
Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila


1
Department of Biology, Taylor University, 236 West Reade Ave., Upland, IN 46989, USA
2
Department of Biology, McMaster University, 1280 Main St. W., Hamilton, ON L8S 4K1, Canada
*
Author to whom correspondence should be addressed.
Vet. Sci. 2017, 4(2), 23; https://doi.org/10.3390/vetsci4020023
Submission received: 25 October 2016
/
Revised: 27 November 2016
/
Accepted: 7 December 2016
/
Published: 24 April 2017
(This article belongs to the Special Issue Comparison of Cardiovascular Systems and Diseases Across Species)
Abstract
:Congenital heart defects, clinically identified in both small and large animals, are multifactorial and complex. Although heritable factors are known to have a role in cardiovascular disease, the full genetic aetiology remains unclear. Model organism research has proven valuable in providing a deeper understanding of the essential factors in heart development. For example, mouse knock-out studies reveal a role for the Integrin adhesion receptor in cardiac tissue. Recent research in Drosophila melanogaster (the fruit fly), a powerful experimental model, has demonstrated that the link between the extracellular matrix and the cell, mediated by Integrins, is required for multiple aspects of cardiogenesis. Here we test the hypothesis that Integrins signal to the heart cells through Src42A kinase. Using the powerful genetics and cell biology analysis possible in Drosophila, we demonstrate that Src42A acts in early events of heart tube development. Careful examination of mutant heart tissue and genetic interaction data suggests that Src42A’s role is independent of Integrin and the Integrin-related Focal Adhesion Kinase. Rather, Src42A acts non-autonomously by promoting programmed cell death of the amnioserosa, a transient tissue that neighbors the developing heart.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
A properly functioning heart is intimately linked to its structure. Defects in this structure may trigger immediate or later-onset heart disease. Congenital heart defects (CHDs) such as patent ductus arteriosus, atrial or ventricular septal defect, and tetralogy of Fallot, have been noted for both large and small animals, including cats [1], dogs [2], horses [3], and cows [4,5]. Development of the heart can effectively be studied at the genetic level to reveal the genes that determine the identity of heart cells and that dictate the response of heart progenitors to signals from neighboring cells and tissues. Clinical observations, especially those which involve domestic purebreds, suggest that the underlying cause of CHDs is often heritable [6,7]. This is supported by studies in the last fifteen years which have begun to link specific CHD’s to genetic aetiologies [8,9]. However, despite the increasing recognition of heritable factors, a full understanding of the genetic aetiology is hindered by the complex and multifactorial nature of these diseases. Even in instances where a specific genetic factor has been correlated to heart development, it remains difficult to definitively establish a causal relationship and determine how this genetic abnormality contributes to disease [10]. The underlying cellular or molecular mechanism remains a mystery.
While few genes have been correlated to CHDs in small and large animals in the clinic, experimental studies in mice have been used to ask targeted questions about likely candidates. Insights from genetic evidence in humans suggest a link between cardiovascular disease and integrin-related factors [10,11,12]. The Integrin heterodimer is a protein adhesion complex that spans the cellular membrane and connects the extracellular matrix to the intracellular cytoskeleton and various signaling cascades. Multiple research groups have studied knock-out mice lacking the alpha (α) or beta (β) subunits of the Integrin receptor. In mice, the β1 subunit is broadly expressed during development and serves as the β-integrin subunit for about half of all known vertebrate heterodimers (reviewed in [13]). Thus, it is not surprising that β1-null mice die very early in development prior to embryo implantation (at E5.5; [14,15]). However, tissue-specific loss of β1 reveals roles for development of multiple organs, including the heart [16,17,18]. Knock-out mice for α4 or α5, two of the β1 partners, also exhibit early embryonic lethality (by E8-10; [19,20,21]), with prominent defects in the epicardium of the heart (α4) and reduced blood vessel integrity (α4 and α5; [20,21]).
While vertebrate models have correlated specific factors like Integrin to CHDs, cell culture and simpler genetic models are powerful tools to map the complex signaling networks surrounding these genes. In particular, here we highlight the fruit fly (Drosophila melanogaster), which has become a well-studied model for cardiovascular development and aging for experimental biologists (reviewed in [22]). Although far simpler anatomically, development of the Drosophila embryonic heart is reminiscent of early vertebrate heart morphogenesis [23,24]. Development of both organs requires specification of the heart cells, medial alignment of two parallel rows of cells, medial-to-midline migration of these cells, and organization and rearrangement of these midline cells into a lumen-containing heart organ (reviewed in [25]). Drosophila is the only well-studied invertebrate model system with a heart comparable to vertebrates. Thus, it is a unique system, valuable because of its simplicity and the powerful genetic and proteomic manipulations and analysis possible (reviewed by [23,26]). The tools utilized in this study include mutant alleles, tissue specific inhibition or overexpression of target genes, labeling and imaging of specific cell types at the single-cell level, and non-invasive live imaging of developing embryos using proteins tagged with fluorescent markers. While alleles or specific mutations in genes may be correlatively linked to heart and vascular diseases in veterinarian animals, it is through the tools employed in model organism research that normal function of these genes can be established and the etiology of the correlated diseases understood.
Consistent with the correlative evidence found in mice studies, recent research in Drosophila has illustrated an essential role for Integrins in heart development [27]. We demonstrated that Integrins are involved in multiple steps of the process including heart cell migration, cardioblast cells (CB) polarization, and assembly of the heart tube [27]. However, Integrin signaling is complex (reviewed in [28]), and it remains less clear how Integrins guide the heart cells to trigger cardial cell migration and tube formation. Using a combination of genetic and cell biological Drosophila techniques, our lab has performed a targeted approach to map the extracellular and intracellular protein network involved in integrin-mediated heart development. This approach has thus far revealed a critical role for the intracellular signaling adaptor Talin [29] and extracellular matrix regulation [30]. Here we extend our study to explore a possible Integrin-dependent role for Src42A, one of two Drosophila Src kinases. Src is a non-receptor tyrosine kinase that transmits cell signals in a variety of contexts, including cell migration [31,32]. Activated Src is recruited to Integrin-tethered focal adhesions [33,34] where it phosphorylates several Integrin complex proteins including FAK and Paxillin [31,35,36]. When examined in cardiomyocytes, in vitro, Src signals participate in mechanical stress signaling that can trigger cardiac hypertrophy [34,37]. Here we use the in vivo Drosophila heart model to test the hypothesis that Src42A transmits Integrin signals to the developing heart. Our experimental approach employs mutant analysis, genetic interaction tests, tissue-specific protein inhibition, and cell biological analysis of living and fixed tissue. Our studies suggest that Src42A acts non-autonomously during heart development by promoting programmed cell death of migration-inhibiting extraembryonic amnioserosa tissue.
2. Materials and Methods
2.1. Drosophila Strains and Genetics
All fly strains and cross schemes were maintained according to standard conditions at 25 °C. All mutant alleles were maintained in a w1118 background over a ftz > lacZ or en > lacZ marked balancer chromosome. The following mutant alleles were tested: src42AE1 (Bloomington Drosophila Stock Center, BL #6408), src42Ak10108 (BL #10969), src42A26-1 [38], fak56DKG00304 (BL #13080), and rhea1 (BL #2296). For overexpression studies, UAS-Src42-YF [39] and UAS-Src42-DN [38] were crossed to dmefGal4 [40]. Yet1 flies express YFP in amnioserosa perimeter cells [41]. In live imaging experiments, dmefGal4, UAS-moesin-mCherry [42], and tup-GFP [43] were all present in a single copy in src42AE1/src42AE1 embryos.
2.2. Immunohistochemistry
Embryo collection, fixation, and immunolabeling protocols were completed as previously described [29,44]. The following primary antibodies were used: rabbit anti-MEF (1:5000; [27]), chicken anti-βGal (1:150; [45]), mouse anti-Hindsight (1:30; Developmental Studies Hybridoma Bank), and chicken anti-GFP (1:1000; Cedarlane). Embryos prepared for light microscopy were treated with a biotinylated secondary antibody (1:150; Jackson ImmunoResearch), incubated with the avidin and biotinynlated peroxidase containing Vectastain ABC system (Vector Laboratories, Burlingame, USA), and reacted with 3,3- Diaminobenzidine Tetra hydrochloride (DAB, Gibco-BRL) and hydrogen peroxide. Images were acquired using a Zeiss Axioskop microscope (with a RETIGA 1300i digital camera, Qimaging, Surrey, Canada) and OpenLab software (PerkinElmer, Woodbridge Canada). For embryos prepared for fluorescent confocal microscopy, the secondary antibodies were Alexa 488 and 594 (Invitrogen). Fluorescent-labelled embryos were imaged on a Leica SP5 confocal microscope (Leica Microsystems, Buffalo Grove, USA). Frontal images displayed are maximum projections of 3–10 slice stacks taken 1 μm apart in the heart chamber. Images of embryo z-sections (transverse images) are single slice images of the heart chamber. All images were processed with ImageJ (Rasband, W.S, NIH, Bethesda USA) and figures were assembled using Adobe Photoshop CC 2015.5 (Adobe Systems Incorporated, San Jose, USA).
2.3. Preparation of Tissue Sections for Electron and Light Microscopy
Embryos were prepared as outlined previously [45]. Embryo sections were examined on a JEOL 1200EXII microscope (electron microscopy) or a Zeiss Axioskop microscope with a Qimaging RETIGA 1300i digital camera (light level microscopy).
2.4. Live Imaging of Embryos
3. Results
3.1. Key Steps in Drosophila Melanogaster Embryonic Heart Development
The Drosophila melanogaster life cycle completes in approximately ten days (Figure 1A). By the end of embryogenesis (24 h), the heart structure is formed and starts to beat. The fly embryonic heart is a linear tube, analogous to the vertebrate heart before looping morphogenesis (Figure 1B’’; see also Figure 2). It is comprised of two parallel rows of muscular cardioblast cells (CBs) that form the mature heart at the dorsal midline of the embryo. Development of the heart initiates during embryogenesis with the specification of two heart fields. These fields align laterally along the embryo (Figure 1B,C), adjacent to the amnioserosa, an extraembryonic cell layer that transiently covers the dorsal surface of the embryo. Another tissue layer, the ectoderm, covers the cardial cells dorsally (Figure 1C). Prior to the heart forming a tube, the two rows of CBs migrate collectively through the embryo and meet at the midline. In Drosophila, CB migration occurs simultaneously with dorsal closure, a process whereby the ectoderm migrates towards the dorsal midline and the transient amnioserosa cells are internalized and undergo apoptosis [47]. Following migration, an initial dorsal contact is made between contralateral CBs (Figure 1C’). The CBs fill the space vacated by the midline amnioserosa cells, immediately ventral to the ectoderm (Figure 1C’–C’’). The CBs become crescent-shaped and adhere ventrally to wrap around and fully enclose the inner lumen and complete the heart tube (Figure 1C’’).
3.2. Src42A Is Required for Drosophila Heart Development
The requirement for specific factors in development can be tested experimentally by removing the factor and observing if development can still proceed normally. In Drosophila, a target protein can be removed by introducing a loss-of-function mutation in the gene that encodes that protein. To test if Src42A is required for Drosophila heart development, we compared the embryonic hearts of src42A mutants to wildtype and rhea mutants (rhea is the gene that encodes for Talin, an essential Integrin signaling adaptor; [29]). The heart was visualized by labeling the embryos with an antibody that binds to dMEF, a muscle-specific transcription factor. In late stage wildtype embryos, CB migration has completed and the cells are aligned along the dorsal midline (Figure 2A). In rhea mutants, migration completes, but no midline space remains between the CB rows, indicating that the heart is unable to form an open tube (Figure 2B, [29]). In contrast, embryos homozygous for the null src42AE1 mutation frequently exhibited an “open heart” phenotype, in which posterior CBs failed to reach the midline (Figure 2C asterisks). This is analogous to a cardia bifida phenotype in mammals. To confirm this was not an allele-specific phenotype perhaps due to genetic abnormalities elsewhere in the genome, several independent src42A mutant alleles were tested. Embryos carrying a partial deletion of src42A (src42A26-1; [38]) had a similar open heart phenotype (Figure 2D). Embryos homozygous for a src42A hypomorphic allele had normal CB alignment (Figure 2E), however transheteroallelic embryos for hypomorphic and null src42A alleles occasionally displayed stalled or delayed posterior CB migration (Figure 2F). This suggests that Src42A is essential for full migration of the CBs, but that low levels of Src42A (as in the hypomorphic mutant) are sufficient for migration to complete. The difference in phenotypes between the rhea and src42A mutants suggests it is unlikely that Src42A is working together with or downstream of the Integrin-Talin signaling complex.
In multiple vertebrate contexts, Src works downstream of Integrins with Focal Adhesion Kinase (FAK) to activate signal transduction [31,32,34]. In Drosophila, there is a single FAK encoded by the gene fak56D. Similar to vertebrate FAK, Fak56D has a focal adhesion targeting domain and a binding site for the SH2 domain of Src, but the function of its sequence-inferred predicted kinase domain remains unclear [48,49,50]. Thus, it is likely that some, but perhaps not all, vertebrate and fly FAK cellular functions are shared. One of the strengths of the Drosophila model is the ability to use genetics to map signaling pathways. For example, Tsai et al. studied the requirement for Fak56D and Src42A in the Drosophila nervous system [51]. Although flies heterozygous for src42AE1 are healthy, Tsai et al. demonstrated that introducing a src42AE1 mutation worsened the phenotype severity of a fak56D hypomorph in the nervous system. This genetic interaction suggests that Src42A and Fak56D work together in a signaling pathway during nervous system development. To test if fak56D similarly works with Src42A during heart development, we assessed flies with disrupted fak56D. fak56D hypomorphic and null Drosophila embryos have no obvious heart phenotype (Figure 2G,H). This is not surprising as flies of these genotypes are viable and healthy [51,52]. Therefore, to further test if Fak56D modulates Src signaling within the heart, fak56D gene dosage was reduced in src42AE1 mutant embryos. Reduction of fak56D did not exacerbate the src42AE1 phenotype (Figure 2I). Similarly, halving the gene dosage of src42A did not induce a phenotype in fak56D homozygous mutant embryos (Figure 2J). This data suggests that Src42A, but not Fak56D, is necessary for proper migration of the cardial cells in Drosophila.
3.3. Src42A Is Required for Cell Migratory Dynamics in the Posterior Heart
Since our fixed-tissue studies demonstrated that CBs fail to reach the midline in src42A mutants, we wanted to explore how these mutants differ from wildtype during the cell migration stage. Migratory cells, whether in cell culture or live organisms, are characterized by a highly dynamic cell membrane that extends filopodia and lamelliopodia in the direction of migration [53,54]. In contrast to mammalian models, Drosophila is highly amenable to in vivo time-lapse analysis of heart development. In the Drosophila heart, CB “leading edge” membrane activity can be visualized in live embryos that express fluorescently tagged proteins. As the CBs migrate, they extend and retract numerous membrane protrusions, which increase as the CBs approach the midline [30]. Our studies on Integrin and Talin function in the heart demonstrated that these genes are required for the highly dynamic leading edge that migrating CBs extend [27,29]. Therefore, we reasoned that if Src42A is functioning downstream of Integrin to control CB migration, lack of Src42A should result in a similar loss of membrane leading edge activity. To directly test this, we compared the membrane activity in src42AE1 mutants to wildtype. Consistent with previous studies, in wildtype embryos numerous membrane protrusions were observed across the length of the CB rows (Figure 3A–A’’). Interestingly, while a reduction in leading edge activity was observed in the posterior of the heart in src42AE1 embryos (similar to integrin or rhea mutants), the CBs at the anterior of the heart retained a highly dynamic membrane activity similar to wildtype (Figure 3B—compare B’’ to B’). Transverse imaging revealed that CBs with high membrane activity extended a protrusion dorsally towards the midline (Figure 3C, arrows) while cells with minimal activity were rounded and did not extend dorsally (Figure 3C’). Thus, it appears that in contrast to integrin and rhea mutants [27,29], src42A nulls retain a highly dynamic leading edge in the anterior of the heart. In the posterior heart, however, CB leading edge activity is minimal, correlating with the inability of the posterior CBs to fully migrate to the midline.
3.4. Heart-Specific Modification of Src42A Function
While the data suggests that Src42A is required for heart development, it was unclear whether Src42A was required in the CBs themselves or if the mutant phenotype observed was due to disruptions in surrounding tissues (e.g., the amnioserosa or ectoderm). A powerful technique in Drosophila is the UAS-Gal4 system which allows temporal and spatial control of transgene expression (reviewed in [55]). Using this system, a constitutively active (YF) or wildtype version of Src42A was first overexpressed in the heart and other body-wall muscle tissue (Figure 2K). Following overexpression of either construct, the heart developed normally (Figure 2K and data not shown), suggesting that tight control of Src42A levels is not essential for heart development. To complement the Src42A overexpression expression, Src42A function was inhibited by expressing a dominant-negative Src42A (Src42A-DN). Src42A-DN lacks kinase activity [56] and ubiquitously expressing Src42A-DN during development results in late pupal lethality (this study). To directly test if the kinase activity of Src42A is required for cardial cell migration, Src42A-DN was expressed in the heart (using a muscle-specific enhancer). In these embryos, the heart cells fully migrated to the midline (Figure 2L), however the cells were frequently mis-positioned and formed small clumps (arrowheads in Figure 2L). This clumping phenotype suggests that Src42A may regulate adhesion between neighboring cells. Such a role would be consistent with a Src kinase-dependent E-cad trafficking mechanism [57,58]. However, the completed migration in the Src42A-DN embryos suggests that Src42A expressed within the heart itself does not regulate cardial cell migration. Another possibility is that Src42A is required within the heart cells for a kinase-independent role. Although Src42A-DN dominantly interferes with endogenous Src42A, it is unclear if it inhibits all Src function. While it interferes with the kinase activity of Src [56], it is possible that other protein-binding or “scaffolding” aspects of Src function remain intact and that these are sufficient for CB migration.
3.5. Delayed Migration in src42A Mutants Correlates with Stalled Internalization of the Amnioserosa
While the mutant data suggests that Src42A is required for heart development, it was intriguing that loss of Src42A only disrupted cardial cell migration in the posterior heart. Furthermore, the heart-specific expression of the Src42A-DN hints that the migration defects may not be due to Src42A signaling in the heart itself. We therefore hypothesized that the src42A mutant phenotype was due to disruptions in surrounding tissues (the amnioserosa or ectoderm). Previous studies have documented that Src42A is required for dorsal closure [38,59] and, consistent with this, a subset of src42A mutant embryos analyzed had incomplete dorsal closure (data not shown). Although somatic muscles positioned close to the midline suggested that dorsal closure was complete in at least some “open heart” src42A mutant embryos (e.g., Figure 1F), subtle phenotypes may not be readily apparent.
Therefore, to test if the CB migration delay was due to disruptions in the amnioserosa, we labeled the amnioserosa using an antibody against Hindsight (Hnt), which is present in the nuclei of the amnioserosa and midgut cells (Figure 4A–B’), and the enhancer trap YET1, which expresses Yellow Fluorescent Protein in the amnioserosa perimeter cells (Figure 4C–D’’). In wildtype embryos, the amnioserosa is visible at stage 16 (Figure 4A), but has internalized and undergone apoptosis by stage 17 (Figure 4A’). Already in stage 16 embryos, there are few amnioserosa cells remaining near the posterior region of the heart. However, in src42AE1 embryos, the amnioserosa cells persist in the posterior region of the embryo, where the CB migration delay is observed (Figure 4B–B’).
Transverse images of wildtype embryos shows that as CB migration proceeds, the CBs move dorsally and eventually over the amnioserosa cells (Figure 4C–C’’). Once CB migration is complete, no amnioserosa cells are between the CBs; any remaining cells have internalized into the embryo and appear round, characteristic of cells entering apoptosis (Figure 4C’’; [60]). This is not the case in src42AE1 embryos: although the amnioserosa cells are rounded suggesting they are apoptotic, they persist at the dorsal midline (Figure 4D–D’’). Thus, it appears that the incomplete amnioserosa internalization is a physical barrier to CB migration and the migration defects observed in src42A mutants are likely not be the result of a requirement of Src42A in the heart itself.
3.6. Src42A Is Not Essential for Development of A Lumen
In integrin and rhea mutants, disruptions in cell migration and leading edge activity is correlated with the inability of the CBs to form a lumen [27,29]. If Src42A is required in the heart itself, we would expect that src42A mutants would form no lumen between CB cells that were able to reach the midline. However, if the migration delays are due to disruptions in amnioserosa internalization, contralateral CBs that meet at the midline would be able to form a lumen. Light level histology of embryo cross-sections was used to test these hypotheses. In late stage wildtype embryos, a lumen is visible between contralateral CBs (Figure 5A, see also Figure 1C’’). Dorsal closure has completed, as evidenced by the thick and continuous ectoderm layer dorsal to the heart (horizontal arrow). Any remaining amnioserosa cells have been internalized and are situated ventral to the heart (asterisks). In src42AE1 embryos, there are a range of phenotypes. Consistent with the images described earlier (Figure 4D–D’’), some embryos have amnioserosa cells remaining at the midline, precluding normal interactions between contralateral CBs (Figure 5B–B’). In many embryos, development is disrupted to such a point that identification of the CBs is impossible (e.g., Figure 5B’). While dorsal closure succeeds, the layer of ectoderm is often thin and variable (Figure 5B–B’’). However, despite these defects in the surrounding tissues, in regions of the embryo where contralateral CBs meet at the midline, a lumen, albeit small, often forms (Figure 5B’’). Taken together with the tissue-specific dominant-negative data, this suggests that Src42A is not required in the CBs to promote lumen formation. Rather, the heart defects observed in src42A mutants are due to disruptions in dorsal closure and amnioserosa internalization. It also remains possible that Src42A has a heart autonomous role in subsequent lumen expansion, a process regulated by different genetic factors than initial lumen formation [61,62].
4. Discussion
CHDs encompass a variety of diseases that range in severity and treatment potential. Correctly and quickly identifying the disease and its etiology aids in determining an appropriate course of action. Knowledge of the causative factor is also important to prevent familial CHDs in pets such as dogs and cats that undergo extensive inbreeding [63]. Despite increasing recognition of the importance of understanding the genetic etiology, clinical studies are limited by the complexity and multifactorial nature of many CHDs. The experiments described here employ the genetic and cell biological strengths of the Drosophila model to explore the mechanism of heart development.
4.1. Genetic Analysis to Map Protein Networks Underlying Heart Development
As demonstrated by our lab and others (reviewed in [25]), the Drosophila heart is a helpful and established model organism to test the requirement and role of specific genes. For almost all genes in the fly genome, multiple mutant alleles are readily available through stock centers (e.g., the Bloomington Drosophila Stock Center) or other experimental research labs. Using three independently derived mutant strains, we demonstrated that a reduction or loss of Src42A results in an inability of the heart to properly form as the cells do not migrate fully to their necessary positions. The ability to test independently derived mutations gives confidence that the heart defect is directly caused by the loss of Src42A, and not by another unknown genetic abnormality.
Since a defective gene impacts the whole organism, the src42A mutant analysis does not reveal whether or not the heart defect is due to a requirement for Src42A in the heart itself or if it is a secondary defect due to Src42A’s role in surrounding tissues. Using the UAS-Gal4 system [55], cell or tissue autonomous roles can be directly tested by expressing transgenes in a temporally and spatially restricted manner. Since neither expression of normal Src42A nor a dominant negative version disrupts heart cell migration, it appears that the requirement of Src42A for heart development is due to Src42A expressed in non-heart tissues. However, since the dominant negative construct only disrupts the kinase domain of Src42A, it remains possible that the other domains of Src42A are required in the heart.
The ready availability of known mutations and the short life-cycle of flies also permits complex breeding schemes to test multiple genes together [64]. The term “genetic interaction” is used to describe mutations in two or more genes that are able to increase or decrease the severity of the phenotype relative to an additive effect of two independently acting genes. A genetic interaction suggests that the product of the two genes work together and such evidence can be used to map complex protein networks. Our combined analysis of src42A and fak56D suggests that Src42A impacts the heart in a Fak56D-independent manner. This is intriguing, as many studies in mammalian systems suggest that Src kinases and FAKs most commonly work as a complex [31,32,33].
4.2. Detailed Cell Biological Analysis Suggests that Src42A Functions in An Integrin-Independent manner
Both Drosophila and rodent (mice) models have demonstrated a requirement for the Integrin adhesion receptor during heart formation [16,17,27], but the complex nature of the mammalian heart precludes detailed analysis when development goes awry. We tested the hypothesis that Integrin signals through Src42A to control formation of the Drosophila heart tube. Several observations suggest that Src42A’s role in heart development is independent of Integrin. First of all, although heart development is disrupted in mutants for src42A, scb or mys (the genes for the αPS3 and βPS1 integrin subunits), and rhea (the gene that encodes Talin, a key Integrin signaling adaptor), the defects in src42A mutants were markedly different. In embryos with a genetic abnormality in src42A, the posterior cardial cells were frequently unable to reach the midline. This correlated with a posterior reduction in membrane dynamics as visualized in live embryos. In contrast, delayed cell migration in scb, mys, and rhea mutants was most frequently seen anteriorly [27,29] and the membrane dynamics were reduced along the entire length of the heart fields [27,29]. Furthermore, when cells in src42A mutants did reach the midline, they were able to form an open heart tube, albeit small. In contrast, the hearts in scb, mys, and rhea mutants remained closed [27,29]. Taken together, the detailed cellular analysis of the mutant embryos suggests that Src42A and Integrin-Talin signaling regulate different aspects of cardiac development.
4.3. A Role for Transient Tissues in Proper Heart Development
Since the tissue-specific expression of Src42A-DN suggests that Src42A kinase is not required in the heart tissue itself, how then is Src42A required for heart development? In mammals, proper blood circulation requires closure of the ductus arteriosus, the vessel that allows blood to bypass the lungs pre-birth. Patent ductus arteriorsus, a failure to close off this blood vessel, leads to increased stress on the heart and lungs and possible congestive heart failure [65,66,67]. A study in swine demonstrated that proper ductus arteriorsus closure involves apoptosis or necrosis of cells within the inner layers of the vessel that are only required in the swine fetus [68]. Similarly, during the final stages of Drosophila embryonic heart development, the transient extraembryonic amnioserosa tissue internalizes and undergoes apoptosis [47]. However, careful analysis of development reveals that removal of the amnioserosa tissue only partially completes in src42A mutants. Rather, the amnioserosa cells persist at the intended site of heart tube formation and trigger secondary heart teratologies, including cardia bifida.
Acknowledgments
We thank the following investigators for providing Drosophila strains: Brian McCabe (dMef-Gal4), Thomas Millard (UAS-moesin-mcherry), Robert Schulz (TUP-GFP), Bruce Reed (YET1), Tetsuya Kojima (src42A26-1, UAS-Src42A-DN), and Tian Xu (UAS-Src42A). Resources of the Bloomington Drosophila Stock Center and Flybase contributed to this work. Emily Viers helped develop the graphics for Figure 1. Research was funded by NSERC CGS-D (JV), NSERC Discovery 46651 (JRJ), and infrastructure supported by the Leaders Opportunity Fund of the Canada Foundation for Innovation.
Author Contributions
Jessica Vanderploeg and J. Roger Jacobs conceived and designed the experiments. Jessica Vanderploeg and J. Roger Jacobs performed the experiments and analyzed the data. Jessica Vanderploeg wrote the paper, with editorial support from J. Roger Jacobs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scansen, B.A.; Schneider, M.; Bonagura, J.D. Sequential segmental classification of feline congenital heart disease. J. Vet. Cardiol. 2015, 17, S10–S52. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.A.; Sisson, D.D. Evaluation of canine congenital heart disease using an echocardiographic algorithm. J. Am. Anim. Hosp. Assoc. 2001, 37, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Buergelt, C.D. Equine cardiovascular pathology: An overview. Anim. Health Res. Rev. 2003, 4, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Buczinski, S.; Rezakhani, A.; Boerboom, D. Heart disease in cattle: Diagnosis, therapeutic approaches and prognosis. Vet. J. 2010, 184, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ohwada, K.; Murakami, T. Morphologies of 469 cases of congenital heart diseases in cattle. J. Jpn. Vet. Med. Assoc. 2000, 53, 205–209. [Google Scholar] [CrossRef]
- MacDonald, K.A. Congenital heart diseases of puppies and kittens. Vet. Clin. N. Am. Small Anim. Pract. 2006, 36, 503–531. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M. Genetics of cardiac disease in the small animal patient. Vet. Clin. N. Am. Small Anim. Pract. 2010, 40, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Andelfinger, G.; Wright, K.N.; Lee, H.S.; Siemens, L.M.; Benson, D.W. Canine tricuspid valve malformation, a model of human Ebstein anomaly, maps to dog chromosome 9. J. Med. Genet. 2003, 40, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, N.; Kumagai, D.; Matsumoto, K.; Inokuma, H.; Furuoka, H.; Kobayashi, Y. Detection of the nonsense mutation of OPA3 gene in Holstein Friesian cattle with dilated cardiomyopathy in Japan. J. Vet. Med. Sci. 2015, 77, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Acksteiner, C.; Baumgarth, J.; Burst, V.; Geissler, H.J.; Margulies, K.B.; Houser, S.; Bloch, W.; Flesch, M. Loss of beta1D-integrin function in human ischemic cardiomyopathy. Basic Res. Cardiol. 2007, 102, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.M.; Moravec, C.S.; McCarthy, P.M.; Altamentova, S.M.; Wong, A.P.; Skrtic, M.; Verma, S.; Weisel, R.D.; Li, R.-K. Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation 2006, 113, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fedak, P.W.M.; Dai, X.; Du, C.; Zhou, Y.-Q.; Henkelman, M.; Mongroo, P.S.; Lau, A.; Yamabi, H.; Hinek, A.; et al. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation 2006, 114, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. In vivo functions of integrins: Lessons from null mutations in mice. Matrix Biol. 2000, 19, 203–209. [Google Scholar] [CrossRef]
- Fässler, R.; Meyer, M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995, 9, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.E.; Sutherland, A.E.; Klimanskaya, I.V.; Andrieux, A.; Meneses, J.; Pedersen, R.A.; Damsky, C.H. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995, 9, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Valencik, M.L.; Keller, R.S.; Loftus, J.C.; McDonald, J.A. A lethal perinatal cardiac phenotype resulting from altered integrin function in cardiomyocytes. J. Card. Fail. 2002, 8, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.S.; Shai, S.Y.; Babbitt, C.J.; Pham, C.G.; Solaro, R.J.; Valencik, M.L.; Loftus, J.C.; Ross, R.S. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am. J. Pathol. 2001, 158, 1079–1090. [Google Scholar] [CrossRef]
- Shai, S.-Y.; Harpf, A.E.; Babbitt, C.J.; Jordan, M.C.; Fishbein, M.C.; Chen, J.; Omura, M.; Leil, T.A.; Becker, K.D.; Jiang, M.; et al. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 2002, 90, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.L.; Yang, J.T.; Hynes, R.O. Mesodermal defects and cranial neural crest apoptosis in alpha5 integrin-null embryos. Development 1997, 124, 4309–4319. [Google Scholar] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development 1993, 119, 1093–1105. [Google Scholar] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 1995, 121, 549–560. [Google Scholar] [PubMed]
- Sujkowski, A.; Wessells, R. Drosophila models of cardiac aging and disease. In Life Extension; Springer: Cham, Switzerland, 2015; pp. 127–150. [Google Scholar]
- Bier, E.; Bodmer, R. Drosophila, an emerging model for cardiac disease. Gene 2004, 342, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.; Eisenberg, C.; Eisenberg, L.; Trusk, T.; Sugi, Y. Epithelial-mesenchymal transformations in early avian heart development. Acta Anatomica 1996, 156, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Medioni, C.; Sénatore, S.; Salmand, P.-A.; Lalevée, N.; Perrin, L.; Sémériva, M. The fabulous destiny of the Drosophila heart. Curr. Opin. Genet. Dev. 2009, 19, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.; Jacobs, J.R. Dissecting the role of the extracellular matrix in heart disease: Lessons from the Drosophila genetic model. Vet. Sci. 2017, 4, 24. [Google Scholar] [CrossRef]
- Vanderploeg, J.; Vazquez Paz, L.L.; MacMullin, A.; Jacobs, J.R. Integrins are required for cardioblast polarisation in Drosophila. BMC Dev. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Morse, E.M.; Brahme, N.N.; Calderwood, D.A. Integrin cytoplasmic tail interactions. Biochemistry 2014, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Vanderploeg, J.; Jacobs, J.R. Talin is required to position and expand the luminal domain of the Drosophila heart tube. Dev. Biol. 2015, 405, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Raza, Q.; Jacobs, J.R. Guidance signalling regulates leading edge behaviour during collective cell migration of cardiac cells in Drosophila. Dev. Biol. 2016, 419, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Chen, Y.-C.; Kuo, C.-T.; Wenshin Yu, H.; Chen, Y.-Q.; Chiou, A.; Kuo, J.-C. Focal adhesion kinase-dependent focal adhesion recruitment of SH2 domains directs SRC into focal adhesions to regulate cell adhesion and migration. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kratimenos, P.; Koutroulis, I.; Marconi, D.; Syriopoulou, V.; Delivoria-Papadopoulos, M.; Chrousos, G.P.; Theocharis, S. Multi-targeted molecular therapeutic approach in aggressive neuroblastoma: The effect of Focal Adhesion Kinase-Src-Paxillin system. Expert Opin. Ther. Targets 2014, 18, 1395–1406. [Google Scholar] [PubMed]
- Wu, Y.; Zhang, K.; Seong, J.; Fan, J.; Chien, S.; Wang, Y.; Lu, S. In-situ coupling between kinase activities and protein dynamics within single focal adhesions. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Willey, C.D.; Balasubramanian, S.; Rodríguez Rosas, M.C.; Ross, R.S.; Kuppuswamy, D. Focal complex formation in adult cardiomyocytes is accompanied by the activation of beta3 integrin and c-Src. J. Mol. Cell. Cardiol. 2003, 35, 671–683. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, D.; Wang, X.; Wan, J.; Liu, C.; Zhang, H. Kindlin-2 phosphorylation by Src at Y193 enhances Src activity and is involved in Migfilin recruitment to the focal adhesions. FEBS Lett. 2015, 589, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Tu, Y.; Guan, J.-L.; Xiao, G.; Wu, C. Kindlin-2 tyrosine phosphorylation and interaction with Src serve as a regulatable switch in the integrin outside-in signaling circuit. J. Biol. Chem. 2014, 289, 31001–31013. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gong, H.; Jiang, G.; Ye, Y.; Wu, J.; You, J.; Zhang, G.; Sun, A.; Komuro, I.; Ge, J.; et al. Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent β-arrestin2 pathways. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Takahashi, F.; Ui-Tei, K.; Kojima, T.; Saigo, K. Requirements of genetic interactions between Src42A, armadillo and shotgun, a gene encoding E-cadherin, for normal development in Drosophila. Development 2005, 132, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, L.G.; Stewart, R.A.; Li, D.-M.; Xu, T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene 2004, 23, 4754–4762. [Google Scholar] [CrossRef] [PubMed]
- Ranganayakulu, G.; Schulz, R.A.; Olson, E.N. Wingless signaling induces nautilus expression in the ventral mesoderm of the Drosophila embryo. Dev. Biol. 1996, 176, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, N.; McMillan, S.C.; Chaudhary, R.; Mok, J.; Reed, B.H. Autophagy promotes caspase-dependent cell death during Drosophila development. Autophagy 2009, 5, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Millard, T.H.; Martin, P. Dynamic analysis of filopodial interactions during the zippering phase of Drosophila dorsal closure. Development 2008, 135, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Tokusumi, T.; Russell, M.; Gajewski, K.; Fossett, N.; Schulz, R.A. U-shaped protein domains required for repression of cardiac gene expression in Drosophila. Differentiation 2007, 75, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell Biol. 1994, 44, 445–487. [Google Scholar] [PubMed]
- MacMullin, A.; Jacobs, J.R. Slit coordinates cardiac morphogenesis in Drosophila. Dev. Biol. 2006, 293, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Reed, B.H.; McMillan, S.C.; Chaudhary, R. The preparation of Drosophila embryos for live-imaging using the hanging drop protocol. J. Vis. Exp. 2009, 25. [Google Scholar] [CrossRef] [PubMed]
- Muliyil, S.; Krishnakumar, P.; Narasimha, M. Spatial, temporal and molecular hierarchies in the link between death, delamination and dorsal closure. Development 2011, 138, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.L.; Rebay, I.; Hynes, R.O. Expression of DFak56, a Drosophila homolog of vertebrate focal adhesion kinase, supports a role in cell migration in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 14978–14983. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, J.; Sawamoto, K.; Okabe, M.; Takagi, Y.; Tezuka, T.; Yoshikawa, S.; Ryo, H.; Okano, H.; Yamamoto, T. Cloning and characterization of Dfak56, a homolog of focal adhesion kinase, in Drosophila melanogaster. J. Biol. Chem. 1999, 274, 29196–29201. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Fessler, L.I.; Edeen, P.T.; Madigan, S.J.; McKeown, M.; Hunter, T. DFak56 is a novel Drosophila melanogaster focal adhesion kinase. J. Biol. Chem. 1999, 274, 35621–35629. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-I.; Kao, H.-H.; Grabbe, C.; Lee, Y.-T.; Ghose, A.; Lai, T.-T.; Peng, K.-P.; Van Vactor, D.; Palmer, R.H.; Chen, R.-H.; et al. Fak56 functions downstream of integrin alphaPS3betanu and suppresses MAPK activation in neuromuscular junction growth. Neural Dev. 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, C.; Zervas, C.G.; Hunter, T.; Brown, N.H.; Palmer, R.H. Focal adhesion kinase is not required for integrin function or viability in Drosophila. Development 2004, 131, 5795–5805. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix—Cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Brand, A.H. The GAL4 system: A versatile system for the expression of genes. Methods Mol. Biol. 2008, 420, 79–95. [Google Scholar] [PubMed]
- Takahashi, F.; Endo, S.; Kojima, T.; Saigo, K. Regulation of cell-cell contacts in developing Drosophila eyes by Dsrc41, a new, close relative of vertebrate c-src. Genes Dev. 1996, 10, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.W.; McLean, G.W.; Wyke, A.W.; Paraskeva, C.; Parkinson, E.K.; Frame, M.C.; Brunton, V.G. The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol. Biol. Cell 2000, 11, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tateno, M.; Nishida, Y.; Adachi-Yamada, T. Regulation of JNK by Src during Drosophila development. Science 2000, 287, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Raza, Q.S.; Vanderploeg, J.; Jacobs, J.R. Matrix Metalloproteinases are required for membrane motility and lumenogenesis during Drosophila heart development. PLOS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, N.; Ordan, E.; Ocorr, K.; Bodmer, R.; Volk, T. Multiplexin promotes heart but not aorta morphogenesis by polarized enhancement of slit/robo activity at the heart lumen. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Hyun, C.; Park, I.-C. Congenital heart diseases in small animals: Part II. Potential genetic aetiologies based on human genetic studies. Vet. J. 2006, 171, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Bodmer, R. Probing the polygenic basis of cardiomyopathies in Drosophila. J. Cell. Mol. Med. 2012, 16, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Hashima, J.N.; Rogers, V.; Langley, S.M.; Ashraf, M.; Sahn, D.J.; Ohtonen, P.; Davis, L.E.; Hohimer, A.R.; Rasanen, J. Fetal ventricular interactions and wall mechanics during ductus arteriosus occlusion in a sheep model. Ultrasound Med. Biol. 2015, 41, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Hofstadler, G.; Tulzer, G.; Altmann, R.; Schmitt, K.; Danford, D.; Huhta, J.C. Spontaneous closure of the human fetal ductus arteriosus—A cause of fetal congestive heart failure. Am. J. Obstet. Gynecol. 1996, 174, 879–883. [Google Scholar] [CrossRef]
- Weichert, J.; Hartge, D.R.; Axt-Fliedner, R. The fetal ductus arteriosus and its abnormalities—A review. Congenit. Heart Dis. 2010, 5, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Tananari, Y.; Maeno, Y.; Takagishi, T.; Sasaguri, Y.; Morimatsu, M.; Kato, H. Role of apoptosis in the closure of neonatal ductus arteriosus. Jpn. Circ. J. 2000, 64, 684–688. [Google Scholar] [CrossRef] [PubMed]
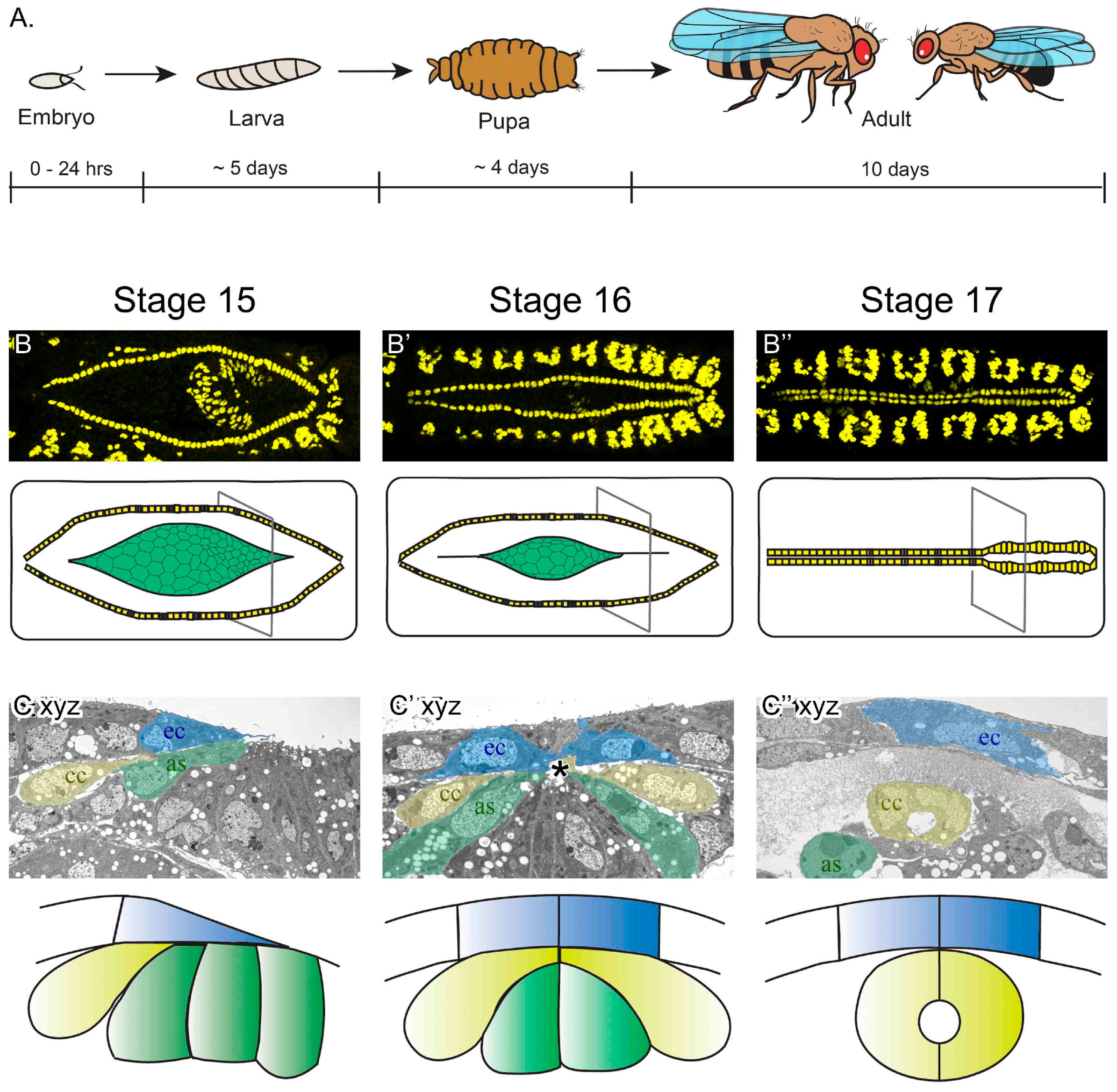
Figure 1.
Drosophila melanogaster embryonic heart development. (A) Although the lifecycle of flies, from egg-laying to adult is approximately 10 days at 25 °C, embryogenesis completes in just 24 hours. It is during embryogenesis that the heart develops into a linear tubular organ. (B–B’’) Images of the dorsal surface of fixed embryos imaged with a confocal fluorescent microscope (top panels are labeled for dMEF, present in the nuclei of somatic and heart muscle). By embryonic stage 15, the heart cells (yellow in schematic) are aligned in two lateral rows, flanking the amnioserosa tissue (green in schematic). As embryogenesis continues, the cardioblast cells (CBs) migrate towards the dorsal midline (stage 15–17). By the end of stage 17, the embryonic heart is a mature vessel, capable of pumping hemolymph throughout the body. The slightly wider heart vessel is at the posterior (to the right), while the outflow tract extends anteriorly (to the left). (C–C’’) In transverse sections of fixed embryos imaged using an electron microscope, migrating cardial cells (yellow) follow the dorsal ectoderm leading edge (blue) and contact the amnioserosa (green) ventrally (stage 15). As CBs approach the midline, they migrate between the ectoderm and amnioserosa to reach their contralateral partner at the midline (asterisk, stage 16). Once at the midline, the CBs adopt a crescent-shape morphology and enclose the lumen (stage 17). At this stage, dorsal closure has completed; the ectoderm covers the dorsal surface of the embryo and the amnioserosa has invaginated and the cells are undergoing apoptosis. Dorsal is at the top of C–C’’ as, amnioserosa; ec, ectoderm; cc, cardial cells.
Figure 1.
Drosophila melanogaster embryonic heart development. (A) Although the lifecycle of flies, from egg-laying to adult is approximately 10 days at 25 °C, embryogenesis completes in just 24 hours. It is during embryogenesis that the heart develops into a linear tubular organ. (B–B’’) Images of the dorsal surface of fixed embryos imaged with a confocal fluorescent microscope (top panels are labeled for dMEF, present in the nuclei of somatic and heart muscle). By embryonic stage 15, the heart cells (yellow in schematic) are aligned in two lateral rows, flanking the amnioserosa tissue (green in schematic). As embryogenesis continues, the cardioblast cells (CBs) migrate towards the dorsal midline (stage 15–17). By the end of stage 17, the embryonic heart is a mature vessel, capable of pumping hemolymph throughout the body. The slightly wider heart vessel is at the posterior (to the right), while the outflow tract extends anteriorly (to the left). (C–C’’) In transverse sections of fixed embryos imaged using an electron microscope, migrating cardial cells (yellow) follow the dorsal ectoderm leading edge (blue) and contact the amnioserosa (green) ventrally (stage 15). As CBs approach the midline, they migrate between the ectoderm and amnioserosa to reach their contralateral partner at the midline (asterisk, stage 16). Once at the midline, the CBs adopt a crescent-shape morphology and enclose the lumen (stage 17). At this stage, dorsal closure has completed; the ectoderm covers the dorsal surface of the embryo and the amnioserosa has invaginated and the cells are undergoing apoptosis. Dorsal is at the top of C–C’’ as, amnioserosa; ec, ectoderm; cc, cardial cells.
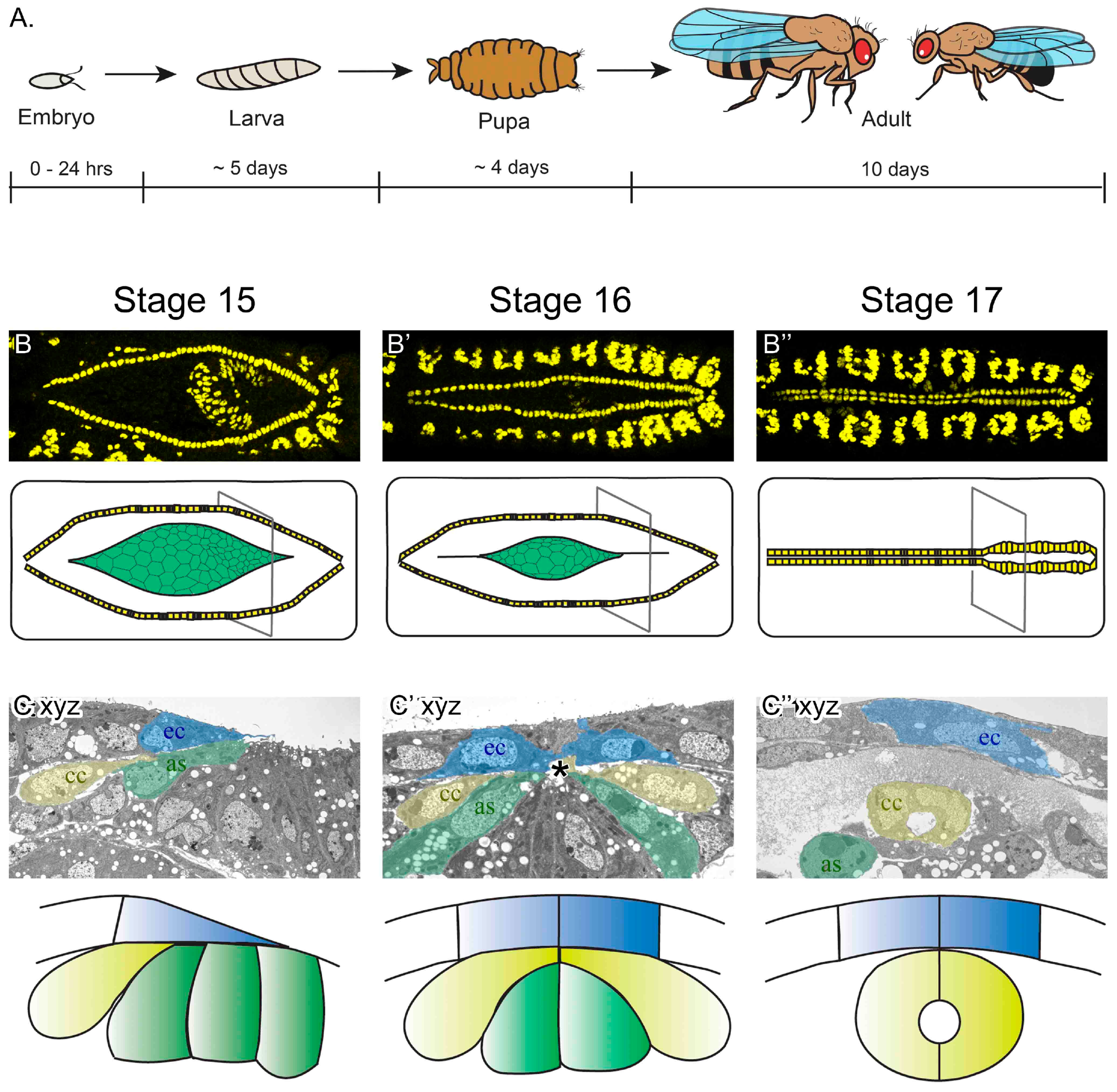
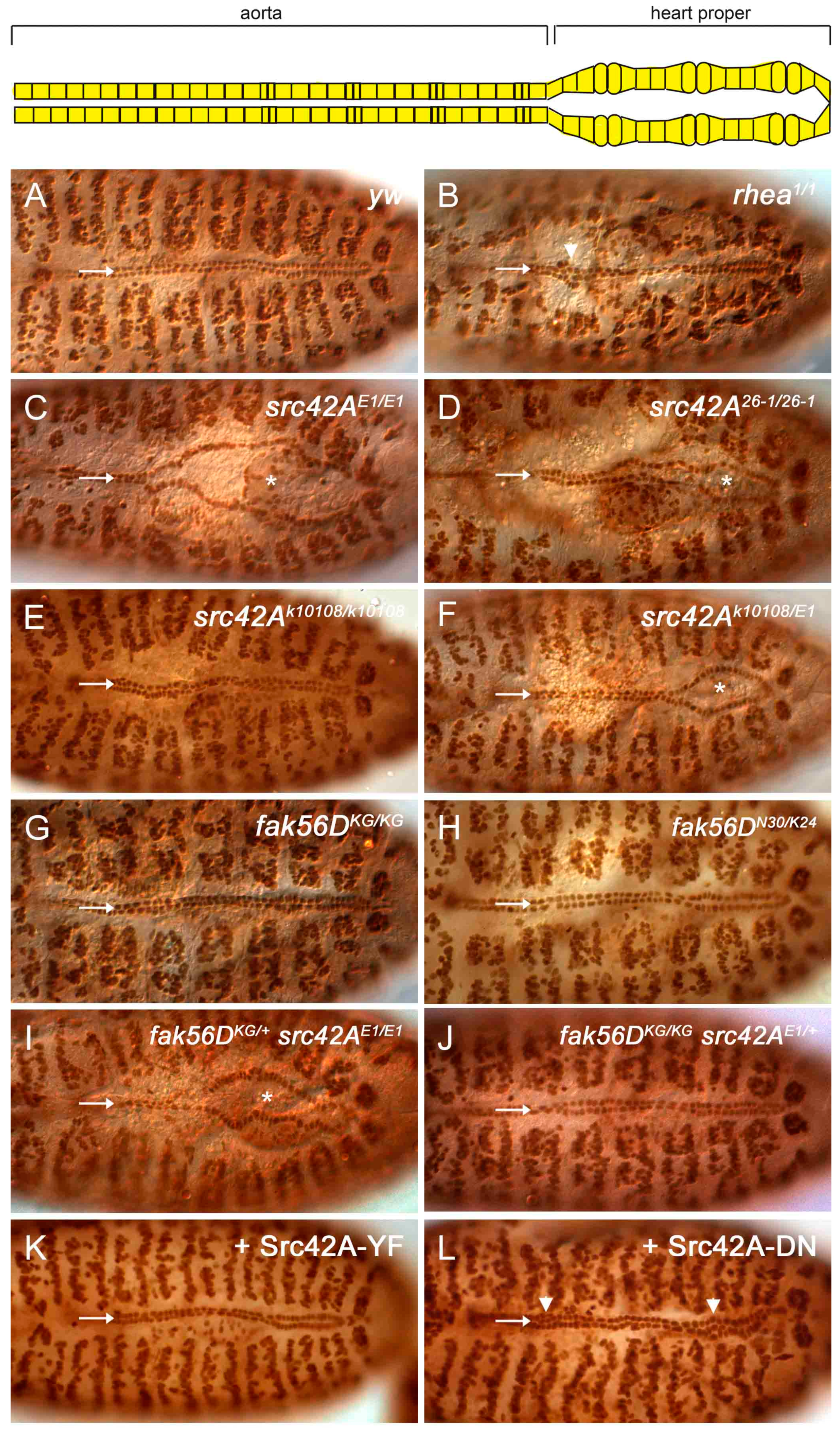
Figure 2.
Loss of Src42A results in incomplete cardioblast migration. (A) By stage 17, the two CB rows are aligned along the dorsal midline in wildtype embryos. The two rows are close, but a narrow gap remains at the midline where the tube lumen is positioned (arrow). (B) In mutants of rhea, the gene encoding the Integrin-adaptor protein Talin, migration completes. However, occasional cell clumping occurs (arrowhead) and the space between the CBs rows is absent (arrow; [29]). (C–F) In src42AE1 and src42A26-1 null mutants, the posterior CBs fail to migrate to the midline (arrow) and the embryos exhibit an “open heart” phenotype (asterisks mark the open heart). The CBs in the src42Ak10108 hypomorph migrate fully to the midline (E), although an open heart phenotype is observed in some src42AE1/src42Ak10108 mutants (F, asterisks). (G–J) Similar to wildtype embryos (A), fak56D mutant embryos have normal CB alignment and migration (G, H). Reducing the gene copy of fak56D did not enhance the delay in CB migration in src42AE1 mutant embryos (I) Reducing the gene copy of src42A did not induce a heart phenotype in fak56D mutant embryos (J). (K) Constitutively active Src42A was overexpressed in the heart and somatic muscles. Similar to wildtype, the heart cells were aligned, migrated to the midline, and maintained a narrow gap between the two rows. (L) Expression of a dominant negative form of Src42A kinase did not prevent CB migration. However, cells were frequently misplaced relative to neighboring cells, resulting in cell clumps (arrowhead). Posterior of the heart is to the right in all panels.
Figure 2.
Loss of Src42A results in incomplete cardioblast migration. (A) By stage 17, the two CB rows are aligned along the dorsal midline in wildtype embryos. The two rows are close, but a narrow gap remains at the midline where the tube lumen is positioned (arrow). (B) In mutants of rhea, the gene encoding the Integrin-adaptor protein Talin, migration completes. However, occasional cell clumping occurs (arrowhead) and the space between the CBs rows is absent (arrow; [29]). (C–F) In src42AE1 and src42A26-1 null mutants, the posterior CBs fail to migrate to the midline (arrow) and the embryos exhibit an “open heart” phenotype (asterisks mark the open heart). The CBs in the src42Ak10108 hypomorph migrate fully to the midline (E), although an open heart phenotype is observed in some src42AE1/src42Ak10108 mutants (F, asterisks). (G–J) Similar to wildtype embryos (A), fak56D mutant embryos have normal CB alignment and migration (G, H). Reducing the gene copy of fak56D did not enhance the delay in CB migration in src42AE1 mutant embryos (I) Reducing the gene copy of src42A did not induce a heart phenotype in fak56D mutant embryos (J). (K) Constitutively active Src42A was overexpressed in the heart and somatic muscles. Similar to wildtype, the heart cells were aligned, migrated to the midline, and maintained a narrow gap between the two rows. (L) Expression of a dominant negative form of Src42A kinase did not prevent CB migration. However, cells were frequently misplaced relative to neighboring cells, resulting in cell clumps (arrowhead). Posterior of the heart is to the right in all panels.
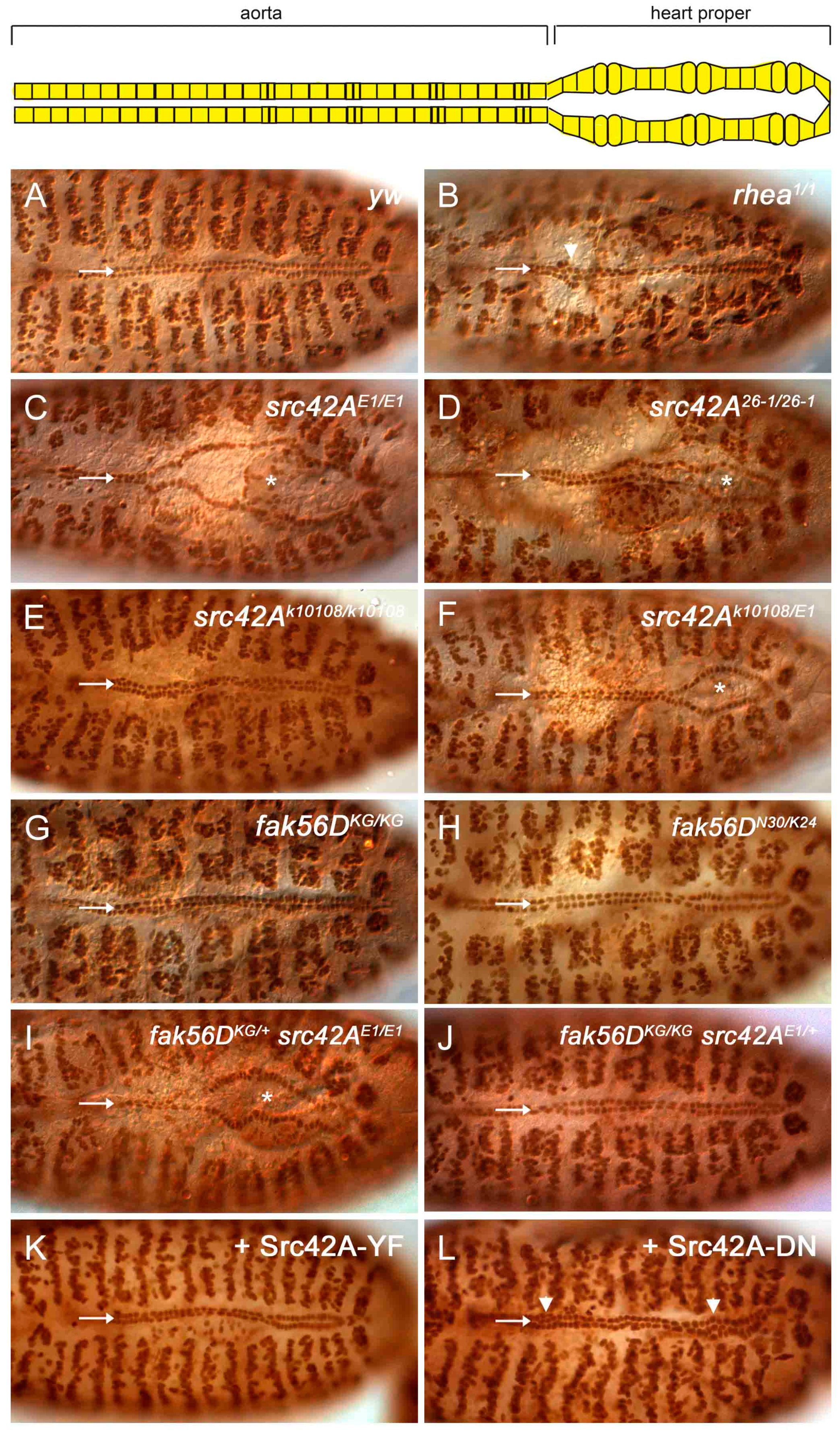
Figure 3.
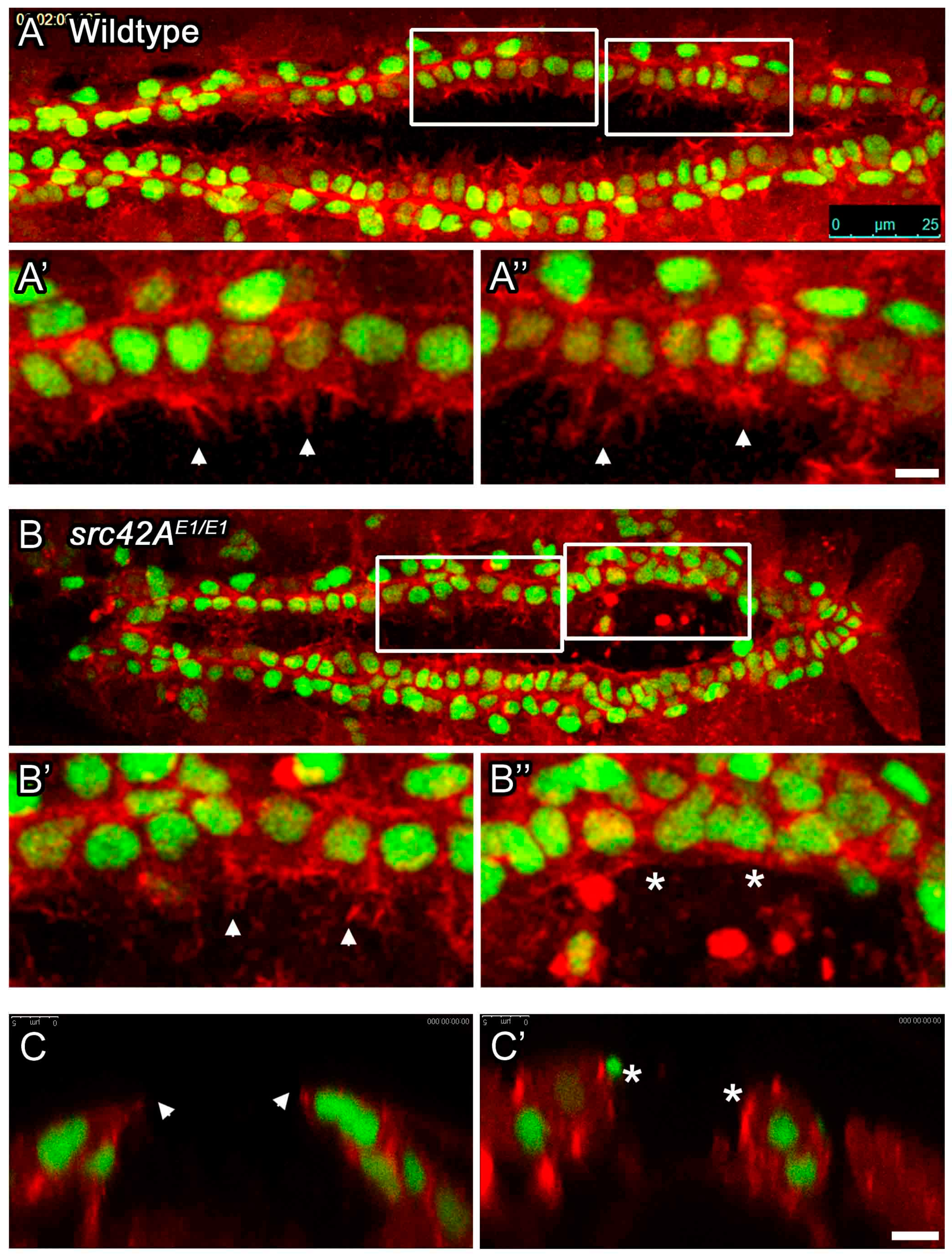
In src42A mutant embryos, cardioblasts with stalled migration lack leading edge membrane activity. The dynamic activity of the leading edge of migrating CBs was monitored in living embryos. Membrane actin was labeled with dMEF-GAL4 regulated UAS-moesin-mCherry (red), while CBs were marked with tup-GFP (green). In wildtype embryos (A), both the anterior and posterior CBs extend highly dynamic processes towards the midline (arrowheads in A’ and A’’). In src42AE1 mutants, anterior CBs extend processes towards the midline (arrowhead in B’, cross-section in C), but posterior CBs remain rounded (C’) and do not extend such processes (asterisks in B’’, C’). Posterior of the heart is to the right (A, B); dorsal at top (C, C’). Calibration: 25 microns in A and B, 5 microns in all other panels.
Figure 3.
In src42A mutant embryos, cardioblasts with stalled migration lack leading edge membrane activity. The dynamic activity of the leading edge of migrating CBs was monitored in living embryos. Membrane actin was labeled with dMEF-GAL4 regulated UAS-moesin-mCherry (red), while CBs were marked with tup-GFP (green). In wildtype embryos (A), both the anterior and posterior CBs extend highly dynamic processes towards the midline (arrowheads in A’ and A’’). In src42AE1 mutants, anterior CBs extend processes towards the midline (arrowhead in B’, cross-section in C), but posterior CBs remain rounded (C’) and do not extend such processes (asterisks in B’’, C’). Posterior of the heart is to the right (A, B); dorsal at top (C, C’). Calibration: 25 microns in A and B, 5 microns in all other panels.
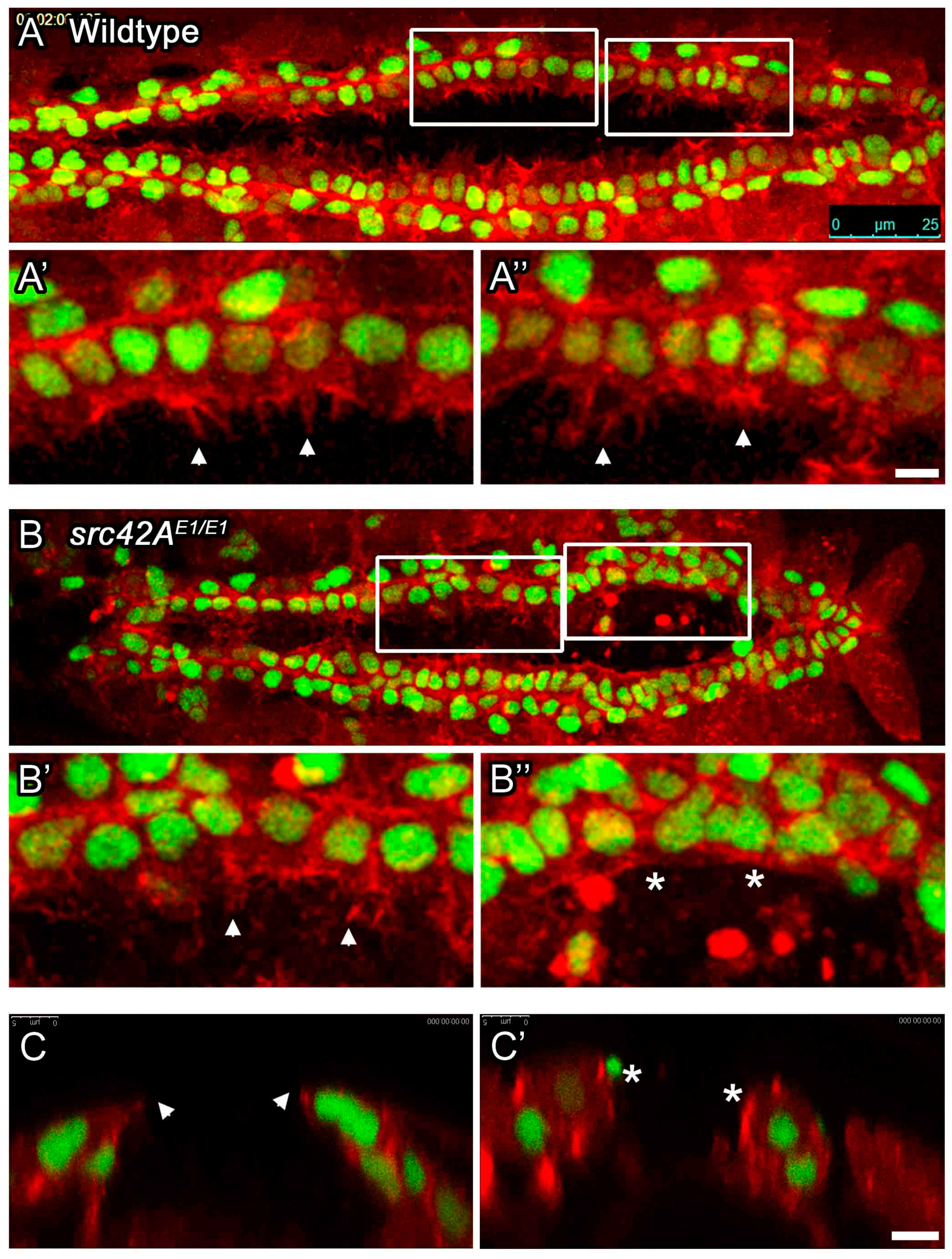
Figure 4.
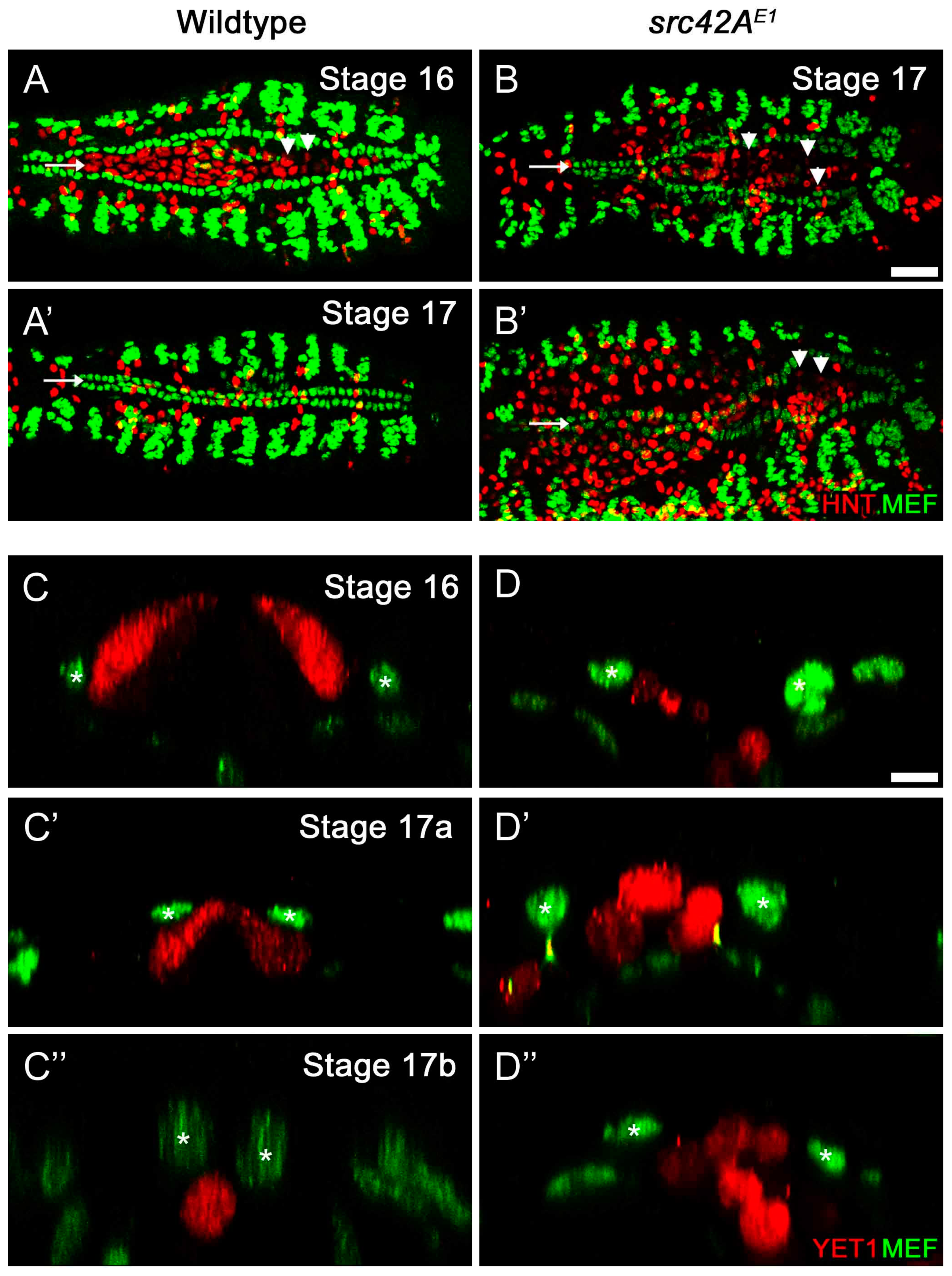
Amnioserosa cells remain at the midline in late stage src42AE1 embryos. (A–B’) In wildtype embryos, the amnioserosa (red) is present at stage 16 (A) but has internalized and undergone apoptosis by the end of embryogenesis (stage 17, A’). In src42AE1 mutants, amnioserosa cells persist in the posterior region of the embryo even at late stages (B–B’; arrowheads identify example amnioserosa cells; brightly red labeled cells in left of B’ are part of the midgut). The nuclei of amnioserosa cells are labeled with αHindsight (HNT, red). The CB nuclei (and somatic muscle nuclei) are labeled with αMEF (green). Posterior of the heart is to the right. Calibration: 25 microns. (C–D’’) Transverse sections through the heart and amnioserosa tissue of wildtype and src42AE1 mutant embryos. In wildtype embryos, migrating CBs (green, asterisks) move dorsally and eventually over the amnioserosa cells (red; C–C’’). Following migration, no amnioserosa cells are between the CBs; any remaining cells have been internalized (C’’). In contrast, late stage src42AE1 embryos are characterized by persistent amnioserosa cells at the midline (D–D’’). Despite having the rounded shape characteristic of cells undergoing apoptosis, the amnioserosa cells do not internalize, but remain along the midline where the heart tube usually forms. CB nuclei (and flanking muscle nuclei in C’ and C’’) are labeled with αMEF (green, asterisks), while YET1 marks the outermost amnioserosa cells (red). Dorsal is at the top. Calibration: 5 microns.
Figure 4.
Amnioserosa cells remain at the midline in late stage src42AE1 embryos. (A–B’) In wildtype embryos, the amnioserosa (red) is present at stage 16 (A) but has internalized and undergone apoptosis by the end of embryogenesis (stage 17, A’). In src42AE1 mutants, amnioserosa cells persist in the posterior region of the embryo even at late stages (B–B’; arrowheads identify example amnioserosa cells; brightly red labeled cells in left of B’ are part of the midgut). The nuclei of amnioserosa cells are labeled with αHindsight (HNT, red). The CB nuclei (and somatic muscle nuclei) are labeled with αMEF (green). Posterior of the heart is to the right. Calibration: 25 microns. (C–D’’) Transverse sections through the heart and amnioserosa tissue of wildtype and src42AE1 mutant embryos. In wildtype embryos, migrating CBs (green, asterisks) move dorsally and eventually over the amnioserosa cells (red; C–C’’). Following migration, no amnioserosa cells are between the CBs; any remaining cells have been internalized (C’’). In contrast, late stage src42AE1 embryos are characterized by persistent amnioserosa cells at the midline (D–D’’). Despite having the rounded shape characteristic of cells undergoing apoptosis, the amnioserosa cells do not internalize, but remain along the midline where the heart tube usually forms. CB nuclei (and flanking muscle nuclei in C’ and C’’) are labeled with αMEF (green, asterisks), while YET1 marks the outermost amnioserosa cells (red). Dorsal is at the top. Calibration: 5 microns.

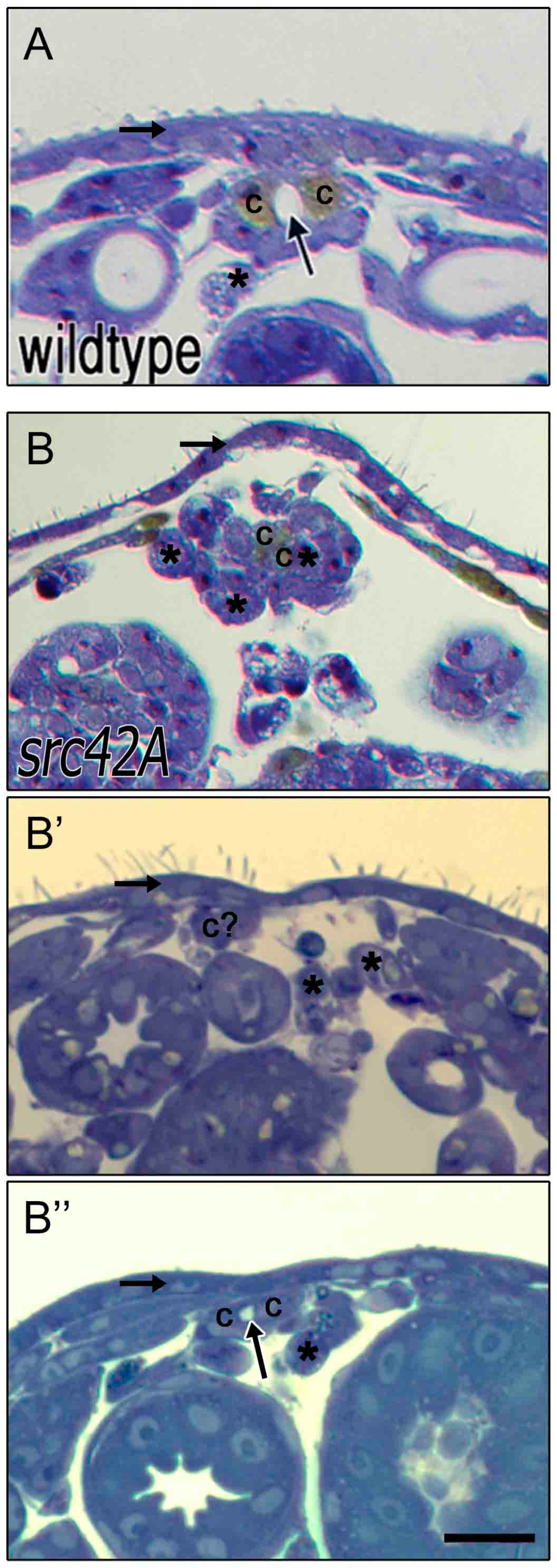
Figure 5.
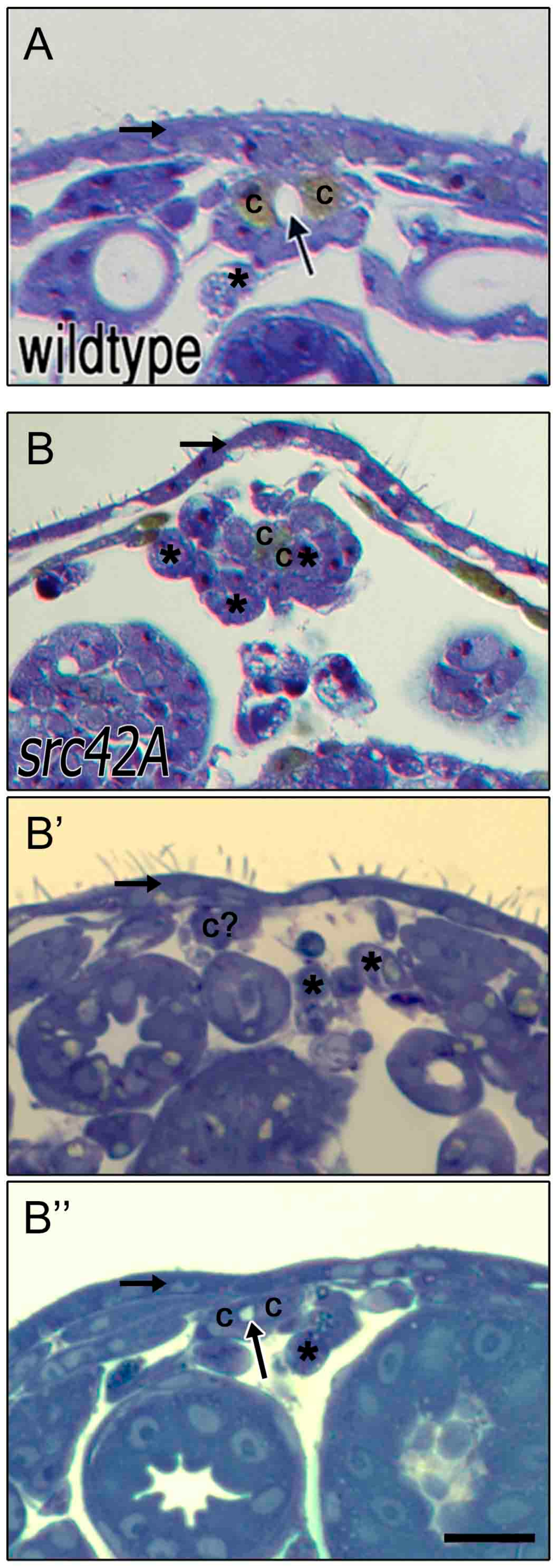
Cardioblasts at the midline are capable of developing an open lumen between contralateral cells. In stage 17 wildtype embryos (A), contralateral CBs (“c”) are present at the dorsal midline and enclose a lumen (arrow). Dorsal to the heart is a thick and continuous ectoderm (horizontal arrow) and few if any amnioserosa cells remain ventrally (asterisks). In src42AE1 mutants, the ectoderm layer is thin and variable (horizontal arrow in B–B’’) and amnioserosa cells persist at or near the midline (asterisks; for some cells marked with an asterisks, identity is uncertain). The CBs are not consistently present at the midline (e.g., c? in B’); however, when two contralateral CBs are present, a small lumen often forms (arrow in B’’). Dorsal at top. Calibration: 10 microns.
Figure 5.
Cardioblasts at the midline are capable of developing an open lumen between contralateral cells. In stage 17 wildtype embryos (A), contralateral CBs (“c”) are present at the dorsal midline and enclose a lumen (arrow). Dorsal to the heart is a thick and continuous ectoderm (horizontal arrow) and few if any amnioserosa cells remain ventrally (asterisks). In src42AE1 mutants, the ectoderm layer is thin and variable (horizontal arrow in B–B’’) and amnioserosa cells persist at or near the midline (asterisks; for some cells marked with an asterisks, identity is uncertain). The CBs are not consistently present at the midline (e.g., c? in B’); however, when two contralateral CBs are present, a small lumen often forms (arrow in B’’). Dorsal at top. Calibration: 10 microns.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vanderploeg, J.; Jacobs, J.R. Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila. Vet. Sci. 2017, 4, 23. https://doi.org/10.3390/vetsci4020023
AMA Style
Vanderploeg J, Jacobs JR. Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila. Veterinary Sciences. 2017; 4(2):23. https://doi.org/10.3390/vetsci4020023
Chicago/Turabian StyleVanderploeg, Jessica, and J. Roger Jacobs. 2017. "Mapping Heart Development in Flies: Src42A Acts Non-Autonomously to Promote Heart Tube Formation in Drosophila" Veterinary Sciences 4, no. 2: 23. https://doi.org/10.3390/vetsci4020023
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.