmoneva, D.A.; Mecham, R.P.; et al. Elastin haploinsufficiency results in progressive aortic valve malformation and latent valve disease in a mouse model. Circ. Res. 2010, 107, 549–557. [Google Scholar]
- Aikawa, E.; Aikawa, M.; Libby, P.; Figueiredo, J.L.; Rusanescu, G.; Iwamoto, Y.; Fukuda, D.; Kohler, R.H.; Shi, G.P.; Jaffer, F.A.; et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation 2009, 119, 1785–1794. [Google Scholar]
- Syväranta, S.; Helske, S.; Laine, M.; Lappalainen, J.; Kupari, M.; Mäyränpää, M.I.; Lindstedt, K.A.; Kovanen, P.T. Vascular endothelial growth factor-secreting mast cells and myofibroblasts: A novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Gry, M.; Rimini, R.; Strömberg, S.; Asplund, A.; Pontén, F.; Uhlén, M.; Nilsson, P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics 2009, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4. [Google Scholar] [CrossRef]
- Zecchin, A.; Pattarini, L.; Gutierrez, M.I.; Mano, M.; Mai, A.; Valente, S.; Myers, M.P.; Pantano, S.; Giacca, M. Reversible acetylation regulates vascular endothelial growth factor receptor-2 activity. J. Mol. Cell Biol. 2014, 6, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 2013, 60, 27–35. [Google Scholar]
- Hofmann, J.J.; Briot, A.; Enciso, J.; Zovein, A.C.; Ren, S.; Zhang, Z.W.; Radtke, F.; Simons, M.; Wang, Y.; Iruela-Arispe, M.L. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012, 139, 4449–4460. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 2004, 230, 239–250. [Google Scholar] [CrossRef] [PubMed]
- De Lange, F.J.; Moorman, A.F.M.; Anderson, R.H.; Männer, J.; Soufan, A.T.; de Gier-de Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.B.; Christoffels, V.M. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Whittaker, P.; Farber, M.; Mendelson, K.; Padera, R.F.; Aikawa, M.; Schoen, F.J. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: Implications for postnatal adaptation, pathology, and tissue engineering. Circulation 2006, 113, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; McCright, B. Functional conservation of Notch1 and Notch2 intracellular domains. FASEB J. 2005, 19, 1311–1313. [Google Scholar] [PubMed]
- High, F.A.; Zhang, M.; Proweller, A.; Tu, L.; Parmacek, M.S.; Pear, W.S.; Epstein, J.A. An essential role for notch in neural crest during cardiovascular development and smooth muscle differentiation. J. Clin. Invest. 2007, 117, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Aherrahrou, Z.; Liptau, H.; Erasmi, A.W.; Hagemann, C.; Wrobel, S.; Borzym, K.; Schunkert, H.; Sievers, H.H.; Erdmann, J. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 2006, 345, 1460–1465. [Google Scholar] [CrossRef]
- Lammich, S.; Okochi, M.; Takeda, M.; Kaether, C.; Capell, A.; Zimmer, A.K.; Edbauer, D.; Walter, J.; Steiner, H.; Haass, C. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an abeta-like peptide. J. Biol. Chem. 2002, 277, 44754–44759. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Lincoln, J. Isolation of murine valve endothelial cells. J. Vis. Exp. 2014, 90, e51860. [Google Scholar]
- Sucosky, P.; Padala, M.; Elhammali, A.; Balachandran, K.; Jo, H.; Yoganathan, A.P. Design of an ex vivo culture system to investigate the effects of shear stress on cardiovascular tissue. J. Biomech. Eng. 2008, 130. [Google Scholar] [CrossRef]
- Sucosky, P.; Balachandran, K.; Elhammali, A.; Jo, H.; Yoganathan, A.P. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Holliday, C.J.; Ankeny, R.F.; Jo, H.; Nerem, R.M. Discovery of shear- and side-specific mrnas and mirnas in human aortic valvular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H856–H867. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sridhar, B.; Leinwand, L.A.; Anseth, K.S. Characterization of cell subpopulations expressing progenitor cell markers in porcine cardiac valves. PLoS One 2013, 8, e69667. [Google Scholar] [CrossRef] [PubMed]
- Stöllberger, C.; Finsterer, J.; Blazek, G. Isolated left ventricular abnormal trabeculation: Follow-up and association with neuromuscular disorders. Can. J. Cardiol. 2001, 17, 163–168. [Google Scholar] [PubMed]
- Dupuis, L.E.; Kern, C.B. Small leucine-rich proteoglycans exhibit unique spatiotemporal expression profiles during cardiac valve development. Dev. Dyn. 2014, 243, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Balaoing, L.R.; Post, A.D.; Liu, H.; Minn, K.T.; Grande-Allen, K.J. Age-related changes in aortic valve hemostatic protein regulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.M.; Nusinow, D.; Brown, C.B.; Violette, K.; Barnett, J.V.; Zhang, B.; Baldwin, H.S.; Caprioli, R.M. Networked-based characterization of extracellular matrix proteins from adult mouse pulmonary and aortic valves. J. Proteome Res. 2011, 10, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B. Bicuspid aortic valve and thoracic aortic aneurysm: Three patient populations, two disease phenotypes, and one shared genotype. Cardiol. Res. Pract. 2012, 2012, 926975. [Google Scholar] [PubMed]
- Sun, L.; Rajamannan, N.M.; Sucosky, P. Design and validation of a novel bioreactor to subject aortic valve leaflets to side-specific shear stress. Ann. Biomed. Eng. 2011, 39, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godby, R.C.; Munjal, C.; Opoka, A.M.; Smith, J.M.; Yutzey, K.E.; Narmoneva, D.A.; Hinton, R.B. Cross Talk between NOTCH Signaling and Biomechanics in Human Aortic Valve Disease Pathogenesis. J. Cardiovasc. Dev. Dis. 2014, 1, 237-256. https://doi.org/10.3390/jcdd1030237
Godby RC, Munjal C, Opoka AM, Smith JM, Yutzey KE, Narmoneva DA, Hinton RB. Cross Talk between NOTCH Signaling and Biomechanics in Human Aortic Valve Disease Pathogenesis. Journal of Cardiovascular Development and Disease. 2014; 1(3):237-256. https://doi.org/10.3390/jcdd1030237
Chicago/Turabian StyleGodby, Richard C., Charu Munjal, Amy M. Opoka, J. Michael Smith, Katherine E. Yutzey, Daria A. Narmoneva, and Robert B. Hinton. 2014. "Cross Talk between NOTCH Signaling and Biomechanics in Human Aortic Valve Disease Pathogenesis" Journal of Cardiovascular Development and Disease 1, no. 3: 237-256. https://doi.org/10.3390/jcdd1030237