Impact of RNA Degradation on Viral Diagnosis: An Understated but Essential Step for the Successful Establishment of a Diagnosis Network

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Virus Strain and Matrix Samples
2.3. RNA Isolation
2.4. In Vitro Transcription Reaction
2.5. cDNA Synthesis and Quantitative Real-Time RT-PCR (qRT-PCR)
2.6. RNA Stability Conditions
2.7. RNA Secondary Structure for M and HA Genes
2.8. Analysis Degradation Sensitibity
2.9. Statistical Analysis
3. Results
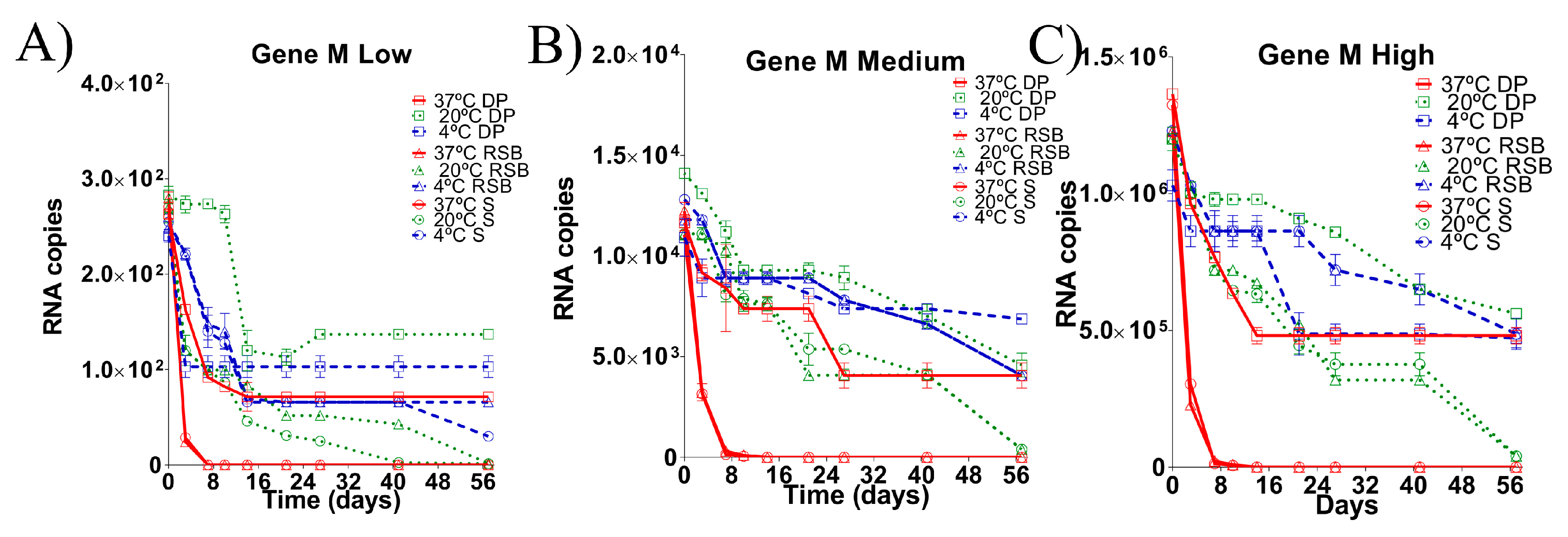
3.1. Stability of the RNA Transcripts for M Gene Stored under Different Conditions
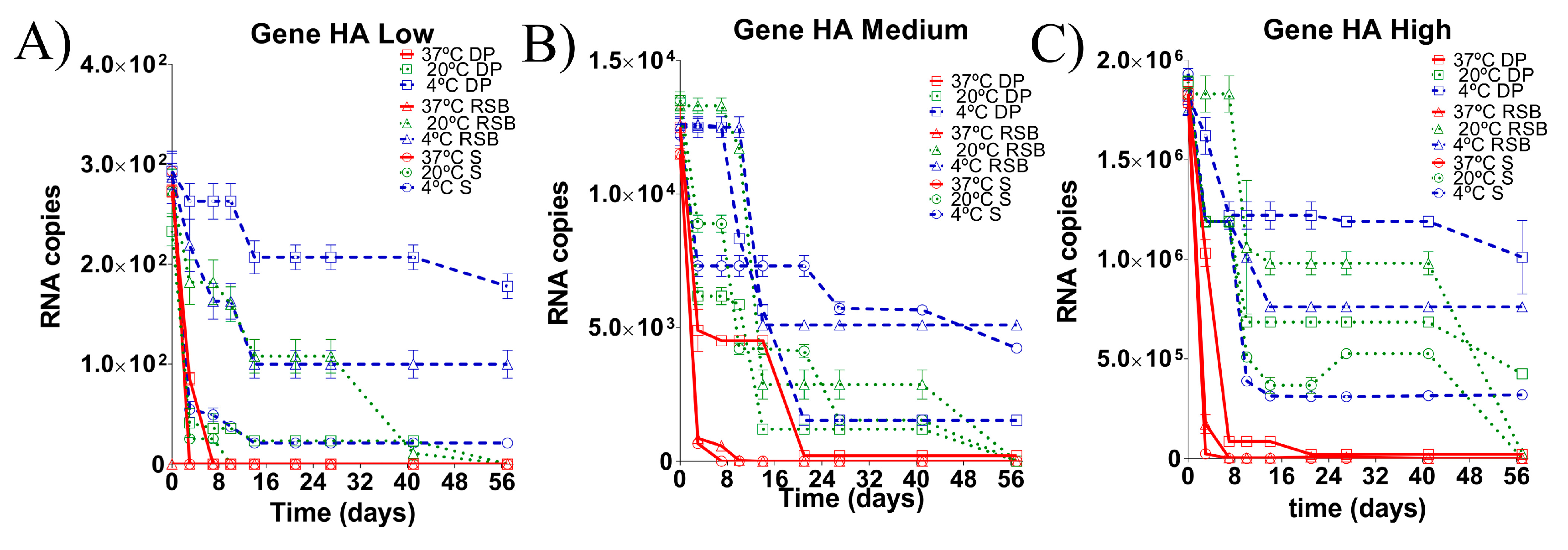
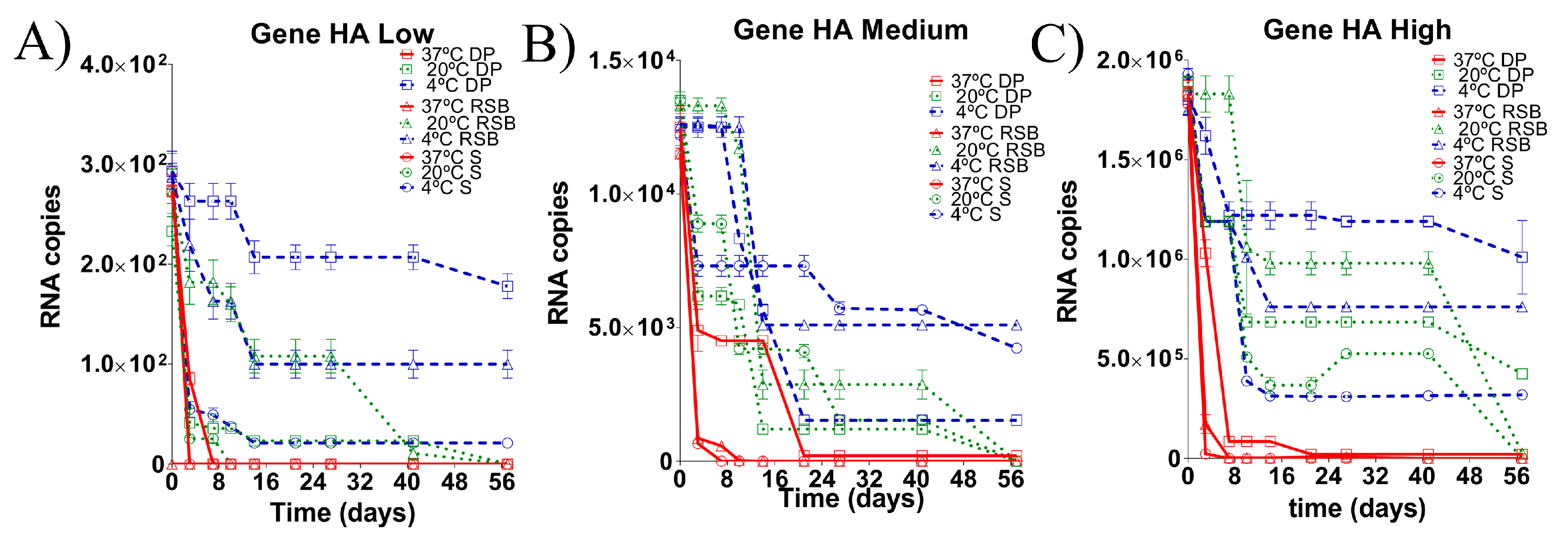
3.2. Stability of the RNA Transcript for HA Gene Stored under Different Conditions
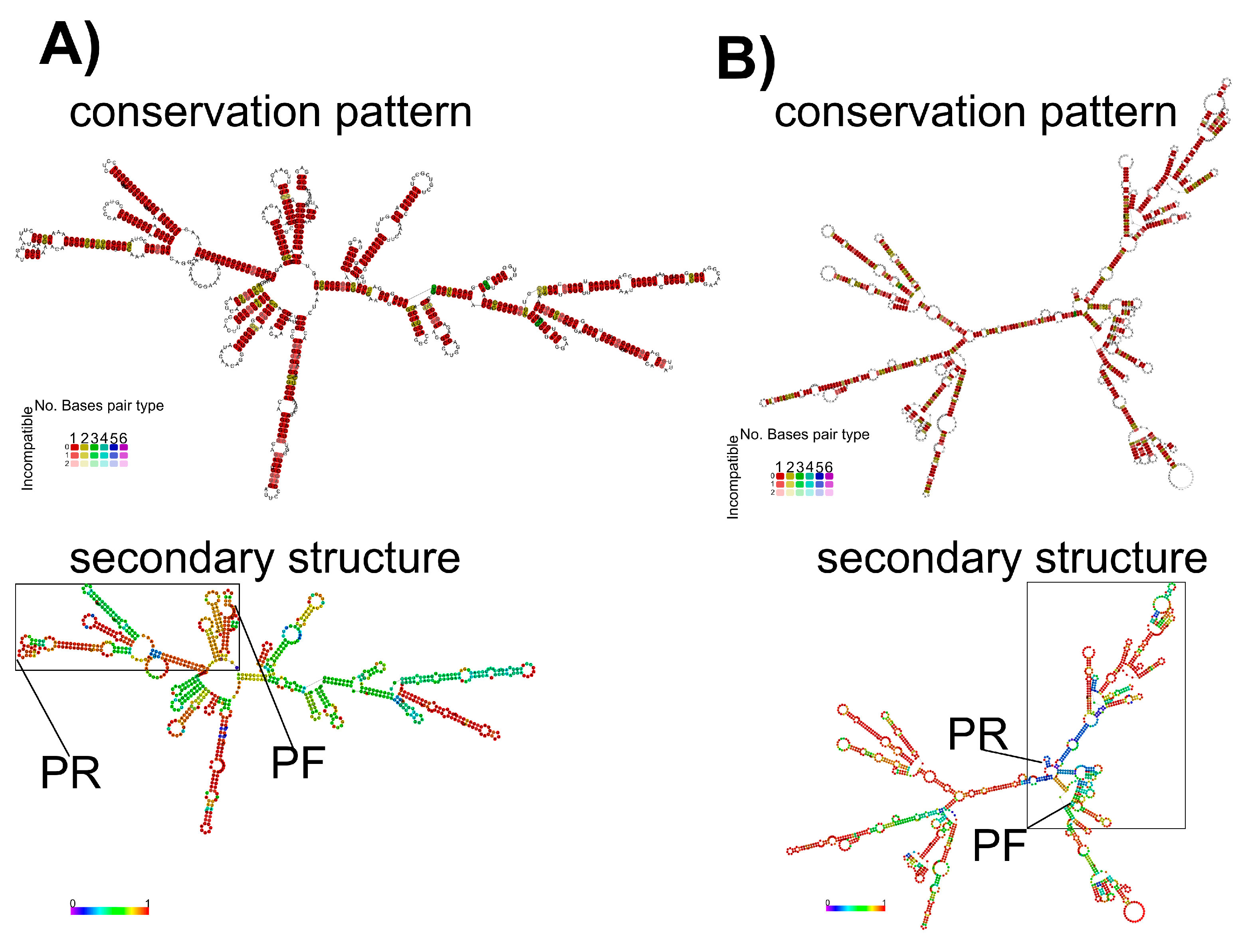
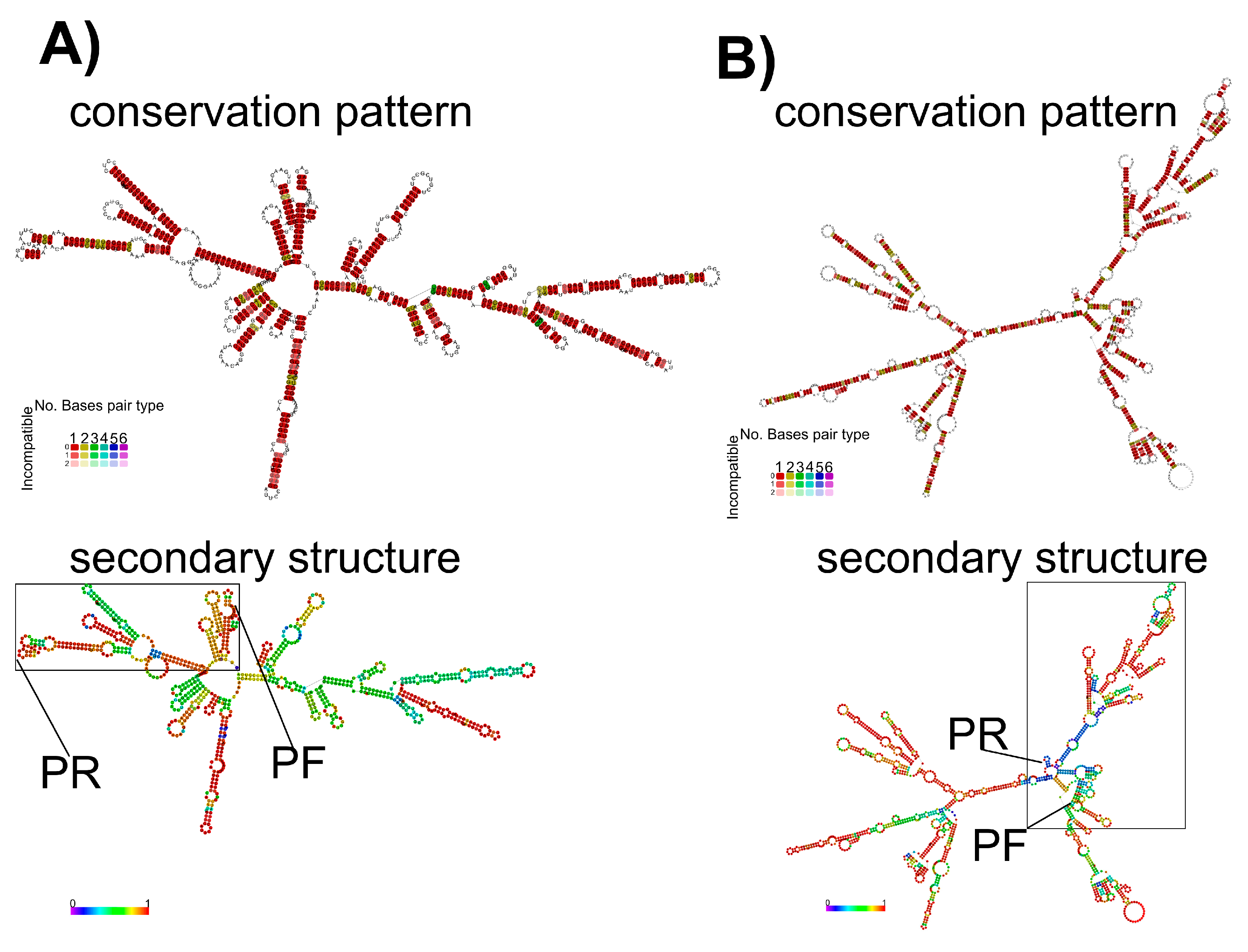
3.3. Secondary RNA Structures Analysis for M and H5 Genes
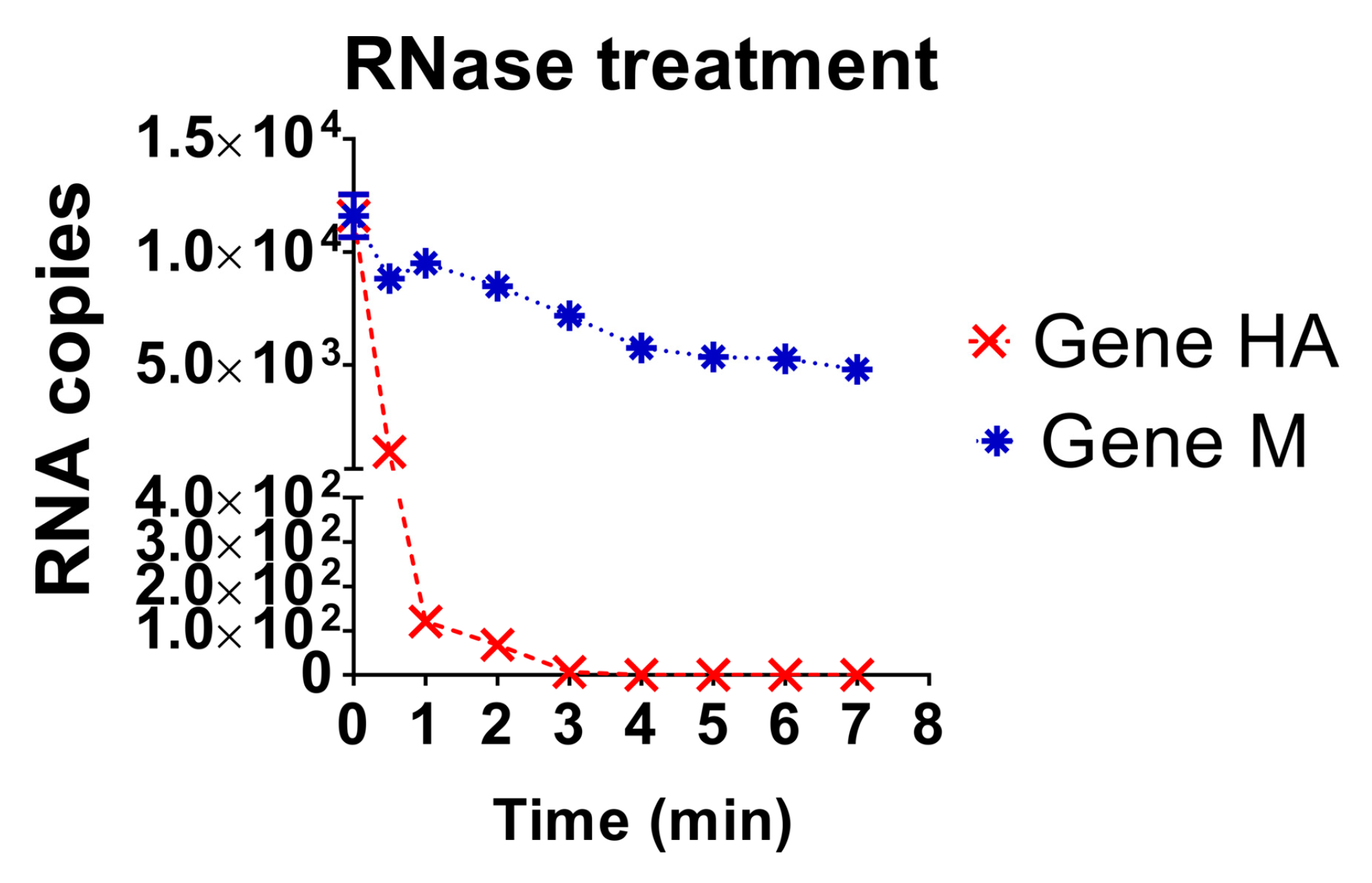
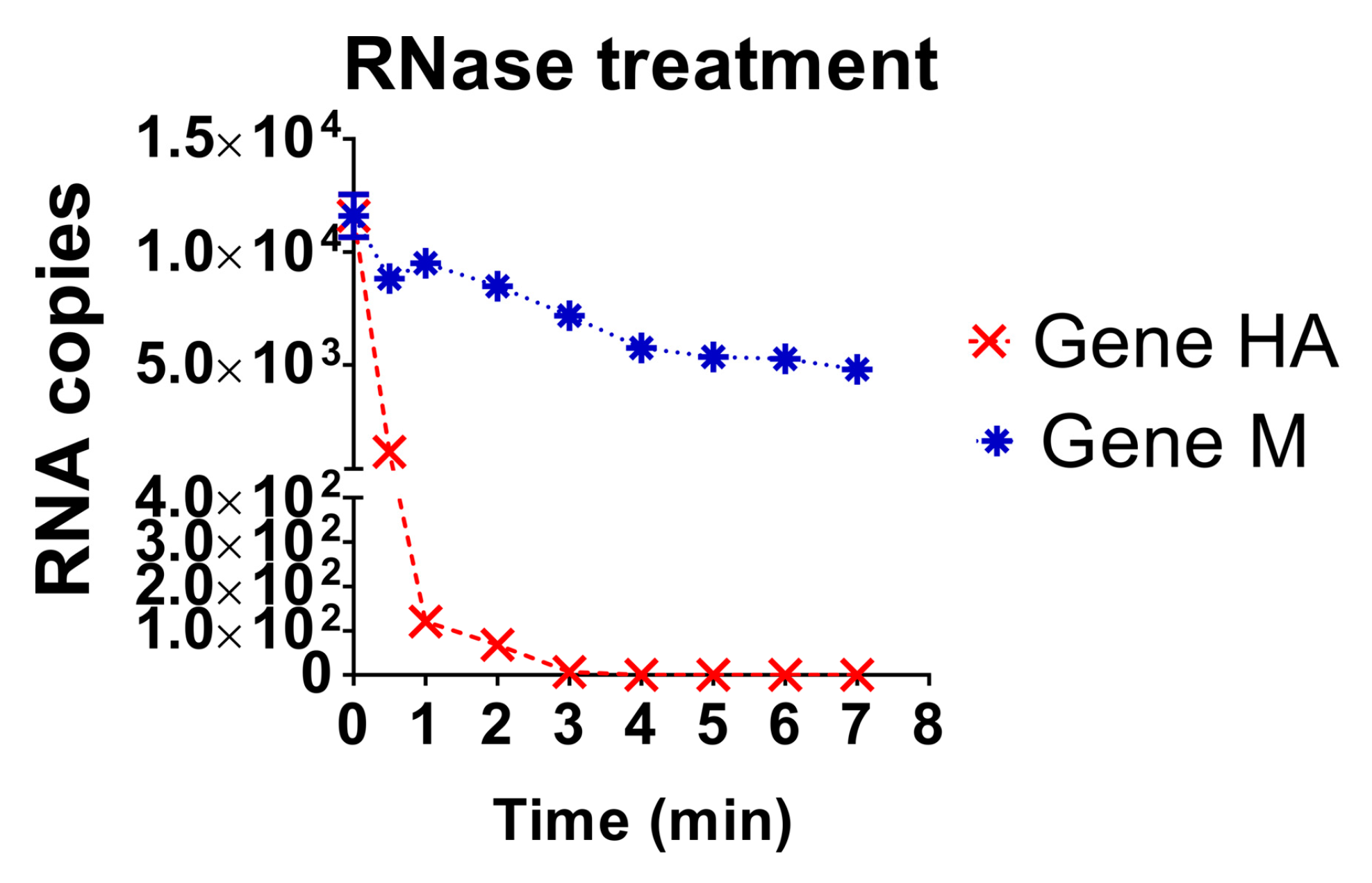
3.4. Evaluation of the Stability of the Diagnostic Target Regions for M and H5 Genes
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Belak, S. Molecular diagnosis of viral diseases, present trends and future aspects a view from the oie collaborating centre for the application of polymerase chain reaction methods for diagnosis of viral diseases in veterinary medicine. Vaccine 2007, 25, 5444–5452. [Google Scholar] [PubMed]
- Postel, A.; Moennig, V.; Becher, P. Classical swine fever in europe—The current situation. Berl. Munchener Tierarztl. Wochenschr. 2013, 126, 468–475. [Google Scholar]
- Nathues, C.; Perler, L.; Bruhn, S.; Suter, D.; Eichhorn, L.; Hofmann, M.; Nathues, H.; Baechlein, C.; Ritzmann, M.; Palzer, A.; et al. An outbreak of porcine reproductive and respiratory syndrome virus in switzerland following import of boar semen. Transbound. Emerg. Dis. 2016, 63, e251–e261. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martinez, I.; Balish, A.; Barrera-Badillo, G.; Jones, J.; Nunez-Garcia, T.E.; Jang, Y.; Aparicio-Antonio, R.; Azziz-Baumgartner, E.; Belser, J.A.; Ramirez-Gonzalez, J.E.; et al. Highly pathogenic avian influenza a(H7N3) virus in poultry workers, Mexico, 2012. Emerg. Infect. Dis. 2013, 19, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Durand, L.O.; Glew, P.; Gross, D.; Kasper, M.; Trock, S.; Kim, I.K.; Bresee, J.S.; Donis, R.; Uyeki, T.M.; Widdowson, M.A.; et al. Timing of influenza a(h5n1) in poultry and humans and seasonal influenza activity worldwide, 2004–2013. Emerg. Infect. Dis. 2015, 21, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Postel, A.; Schmeiser, S.; Perera, C.L.; Rodriguez, L.J.; Frias-Lepoureau, M.T.; Becher, P. Classical swine fever virus isolates from Cuba form a new subgenotype 1.4. Vet. Microbiol. 2013, 161, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Morales, A.; Martinez-Perez, O.; Dolz, R.; Valle, R.; Perera, C.L.; Bertran, K.; Frias, M.T.; Majo, N.; Ganges, L.; Perez, L.J. Spatiotemporal phylogenetic analysis and molecular characterisation of infectious bursal disease viruses based on the VP2 hyper-variable region. PLoS ONE 2013, 8, e65999. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.J.; de Arce, H.D.; Cilloni, F.; Salviato, A.; Marciano, S.; Perera, C.L.; Salomoni, A.; Beato, M.S.; Romero, A.; Capua, I.; et al. An SYBR green-based real-time RT-PCR assay for the detection of h5 hemagglutinin subtype avian influenza virus. Mol. Cell. Probes 2012, 26, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Thur, B.; Liu, L.; Gerber, M.; Stettler, P.; Moser, C.; Bossy, S. Rescue of infectious classical swine fever and foot-and-mouth disease virus by rna transfection and virus detection by RT-PCR after extended storage of samples in trizol. J. Virol. Methods 2000, 87, 29–39. [Google Scholar] [CrossRef]
- Ginocchio, C.C.; Wang, X.P.; Kaplan, M.H.; Mulligan, G.; Witt, D.; Romano, J.W.; Cronin, M.; Carroll, R. Effects of specimen collection, processing, and storage conditions on stability of human immunodeficiency virus type 1 RNA levels in plasma. J. Clin. Microbiol. 1997, 35, 2886–2893. [Google Scholar] [PubMed]
- Hoffmann, B.; Depner, K.; Schirrmeier, H.; Beer, M. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J. Virol. Methods 2006, 136, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, I.; Cayarga, A.A.; Hernandez, Y.P.; de la Rosa, I.G.; Gonzalez, Y.J.; Leon, C.S.; Alvarez, R.R. Long-term conservation of HCV RNA at 4 °C using a new RNA stabilizing solution. J. Virol. Methods 2010, 168, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a real-time reverse transcriptase PCR assay for type a influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Chang, P.C.; Shien, J.H.; Cheng, M.C.; Shieh, H.K. Identification and subtyping of avian influenza viruses by reverse transcription-PCR. J. Virol. Methods 2001, 97, 13–22. [Google Scholar] [CrossRef]
- De Arce, H.D.; Perez, L.J.; Frias, M.T.; Rosell, R.; Tarradas, J.; Nunez, J.I.; Ganges, L. A multiplex RT-PCR assay for the rapid and differential diagnosis of classical swine fever and other pestivirus infections. Vet. Microbiol. 2009, 139, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Capua, I.; Terregino, C.; Cattoli, G.; Toffan, A. Increased resistance of vaccinated turkeys to experimental infection with an H7N3 low-pathogenicity avian influenza virus. Avian Pathol. 2004, 33, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Bernhart, S.H.; Lorenz, R. The viennarna web services. Methods Mol. Biol. 2015, 1269, 307–326. [Google Scholar] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Belak, S.; Karlsson, O.E.; Blomstrom, A.L.; Berg, M.; Granberg, F. New viruses in veterinary medicine, detected by metagenomic approaches. Vet. Microbiol. 2013, 165, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Gebreyes, W.A.; Dupouy-Camet, J.; Newport, M.J.; Oliveira, C.J.; Schlesinger, L.S.; Saif, Y.M.; Kariuki, S.; Saif, L.J.; Saville, W.; Wittum, T.; et al. The global one health paradigm: Challenges and opportunities for tackling infectious diseases at the human, animal, and environment interface in low-resource settings. PLoS Negl. Trop. Dis. 2014, 8, e3257. [Google Scholar] [CrossRef] [PubMed]
- Marais, B.; Crawford, J.; Iredell, J.; Ward, M.; Simpson, S.; Gilbert, L.; Griffiths, P.; Kamradt-Scott, A.; Colagiuri, R.; Jones, C.; et al. One world, one health: Beyond the millennium development goals. Lancet 2012, 380, 805–806. [Google Scholar] [CrossRef]
- Wiegers, A.L. The quality assurance of proficiency testing programs for animal disease diagnostic laboratories. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc. 2004, 16, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Plebani, M. Harmonization in laboratory medicine: The complete picture. Clin. Chem. Lab. Med. 2013, 51, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.E. Central or national veterinary diagnostic laboratories. Rev. Sci. Tech. Int. Off. Epizoot. 1998, 17, 411–417. [Google Scholar] [CrossRef]
- Zepeda, C. The role of diagnostic laboratories in support of animal disease surveillance systems. Dev. Biol. 2007, 128, 139–143. [Google Scholar]
- Escorcia, M.; Attene-Ramos, M.S.; Estrada, M.J.; Nava, G.M. Improving global influenza surveillance: Trends of a(H5N1) virus in africa and asia. BMC Res. Notes 2012, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Taubenberger, J.K. Methods for molecular surveillance of influenza. Exp. Rev. Anti-Infect. Ther. 2010, 8, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Domanska-Blicharz, K.; Minta, Z.; Smietanka, K.; Marche, S.; van den Berg, T. H5N1 high pathogenicity avian influenza virus survival in different types of water. Avian Dis. 2010, 54, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Mathy, N.; Benard, L.; Pellegrini, O.; Daou, R.; Wen, T.; Condon, C. 5′-to-3′ exoribonuclease activity in bacteria: Role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 2007, 129, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Baldwin, D.A.; Scearce, L.M.; Oberholtzer, J.C.; Tobias, J.W.; Mourelatos, Z. Microarray-based, high-throughput gene expression profiling of micrornas. Nat. Methods 2004, 1, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.G.; Tsoras, M.; Thompson, S.D.; Martinez, H.; Iverson, B.; Nuñez, R. Ambient temperature stabilization of purified rna in gentegra™ for use in affymetrix human exon 1.0 st arrays. BioTechniques 2010, 48, 2. [Google Scholar] [CrossRef]
- Martinez, H.; Beaudry, G.; Veer, J.; Robitaille, M.; Wong, D.; Iverson, B. Ambient temperature storage of RNA in gentegra™ for use in RT-qPCR. BioTechniques 2010, 48, 2. [Google Scholar] [CrossRef]
- Opitz, L.; Salinas-Riester, G.; Grade, M.; Jung, K.; Jo, P.; Emons, G.; Ghadimi, B.M.; Beissbarth, T.; Gaedcke, J. Impact of RNA degradation on gene expression profiling. BMC Med. Genom. 2010, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Schaefer, A.; Steiner, I.; Kempkensteffen, C.; Stephan, C.; Erbersdobler, A.; Jung, K. Robust microrna stability in degraded RNA preparations from human tissue and cell samples. Clin. Chem. 2010, 56, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Henke, W.; Herdel, K.; Jung, K.; Schnorr, D.; Loening, S.A. Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 1997, 25, 3957–3958. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Relova, D.; Rios, L.; Acevedo, A.M.; Coronado, L.; Perera, C.L.; Pérez, L.J. Impact of RNA Degradation on Viral Diagnosis: An Understated but Essential Step for the Successful Establishment of a Diagnosis Network. Vet. Sci. 2018, 5, 19. https://doi.org/10.3390/vetsci5010019
Relova D, Rios L, Acevedo AM, Coronado L, Perera CL, Pérez LJ. Impact of RNA Degradation on Viral Diagnosis: An Understated but Essential Step for the Successful Establishment of a Diagnosis Network. Veterinary Sciences. 2018; 5(1):19. https://doi.org/10.3390/vetsci5010019
Chicago/Turabian StyleRelova, Damarys, Liliam Rios, Ana M. Acevedo, Liani Coronado, Carmen L. Perera, and Lester J. Pérez. 2018. "Impact of RNA Degradation on Viral Diagnosis: An Understated but Essential Step for the Successful Establishment of a Diagnosis Network" Veterinary Sciences 5, no. 1: 19. https://doi.org/10.3390/vetsci5010019