
Manipulation of Innate Immunity for Cancer Therapy in Dogs

1
Flint Animal Cancer Center, Department of Clinical Sciences, Colorado State University, Ft. Collins, CO 80525, USA
2
The Center for Immune and Regenerative Medicine, Department of Clinical Sciences, Colorado State University, Ft. Collins, CO 80525, USA
*
Author to whom correspondence should be addressed.
Vet. Sci. 2015, 2(4), 423-439; https://doi.org/10.3390/vetsci2040423
Submission received: 9 October 2015
/
Revised: 20 November 2015
/
Accepted: 23 November 2015
/
Published: 1 December 2015
(This article belongs to the Special Issue Comparative Pathogenesis of Cancers in Animals and Humans)
Abstract
:Over the last one to two decades, the field of cancer immunotherapy has rapidly progressed from early preclinical studies to a successful clinical reality and fourth major pillar of human cancer therapy. While current excitement in the field of immunotherapy is being driven by several major breakthroughs including immune checkpoint inhibitors and adoptive cell therapies, these advances stem from a foundation of pivotal studies demonstrating the immune systems role in tumor control and eradication. The following will be a succinct review on veterinary cancer immunotherapy as it pertains to manipulation of the innate immune system to control tumor growth and metastasis. In addition, we will provide an update on recent progress in our understanding of the innate immune system in veterinary tumor immunology, and how these gains may lead to novel therapies for the treatment of cancer in companion animals.
1. Introduction
Immunotherapy for cancer has rapidly moved from a research concept to successful clinical reality in cancer treatment for humans in the span of just 10 years [1,2,3,4,5,6]. Several major breakthroughs are driving the current excitement around cancer immunotherapy, including the discovery that blockade of immune checkpoint molecules can stimulate durable tumor remissions, as well as the administration of engineered T cells designed to target specific antigens on tumor cells. In addition, our understanding of the critical role of the innate immune system in regulating adaptive immune responses to cancer has also increased rapidly [7,8,9]. Thus, we are poised now to add immunotherapy as a fourth major pillar of cancer therapy, in addition to surgery, chemotherapy, and radiation therapy. The following review will provide historical perspective on the use of biological response modifiers to activate innate immunity for tumor control, as well as discuss more recent studies using molecules targeting specific pathways in innate immune activation to induce non-specific anti-tumor immunity.
2. Dual Roles of Innate Immunity in Cancer Control
The innate immune system plays a much more nuanced role in cancer immunity than the adaptive immune system (T cells and B cells), which is considered in most cases to actively suppress tumor growth. It is now apparent that the innate immune system can in many cases suppress adaptive immune responses and remove immune checks on tumor growth (Figure 1) [1,2]. In addition, cells of the innate immune system, especially tumor-associated macrophages, can directly interact with tumor cells to stimulate tumor cell growth, genetic instability, and metastasis [3,4,5,6]. Macrophages can also modify the tumor microenvironment to enhance tumor growth, including increasing angiogenesis, stimulating local release of reactive oxygen species, and promoting tumor invasion through extracellular basement membranes. Inflammatory monocytes recruited to sites of early tumor metastases promote the early survival of metastatic tumor cells, in part through enhanced angiogenesis [7,8]. We have reported that increased numbers of circulating monocytes are associated with shorter survival times in dogs with osteosarcoma and animals with B cell lymphoma [9,10]. Thus, on balance macrophages and monocytes can be considered as major drivers of tumor growth and progression.
However, it is also clear that macrophages can be activated therapeutically to control tumor growth (Figure 2 and Figure 3). Earlier studies demonstrated that activated macrophages could kill tumor cells, primarily via the secretion of TNF-α. For example, canine alveolar macrophages activated by the Nod-like receptor (NLR) agonist muramyl tripeptide (MTP) were shown to kill canine osteosarcoma cells in vitro [11,12].Other immune activating molecules, including certain Toll-like receptor ligands (TLR), can also induce macrophage killing of tumor cells [13]. Administration of cytokines such as IL-12 and INF-γ can also activate macrophages to become tumoricidal.
A mixed population of immature myeloid cells (comprised primarily monocytes and neutrophils) collectively known as myeloid derived suppressor cells (MDSCs) contribute in a major way to global suppression of tumor immunity [14,15,16,17,18]. Large numbers of MDSCs are found in cancer patients and individuals with chronic infections [19,20,21]. Expanded circulating populations of MDSCs have been described in dogs with cancer [22,23]. MDSCs infiltrate the bone marrow and blood stream, as well as secondary lymphoid tissues (spleen and peripheral lymph nodes), and tumor tissues, where they potently suppress T cell and NK cell responses [14,15]. The mechanisms by which MDSCs suppress T cells vary by species, but include production of immune suppressive metabolites (e.g., reactive nitrogen and oxygen intermediates), production of immunologically active enzymes (arginase, indoleamine dioxygenase, aminopeptidases), nitrosylation of T cell receptors, production of immune suppressive cytokines (e.g., TGF-β, IL-10) and by production of immune suppressive prostaglandin E [24]. In dogs, MDSCs are reported to suppress T cell function by production of arginase, which leads to local depletion of arginine, an essential amino acid required for normal T cell function [22,23]. Myeloid derived suppressor cells are therefore very attractive targets for immunotherapeutic manipulation of both the innate and adaptive immune systems.
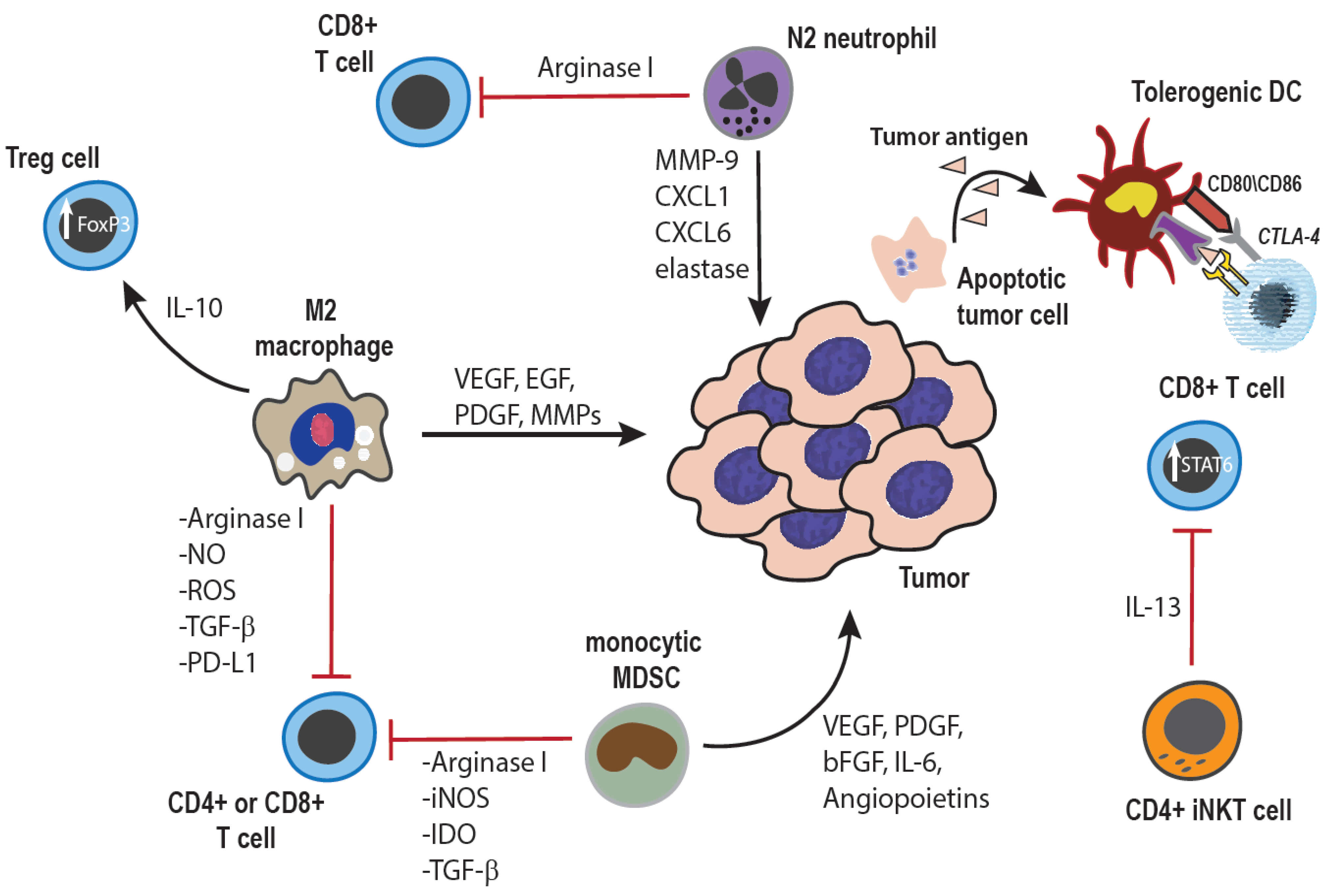
Figure 1.
Tumor-promoting effects of innate immune cells: Cells of the innate immune system can promote tumor growth through both direct interactions with tumor cells, as well as indirectly through modulation of the adaptive immune response. Macrophages within the tumor microenvironment can be polarized towards an anti-inflammatory, M2 phenotype, and secrete cytokines, growth factors, and enzymes which promote angiogenesis as well as tumor cell survival, proliferation, invasion, and metastasis [3,25]. Additionally, both macrophages and monocytes can inhibit anti-tumor T cell responses through production of anti-inflammatory cytokines, induction of Treg cells, expression of negative co-stimulatory ligands such as PD-L1 or PD-L2, and depletion of extra-cellular arginine, an amino acid essential for normal T cell function and proliferation [7,14,25,26,27]. Similarly, tumor-associated neutrophils can also be polarized towards a tumor-promoting N2 phenotype, elaborating similar growth factors and pro-angiogenic cytokines, as well as suppressing T cell responses by mechanisms similar to those used by monocytes and macrophages [25,28]. The immune suppressive cytokine milieu within the tumor microenvironment can also inhibit DC maturation, resulting in tumor antigen-specific anergy of T cells [29]. Lastly, specific CD4+ subsets of invariant NK T cells are also known to produce immune suppressive cytokines, which inhibit CD8+ anti-tumor T cell responses [30].
Figure 1.
Tumor-promoting effects of innate immune cells: Cells of the innate immune system can promote tumor growth through both direct interactions with tumor cells, as well as indirectly through modulation of the adaptive immune response. Macrophages within the tumor microenvironment can be polarized towards an anti-inflammatory, M2 phenotype, and secrete cytokines, growth factors, and enzymes which promote angiogenesis as well as tumor cell survival, proliferation, invasion, and metastasis [3,25]. Additionally, both macrophages and monocytes can inhibit anti-tumor T cell responses through production of anti-inflammatory cytokines, induction of Treg cells, expression of negative co-stimulatory ligands such as PD-L1 or PD-L2, and depletion of extra-cellular arginine, an amino acid essential for normal T cell function and proliferation [7,14,25,26,27]. Similarly, tumor-associated neutrophils can also be polarized towards a tumor-promoting N2 phenotype, elaborating similar growth factors and pro-angiogenic cytokines, as well as suppressing T cell responses by mechanisms similar to those used by monocytes and macrophages [25,28]. The immune suppressive cytokine milieu within the tumor microenvironment can also inhibit DC maturation, resulting in tumor antigen-specific anergy of T cells [29]. Lastly, specific CD4+ subsets of invariant NK T cells are also known to produce immune suppressive cytokines, which inhibit CD8+ anti-tumor T cell responses [30].
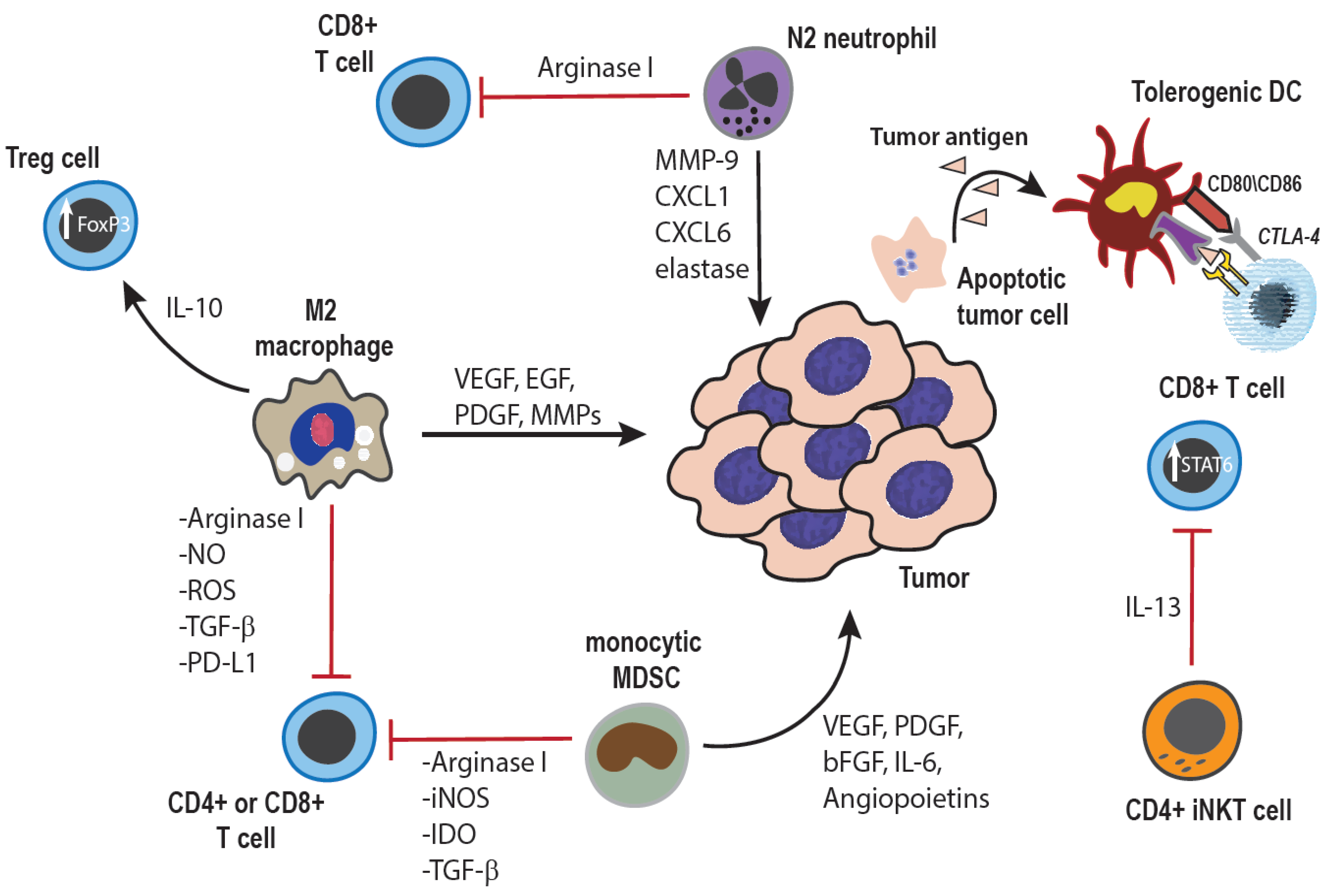
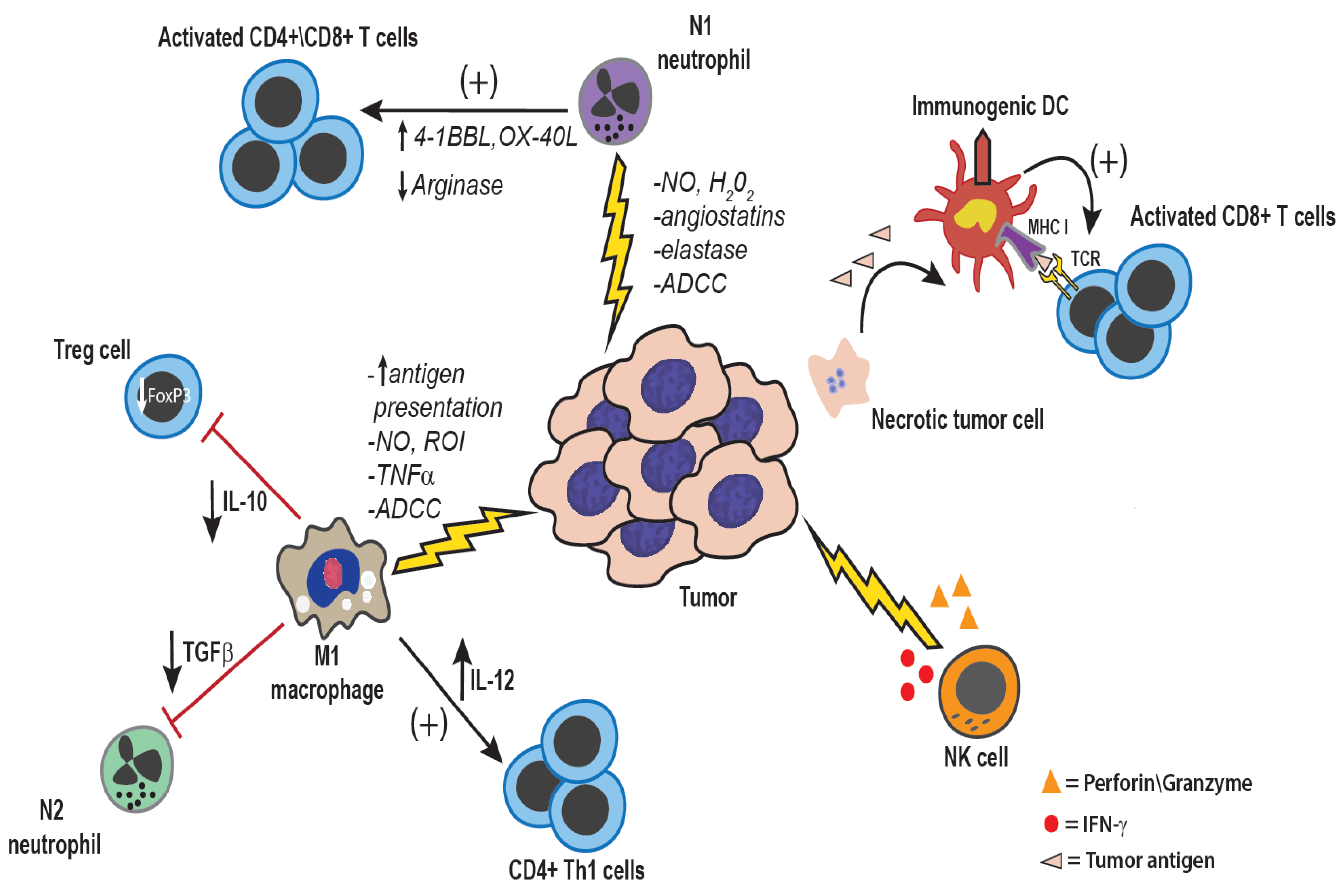
Figure 2.
Anti-tumor effects of innate immune cells: Both tumor-associated macrophages and neutrophils can be polarized to a more pro-inflammatory anti-tumor phenotype, either inherently within certain tumor types, or through therapeutic manipulation. Direct anti-tumor mechanisms of macrophages and neutrophils are mediated by production of reactive nitrogen and oxygen intermediates, cytokines such as TNF-α, and enzymes such as elastase [25,31]. Additionally, through the production of IL-12, macrophages can activate NK cells as well as induce a Th1 type anti-tumor immune response [32]. NK cells are also potent anti-tumor innate immune effector cells. NK cells are activated in response to reduced expression of MHC I and by ligation of activating receptors such as NKG2D [33]. NK cells mediate direct tumor cell killing via perforin and granzyme, or expression of FasL and TNF-related apoptosis-inducing ligand (TRAIL) [33]. Additionally, NK cells are an important source of IFN-γ within the tumor microenvironment, which serves to activate macrophages, and DCs, and up-regulated MHC I and MHC II expression on tumor cells and antigen-presenting cells, respectively [33].
Figure 2.
Anti-tumor effects of innate immune cells: Both tumor-associated macrophages and neutrophils can be polarized to a more pro-inflammatory anti-tumor phenotype, either inherently within certain tumor types, or through therapeutic manipulation. Direct anti-tumor mechanisms of macrophages and neutrophils are mediated by production of reactive nitrogen and oxygen intermediates, cytokines such as TNF-α, and enzymes such as elastase [25,31]. Additionally, through the production of IL-12, macrophages can activate NK cells as well as induce a Th1 type anti-tumor immune response [32]. NK cells are also potent anti-tumor innate immune effector cells. NK cells are activated in response to reduced expression of MHC I and by ligation of activating receptors such as NKG2D [33]. NK cells mediate direct tumor cell killing via perforin and granzyme, or expression of FasL and TNF-related apoptosis-inducing ligand (TRAIL) [33]. Additionally, NK cells are an important source of IFN-γ within the tumor microenvironment, which serves to activate macrophages, and DCs, and up-regulated MHC I and MHC II expression on tumor cells and antigen-presenting cells, respectively [33].
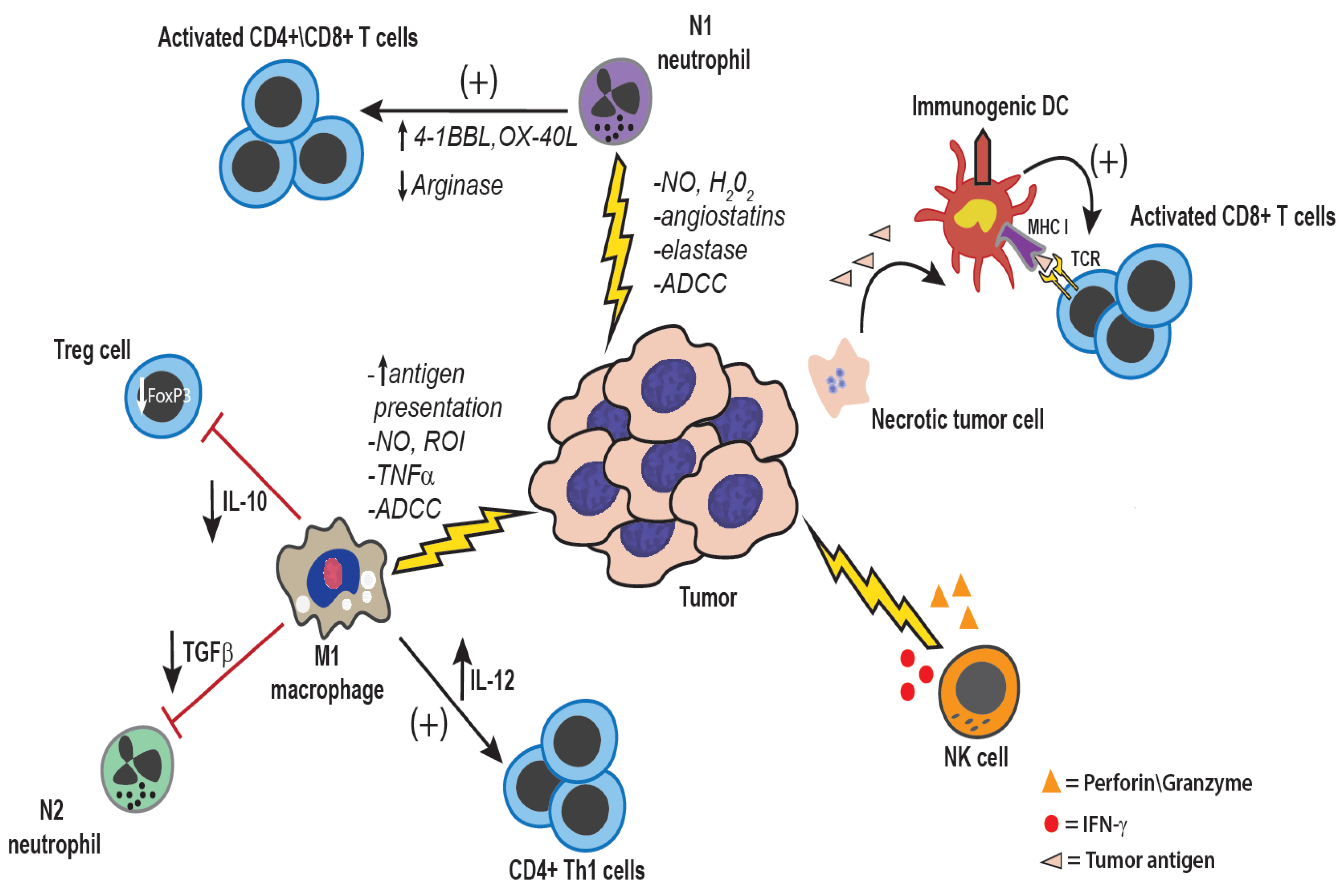
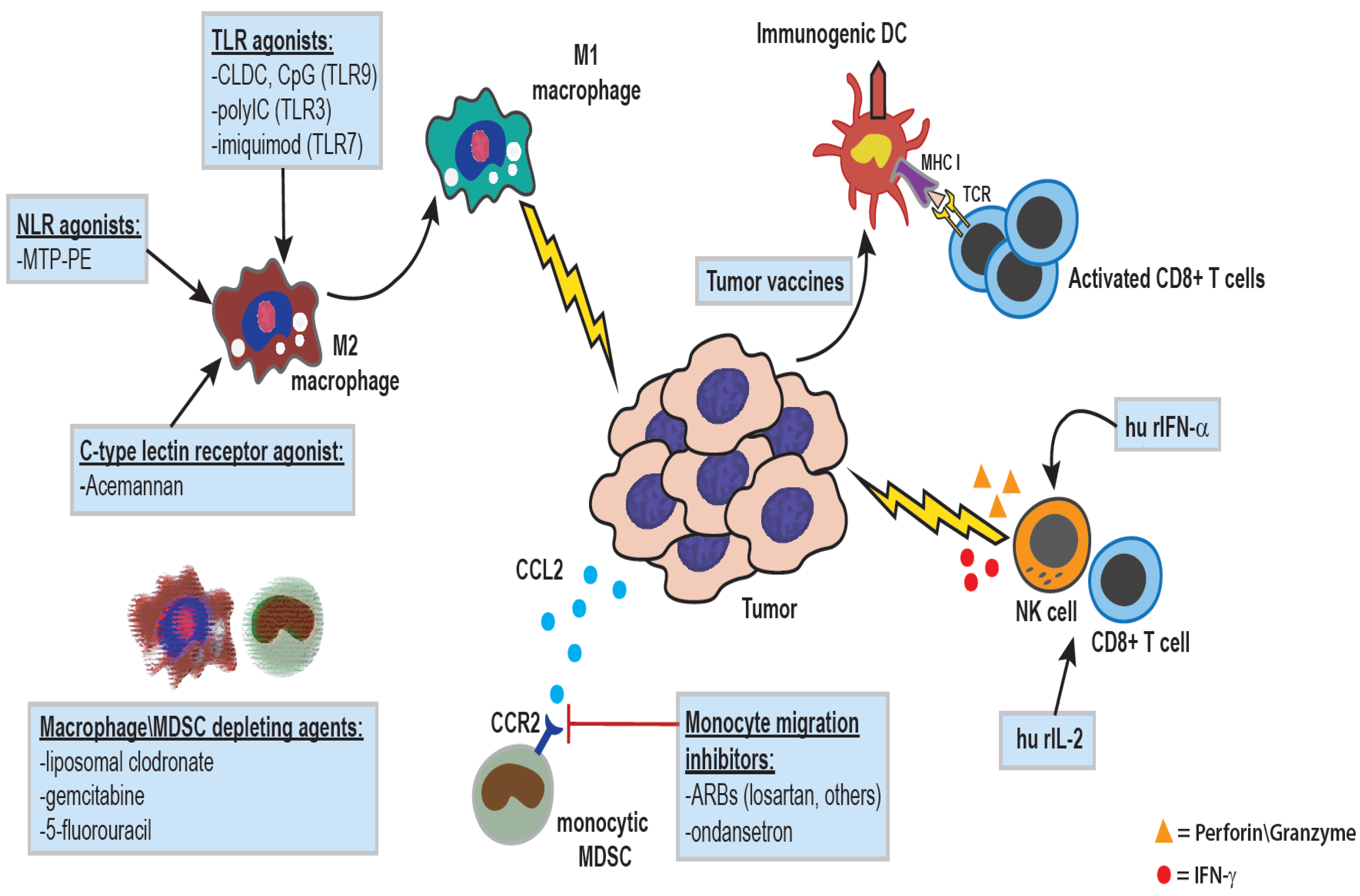
Figure 3.
Therapeutic manipulations of the innate immune system for treatment of cancer: The administration of agonists for various pattern-recognition receptors, including Toll-like receptors (cationic liposome-DNA complexes (CLDC), pIC, or imiquimod), Nod-like receptors (liposomal muramyl tripeptide), or lectin receptors (acemannan), can result in macrophage activation and polarization towards a pro-inflammatory anti-tumor phenotype. IL-2 is a potent activator of NK and T cells, and human recombinant IL-2 has been used in the treatment of multiple canine cancer types including melanoma, metastatic osteosarcoma, lymphoma, and soft tissue sarcoma. Type I interferons such as IFN-α serve to activate and enhance DC maturation, and increase cytotoxicity of CD8+ T cells and NK cells, and recombinant human IFN-α has been administered to dogs with various epithelial neoplasms. Macrophages and monocytes can also be targeted with various drugs as a means of augmenting tumor angiogenesis and restoring anti-tumor immunity. Drugs such as liposomal clodronate or conventional chemotherapeutics like gemcitabine and 5-fluorouracil can result in systemic depletion of macrophages\monocytes [34], while work in our lab has shown that small molecules drugs such as ondansetron, and angiotensin-receptor blockers like losartan, can function to inhibit monocyte migration.
Figure 3.
Therapeutic manipulations of the innate immune system for treatment of cancer: The administration of agonists for various pattern-recognition receptors, including Toll-like receptors (cationic liposome-DNA complexes (CLDC), pIC, or imiquimod), Nod-like receptors (liposomal muramyl tripeptide), or lectin receptors (acemannan), can result in macrophage activation and polarization towards a pro-inflammatory anti-tumor phenotype. IL-2 is a potent activator of NK and T cells, and human recombinant IL-2 has been used in the treatment of multiple canine cancer types including melanoma, metastatic osteosarcoma, lymphoma, and soft tissue sarcoma. Type I interferons such as IFN-α serve to activate and enhance DC maturation, and increase cytotoxicity of CD8+ T cells and NK cells, and recombinant human IFN-α has been administered to dogs with various epithelial neoplasms. Macrophages and monocytes can also be targeted with various drugs as a means of augmenting tumor angiogenesis and restoring anti-tumor immunity. Drugs such as liposomal clodronate or conventional chemotherapeutics like gemcitabine and 5-fluorouracil can result in systemic depletion of macrophages\monocytes [34], while work in our lab has shown that small molecules drugs such as ondansetron, and angiotensin-receptor blockers like losartan, can function to inhibit monocyte migration.

Conventional NK cells, when appropriately activated, can exert powerful tumor suppressive activity (Figure 2) [33,35,36]. For example, in vivo administration of molecules that elicit production of type I interferons (e.g., IFN-α and IFN-β) can activate and expand NK cell populations, which control tumor growth by producing IFN-γ and by directly inducing tumor lysis [36]. A subpopulation of NK cells known as Natural Killer T cells (NKT cells) can also be directly activated by administration of the CD1 ligand alpha-galactosyl ceramide. Depletion of NK cells or NK cell dysfunction is associated with increased spontaneous generation of tumors [37,38]. However, not all NK cells control tumor growth, as certain subpopulations of NK cells can also suppress tumor immunity by producing immune suppressive cytokines (e.g., IL-10, IL-13) and promoting the growth of regulatory T cells (Tregs) (Figure 1) [39,40].
The role of neutrophils in the regulation of tumor immunity remains incompletely defined. Recent studies in mice have suggested that neutrophils, similar to tumor-associated macrophages, can be polarized towards either a pro-tumor or anti-tumor phenotype, termed N1 or N2, respectively [28]. Consistent with this paradigm, some studies have demonstrated a protective effect of tumor-associated neutrophils in tumor immunity via stimulation of T cell activation and proliferation, while others have shown that neutrophils may subvert tumor immune responses through mechanisms similar to those reported for MDSCs [25,41,42,43]. Recent studies in dogs suggest that neutrophils may be a major component of the expanded MDSC population observed in dogs with cancer [23]. Our studies and others have found an association between increased numbers of circulating neutrophils and poor outcomes in dogs with lymphoma and OS [10].
3. Activation of Innate Immunity for Cancer Immunotherapy Using Biological Response Modifiers
Cancer immunotherapy has a relatively long history in veterinary medicine. In humans, non-specific immunotherapy dates back to the pioneering studies of William Coley in the late 1800s, who demonstrated that injection of live bacteria or bacterial extracts could induce tumor regression in human sarcoma patients [44]. Many of the earliest immunotherapy studies using so-called biological response modifiers were done in dogs with cancer. For example, immunotherapy using live, attenuated Mycobacterium bovis (strain Bacillus Calmette-Guerin; or BCG) was conducted in dogs beginning in the 1970s [45,46]. Injection of BCG in rodent models was shown to activate a number of innate immune pathways, including macrophages, monocytes, neutrophils, and NK cells. In dogs, BCG was administered by a variety of routes to dogs with osteosarcoma (OS) to elicit non-specific tumor immunity, with or without administration of irradiated tumor cells as vaccines. Significant increases in survival times in dogs with OS were observed following BCG treatment, and the effects were attributed to activation of macrophage tumoricidal activity [45]. Specifically, the median survival time (MST) of BCG-treated dogs with no radiographic evidence of pulmonary metastasis at time of amputation was greater than 41 weeks (287 days), which was significantly longer than control dogs receiving amputation alone (MST of 11 weeks/77 days) [45], and is comparable to those reported for the current standard of care adjuvant chemotherapies of carboplatin and doxorubicin [47,48,49]. Importantly, 2 of the 6 BCG-treated dogs were still alive with no radiographic evidence of metastasis 322 and 371 days after amputation [45].
4. Activation of Innate Immunity by Nod-Like Receptor Antagonists
In the 1980s, a more fully defined cancer immunotherapeutic derived from biological sources was developed. This new compound (MTP-PE) consisted of the Nod-like receptor ligand muramyl tripeptide, delivered within phosphatidyl ethanolamine liposomes [50,51]. MTP was first identified as a bacterial peptide that stimulated neutrophil migration and activation, and later its ability to activate monocytes and macrophages was also discovered. Only recently was the actual receptor for MTP identified. Nod-like receptors (NLRs) are intracellular receptors that recognize diverse ligands, including products of bacterial degradation (e.g., peptidoglycans), viral nucleic acids, uric acid, and even changes in intracellular K+ concentration [52,53].
In vitro incubation of tumor cells and PBMC with L-MTP-PE induced tumor cytolysis, in a process that was shown to be dependent on TNF-α production by monocytes and macrophages. Intravenous administration of L-MTP-PE to dogs induced rapid release of TNF-α into circulation, and also induced TNF-α production by alveolar macrophages [54]. Clinical trials of L-MTP-PE were initiated in dogs with OS, initially as a single agent for adjuvant therapy to prevent metastasis, and was shown to significantly prolong survival compared to amputation alone, with a MST of 222 days for dogs receiving L-MTP-PE, as compared to 77 days for dogs receiving control, empty liposomes [11,12,55]. When administered in succession with cisplatin chemotherapy, L-MTP-PE further improved median survival times to 14.4 months (~439 days), as compared to 9.8 months (~299 days) for dogs receiving cisplatin alone; however, when the two drugs were administered concurrently, the additive benefit was lost [12]. L-MTP-PE is now approved for treatment of pediatric OS in Europe, but has not been approved in the US and is no longer available for routine clinical use.
5. Activation of Anti-Tumor Immunity by Biological Molecules
The first Toll-like receptors (TLRs) were discovered in the 1990s and an explosion of research led to the identification of at least 11 different TLRs. TLRs function primarily to recognize pathogen molecules, including viral, bacterial, and fungal pathogens, and are expressed by many different cell types, especially antigen presenting cells (macrophages, monocytes, dendritic cells, neutrophils) of the innate immune system. Early immunotherapy studies by William Coley in the late 1800s using complex mixtures of bacterial products to stimulate innate immune activation provided the first evidence that bacterial products could trigger immune activation sufficient to cause tumor regression or sustained tumor stasis [44]. We now know that the activity of Coley’s toxin was mediated by simultaneous activation of multiple TLRs, which provides a very potent signal to innate immune cells [56].
6. Immunotherapy by Activation of C-Type Lectin Receptors Using Plant Extracts
One of the earliest cancer immune therapeutics evaluated in veterinary medicine consisted of extracts of the Aloe plant, known as Acemannan. This product consists of mannan polymers interspersed with O-acetyl groups. Though not proven, it is likely this compound activates innate immune cells via the dectin receptor, a C-type lectin known to bind to beta-linked glucans from plants and fungi [56]. Clinically, Acemannan was shown to induce tumor regression following direct intra-tumoral injection in dogs with fibrosarcoma, and to induce systemic immune activation following i.p. administration [57,58]. However, there are no data to suggest that the drug has a systemic effect in preventing local tumor recurrence or tumor metastasis. Additionally, it should be noted that while acemannan showed no direct toxicity following repeated injections in normal tissues, the marked intra-tumoral inflammation and secondary necrosis elicited by the drug can be extensive, subsequently requiring surgical excision of the tumor [58]. Thus, location of the primary tumor and complexity of surgical removal should be considered prior to initiating therapy with acemannan. The drug is still commercially available from Carrington Laboratories (now marketed by VPL, Inc., Phoenix, AZ, USA).
7. Activation of Anti-Tumor Immunity in Dogs with TLR Agonists
TLR-9 is a key sensor of bacterial infections (both Gram+ and Gram−) and recognizes certain CpG motifs present in all DNA of bacterial origin [56]. The TLR9 molecule is expressed in an endosomal location, such that only internalized bacterial DNA is sensed. TLR9 can be activated by administration of short oligonucleotides containing CpG motifs (CpG oligos) and activation leads to strong activation of DCs, monocytes, and macrophages, with release of a variety of pro-inflammatory cytokines, including TNF-α, IL-12, IL-6, and type I interferons. Immunotherapy with CpG oligos demonstrated impressive anti-cancer activity in mouse models in the late 1990s [59,60,61,62]. However, human clinical trials failed to demonstrate sufficient activity and commercial products for cancer immunotherapy have not been vigorously pursued. However, more recent studies suggest that local intra-tumoral injection of CpG oligonucleotides, combined with local administration of antibodies that inhibit immune checkpoint molecules, may have a new role to play in innate immune control of cancer [63].
Larger DNA molecules (e.g., plasmid DNA) can also activate TLR9 when complexed first to charged liposomes, which facilitate intracellular entry and endosomal uptake. These cationic liposome-DNA complexes (CLDC) have been shown to potently stimulate innate immunity and NK cell mediated anti-tumor activity in mouse models [64,65]. Importantly, CLDC have also demonstrated an ability to strongly activate innate immunity in dogs following i.v. administration. For example, i.v. infusion of labeled CLDC resulted in significant uptake and activation of circulating monocytes, characterized by strong up regulation of MHC class II and CD86 expression along with activation of NK cells, as evidenced by enhanced in vitro spontaneous cytolysis of MHC-mismatched target cells [66]. Other effects of CLDC infusion in dogs include binding to tumor blood vessels, inhibition of tumor angiogenesis and up regulation of MHCII expression by T cells [67]. Clinically, CLDC infusion was shown to significantly prolong survival in dogs with established OS pulmonary metastases, with CLDC-treated dogs having a MST of 82 days, as compared to median survival times of 58 days for untreated control dogs, and 61 days for dogs receiving cisplatin and/or doxorubicin in the metastatic setting [66,68]. Additionally, CLDC infusion was also shown to induce tumor growth stabilization in dogs with soft tissue sarcoma, with 8 of 13 dogs exhibiting stable disease, and one dog each exhibiting complete and partial responses [67]. Other studies have shown that s.c. administration of CLDC can trigger full regression of adult-onset papillomatosis in dogs [69]. Commercially, CLDC is now approved in Europe as an immunotherapeutic for use in poultry and is currently being investigated as a mucosal immune stimulant in companion animals.
The use of TLR 3 agonists has also been explored in cancer immunotherapy in rodent models and in limited human clinical trials. These studies have primarily utilized a synthetic analog of single-stranded RNA known as polyinosinic-polycytidylic acid, or pIC. This molecule activates the intracellular TLR3 receptor and triggers release of type I interferons (IFN-α and IFN-β) as well as other pro-inflammatory cytokines. Complexes of pIC and cationic liposomes are also capable of activating innate immunity in cats and dogs [70].

{kind=link}
{kind=link}
{kind=link}
| Cytokine\Immune Molecule | Cellular Source | Anti-Tumor Mechanism(s) |
|---|---|---|
| IFN-γ |
|
|
| IL-12 |
| |
| IL-2 |
| |
| IFN-α/β |
|
|
| TNF-α |
|
|
| Perforin and granzyme |
|
|
| FasFasL |
| |
| TRAIL |
|
|
8. Innate Immune Activation by Recombinant Cytokines
The innate immune system can also be activated by administration of cytokines, including IL-2, IL-12, IFN-γ, IFN-α, and TNF-α (Table 1). TNF-α was one of the first cytokines identified with antitumor activity, and was widely investigated as a stand-alone immunotherapeutic. However, systemic administration of TNF-α was accompanied by substantial toxicity, which precluded its use in humans or in companion animals. IL-2 was investigated in dogs as a means of activating spontaneous NK cell activity [82,83,84]. These studies utilized human recombinant IL-2 and demonstrated in vitro activation of NK cell activity. A safe dose for i.v. administration of huIL-2 was also determined, though formal clinical trials in cancer-bearing animals were not pursued. Inhalational administration of human IL-2 has also been shown to generate significant antitumor activity in dogs with lung metastases [85,86]. Interferon-α has been widely used for immunotherapy of cancer in dogs and in cats. Studies in rodents and humans indicate that IFN-α is capable of triggering NK cell proliferation and activation, which likely accounts for its antitumor activity. While IFN-α is FDA approved for the treatment of various human cancers, including chronic myeloid leukemia, melanoma, and multiple myeloma [75], randomized clinical trials have not yet been conducted in dogs or cats treated with IFN-α, though the drug is anecdotally administered for certain types of canine and feline cancers, including squamous cell carcinoma and papillomatosis. Currently, a canine IFN-γ product is not available, and human IFN-γ does not cross react with dog or cat innate immune cells.
9. Reversal of Immune Suppression by Macrophage Depletion or Monocyte Migration Blockade
Most previous work in tumor immunotherapy has focused on direct activation of innate immune cells to elicit tumor immune control. However, it is now clear that certain myeloid cells (especially tumor associated macrophages and circulating immature monocytes and neutrophils) can exert a profound suppressive influence on tumor immunity [14,15,17]. In cancer patients, low level sustained inflammation drives the release of immature monocytes and neutrophils from the bone marrow, and also prevents their maturation once they reach peripheral tissues [24]. The primary immunological targets of these so-called myeloid derived suppressor cells (MDSCs) are T cells and NK cells, though MDSCs can also directly affect tumor behavior as well. For example, MDSCs interfere with multiple steps in the T cell activation and expansion cascade, through a variety of mediators including production of reactive nitrogen and oxygen intermediates, production of indoleamine dioxygenase, arginase, and immune suppressive cytokines (TGF-β, IL-10, VEGF) [24] (Figure 1).
These expanded populations of MDSCs therefore make an attractive target for immunotherapeutic intervention in cancer; to help restore T cell and NK cell anti-tumor function. For example; studies in rodent models have demonstrated that the biochemical pathways mediating immune suppression by macrophages can be interrupted by specific pathway inhibitors; though these inhibitors have not been evaluated clinically in dogs with cancer [87]. Alternatively; MDSCs can also be targeted for specific elimination pharmacologically. Currently; the most effective methods of eliminating MDSCs (and tumor-associated macrophages and monocytes) take advantage of the fact that these cells are highly phagocytic; and actively phagocytose particles in the size range of 100 nm to 1 μm in diameter. Thus; when the bisphosphonate drug clodronate; which induces macrophage apoptosis by competing for ATP energy stores; is encapsulated within liposomes and administered to animals; macrophages phagocytose the liposome-encapsulated drug; resulting in cytoplasmic drug release and rapid induction of macrophage apoptosis [88]. Our studies in rodent models demonstrated that treatment of tumor-bearing mice with liposomal clodronate induced efficient depletion of MDSCs and tumor macrophages [89]. Importantly; MDSC depletion with liposomal clodronate also induced tumor growth arrest; associated with spontaneous T cell and NK cell activation.
Liposomal clodronate (LC) has also been evaluated as a cancer immunotherapeutic in dogs. In one study, i.v. infusion of liposomal clodronate was shown to induce tumor regression in some dogs with histiocytic sarcoma that had previously failed conventional prednisone and/or lomustine chemotherapy, with 1 of the 5 treated dogs surviving for five months following LC treatment [90]. In vitro, liposomal clodronate was also shown to induce direct killing of canine histiocytic sarcoma cells, by virtue of their phagocytic properties, whereas most tumor cells of other lineages (which are not phagocytic) were not affected. In a second study, liposomal clodronate was also administered to dogs with soft tissue sarcomas and effects on tumor macrophages and tumor angiogenesis were assessed prior to treatment and again following 3 treatments [91]. We found that treatment with liposomal clodronate was associated with significant depletion of tumor-associated macrophages, as well as a significant reduction in tumor microvessel density; however, these reductions in tumor macrophages and angiogenesis did not translate to objective tumor responses [91]. Of not, adverse effects related to liposomal clodronate treatment in these two studies were limited and included fever, and transient neutrophilia [90,91]. Thus, these studies illustrate that macrophages and other immunosuppressive myeloid cell populations can be efficiently depleted in dogs by liposomal clodronate.
An alternative strategy for eliminating tumor macrophages is to selectively block the migration and recruitment of inflammatory monocytes to tumor tissues. Tumor production of certain chemokines, especially the chemokine CCL2, is a major driver of monocyte mobilization of inflammatory (CCR2+) monocytes from the bone marrow and recruitment into tissues, where the monocytes mature into tumor macrophages, which support the growth of tumor metastases [7]. Sustained administration of drugs that block the CCR2 receptor can over time induce macrophage depletion and slow tumor growth in rodent models (Dow, S., unpublished data). Moreover, we have recently observed that highly metastatic tumors in dogs (e.g., hemangiosarcoma and OS) are associated with intense monocyte infiltrates and local production of CCL2 (Regan, D., et al., manuscript in preparation). Importantly, we have also observed that certain classes of drugs (e.g., angiotensin-receptor blocking agents such as losartan) can also block CCL2 dependent migration of canine monocytes in vivo and in vitro. Thus, in the future it may be possible to combine myeloid targeted immunotherapeutics with other treatment modalities (e.g., chemotherapy or radiation therapy) to help augment anti-tumor immunity [92].
10. Conclusions and Implications
Recent successes in immunotherapy for treatment of cancer in humans have generated a considerable reawakening of interest in tumor immunotherapy in veterinary medicine as well. At present, the best opportunities for use of immunotherapy in treatment of companion animal cancer include administration of activators of innate immunity, including both TLR and NLR agonists. In addition, alternative routes of delivering activating ligands are likely to figure more prominently in cancer immunotherapy. There is also a great deal of interest in the use of monoclonal antibodies targeting immune checkpoint molecules such as PD-1 and CTLA-4, which will hopefully become available in the near future. The combination of innate immune targeted therapy with cancer vaccines and checkpoint molecule inhibitors will undoubtedly be explored as well. Therefore it is not unrealistic to expect that immunotherapy will soon gain accepted status as the fourth major component of tumor therapy in companion animals as wells as in humans.
Acknowledgments
The authors gratefully acknowledge the Shipley Foundation, which provides funding support for this and other projects within the Tumor Immunology Laboratory of the Flint Animal Cancer Center. Daniel Regan is currently supported by a National Institute of Health Biomedical Research Training Grant for Veterinarians (5T32OD010437-14). The authors also wish to acknowledge the support of the faculty and staff of the Flint Animal Cancer Center at Colorado State University.
Author Contributions
Steven Dow wrote the first draft of the manuscript. Daniel Regan generated and assembled the figures, and edited the first draft of the manuscript. Daniel Regan and Steven Dow co-wrote and co-edited the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pardoll, D. Cancer and the immune system: Basic concepts and targets for intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Rao, S.; Lafferty, M.H.; Thamm, D.H.; Morley, P.S.; Withrow, S.J.; Dow, S.W. Association of blood monocyte and lymphocyte count and disease-free interval in dogs with osteosarcoma. J. Vet. Intern. Med. 2010, 24, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Thamm, D.H.; Eickhoff, J.; Avery, A.C.; Dow, S.W. Increased monocyte chemotactic protein-1 concentration and monocyte count independently associate with a poor prognosis in dogs with lymphoma. Vet. Comp. Oncol. 2011, 9, 55–64. [Google Scholar] [CrossRef] [PubMed]
- MacEwen, E.G.; Kurzman, I.D.; Rosenthal, R.C.; Smith, B.W.; Manley, P.A.; Roush, J.K.; Howard, P.E. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J. Natl. Cancer Inst. 1989, 81, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Kurzman, I.D.; MacEwen, E.G.; Rosenthal, R.C.; Fox, L.E.; Keller, E.T.; Helfand, S.C.; Vail, D.M.; Dubielzig, R.R.; Madewell, B.R.; Rodriguez, C.O., Jr. Adjuvant therapy for osteosarcoma in dogs: Results of randomized clinical trials using combined liposome-encapsulated muramyl tripeptide and cisplatin. Clin. Cancer Res. 1995, 1, 1595–1601. [Google Scholar] [PubMed]
- Janotova, T.; Jalovecka, M.; Auerova, M.; Švecová, I.; Bruzlová, P.; Maierová, V.; Kumžáková, Z.; Cunátová, Š.; Vlčková, Z.; Caisová, V.; et al. The use of anchored agonists of phagocytic receptors for cancer immunotherapy: B16-F10 murine melanoma model. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol. Immunother. 2002, 51, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 2006, 55, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Sherger, M.; Kisseberth, W.; London, C.; Olivo-Marston, S.; Papenfuss, T.L. Identification of myeloid derived suppressor cells in the peripheral blood of tumor bearing dogs. BMC Vet. Res. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Goulart, M.R.; Pluhar, G.E.; Ohlfest, J.R. Identification of myeloid derived suppressor cells in dogs with naturally occurring cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Maenhout, S.K.; Thielemans, K.; Aerts, J.L. Location, location, location: Functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Gagliani, N.; Huber, S.; Flavell, R.A. Innate immune cells in inflammation and cancer. Cancer Immunol. Res. 2013, 1, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” vs. “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Massari, F.; Amantini, C.; Nabissi, M.; Maines, F.; Burattini, L.; Berardi, R.; Santoni, G.; Montironi, R.; Tortora, G.; et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2015. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Larmonier, N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. 2014, 74, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Tarek, N.; Lee, D.A. Natural killer cells for osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 341–353. [Google Scholar] [PubMed]
- Mentlik James, A.; Cohen, A.D.; Campbell, K.S. Combination immune therapies to enhance anti-tumor responses by NK cells. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Dunn, G.P.; Lacoursiere, D.Y.; Schreiber, R.D.; Bui, J.D. Cancer immunoediting of the NK group 2D ligand H60a. J. Immunol. 2011, 187, 3538–3545. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Terabe, M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunol. Immunother. 2008, 57, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Terabe, M.; Donaldson, D.D.; Forni, G.; Berzofsky, J.A. Natural immunosurveillance against spontaneous, autochthonous breast cancers revealed and enhanced by blockade of IL-13-mediated negative regulation. Cancer Immunol. Immunother. 2008, 57, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Hegmans, J.P.; Aerts, J.G. Immunomodulation in cancer. Curr. Opin. Pharmacol. 2014, 17, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hawes, M.C.; Wen, F.; Elquza, E. Extracellular DNA: A bridge to cancer. Cancer Res. 2015, 75, 4260–4264. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Dr William Coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [PubMed]
- Owen, L.N.; Bostock, D.E. Effects of intravenous BCG in normal dogs and in dogs with spontaneous osteosarcoma. Eur. J. Cancer 1974, 10, 775–780. [Google Scholar] [CrossRef]
- Owen, L.N.; Bostock, D.E.; Lavelle, R.B. Studies on chemotherapy and immunotherapy in canine lymphosarcoma and osteosarcoma. Bibl. Haematol. 1975, 43, 522–523. [Google Scholar] [PubMed]
- Bacon, N.J.; Ehrhart, N.P.; Dernell, W.S.; Lafferty, M.; Withrow, S.J. Use of alternating administration of carboplatin and doxorubicin in dogs with microscopic metastases after amputation for appendicular osteosarcoma: 50 cases (1999–2006). J. Am. Vet. Med. Assoc. 2008, 232, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Weinstein, M.J.; Springfield, D.S.; Rand, W.M. Results of surgery and doxorubicin chemotherapy in dogs with osteosarcoma. J. Am. Vet. Med. Assoc. 1995, 206, 1555–1560. [Google Scholar] [PubMed]
- Saam, D.E.; Liptak, J.M.; Stalker, M.J.; Chun, R. Predictors of outcome in dogs treated with adjuvant carboplatin for appendicular osteosarcoma: 65 cases (1996–2006). J. Am. Vet. Med. Assoc. 2011, 238, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, E.S.; Maeda, M.; Jaffe, N. Liposome-encapsulated muramyl tripeptide: A new biologic response modifier for the treatment of osteosarcoma. Cancer Treat Res. 1993, 62, 101–107. [Google Scholar] [PubMed]
- Kleinerman, E.S.; Jia, S.F.; Griffin, J.; Seibel, N.L.; Benjamin, R.S.; Jaffe, N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: The cytokine cascade and monocyte activation following administration. J. Clin. Oncol. 1992, 10, 1310–1316. [Google Scholar] [PubMed]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.R.; Beck, P.L.; Muruve, D.A. Nucleotide-binding oligomerization domain-like receptors and inflammasomes in the pathogenesis of non-microbial inflammation and diseases. J. Innate Immun. 2012, 4, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Kurzman, I.D.; Shi, F.; MacEwen, E.G. In vitro and in vivo canine mononuclear cell production of tumor necrosis factor induced by muramyl peptides and lipopolysaccharide. Vet. Immunol. Immunopathol. 1993, 38, 45–56. [Google Scholar] [CrossRef]
- MacEwen, E.G.; Kurzman, I.D.; Vail, D.M.; Dubielzig, R.R.; Everlith, K.; Madewell, B.R.; Rodriguez, C.O., Jr.; Phillips, B.; Zwahlen, C.H.; Obradovich, J.; et al. Adjuvant therapy for melanoma in dogs: Results of randomized clinical trials using surgery, liposome-encapsulated muramyl tripeptide, and granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 1999, 5, 4249–4258. [Google Scholar] [PubMed]
- Ishii, K.J.; Coban, C.; Akira, S. Manifold mechanisms of Toll-like receptor-ligand recognition. J. Clin. Immunol. 2005, 25, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Pierce, K.; King, G.; Yates, K.M.; Hall, J.; Tizard, I. Efficacy of acemannan in treatment of canine and feline spontaneous neoplasms. Mol. Biother. 1991, 3, 207–213. [Google Scholar] [PubMed]
- King, G.K.; Yates, K.M.; Greenlee, P.G.; Pierce, K.R.; Ford, C.R.; McAnalley, B.H.; Tizard, I.R. The effect of Acemannan Immunostimulant in combination with surgery and radiation therapy on spontaneous canine and feline fibrosarcomas. J. Am. Anim. Hosp. Assoc. 1995, 31, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. CpG DNA in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 2000, 247, 157–170. [Google Scholar] [PubMed]
- Walker, P.S.; Scharton-Kersten, T.; Krieg, A.M.; Love-Homan, L.; Rowton, E.D.; Udey, M.C.; Vogel, J.C. Immunostimulatory oligodeoxynucleotides promote protective immunity and provide systemic therapy for leishmaniasis via IL-12- and IFN-gamma-dependent mechanisms. Proc. Natl. Acad. Sci. USA 1999, 96, 6970–6975. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. DNA-based immune enhancers. Curr. Opin. Drug Discov. Devel. 2000, 3, 214–221. [Google Scholar] [PubMed]
- Jahrsdorfer, B.; Weiner, G.J. CpG oligodeoxynucleotides for immune stimulation in cancer immunotherapy. Curr. Opin. Investig. Drugs 2003, 4, 686–690. [Google Scholar] [PubMed]
- Marabelle, A.; Kohrt, H.; Caux, C.; Levy, R. Intratumoral immunization: A new paradigm for cancer therapy. Clin. Cancer Res. 2014, 20, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.W.; Fradkin, L.G.; Liggitt, D.H.; Willson, A.P.; Heath, T.D.; Potter, T.A. Lipid-DNA complexes induce potent activation of innate immune responses and antitumor activity when administered intravenously. J. Immunol. 1999, 163, 1552–1561. [Google Scholar] [PubMed]
- Dow, S. Liposome-nucleic acid immunotherapeutics. Expert Opin. Drug Deliv. 2008, 5, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.; Elmslie, R.; Kurzman, I.; MacEwen, G.; Pericle, F.; Liggitt, D. Phase I study of liposome-DNA complexes encoding the interleukin-2 gene in dogs with osteosarcoma lung metastases. Hum. Gene Ther. 2005, 16, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Kamstock, D.; Guth, A.; Elmslie, R.; Kurzman, I.; Liggitt, D.; Coro, L.; Fairman, J.; Dow, S. Liposome-DNA complexes infused intravenously inhibit tumor angiogenesis and elicit antitumor activity in dogs with soft tissue sarcoma. Cancer Gene Ther. 2006, 13, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, G.K.; Straw, R.C.; Jameson, V.J.; Walters, L.M.; Lafferty, M.H.; Powers, B.E.; Withrow, S.J. Evaluation of single-agent chemotherapy for treatment of clinically evident osteosarcoma metastases in dogs: 45 cases (1987-1991). J. Am. Vet. Med. Assoc. 1993, 202, 304–306. [Google Scholar] [PubMed]
- Dow, S.; Kurihara, J.; Regan, D. Cationic Lipid-DNA complexes induced spontaneous regression of adult-onset papillomatosis in dogs. 2015; in preparation. [Google Scholar]
- Veir, J.K.; Lappin, M.R.; Dow, S.W. Evaluation of a novel immunotherapy for treatment of chronic rhinitis in cats. J. Feline Med. Surg. 2006, 8, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-gamma in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, R.; Ten Hagen, T.L.; Eggermont, A.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Helfand, S.C.; Modiano, J.F.; Nowell, P.C. Immunophysiological studies of interleukin-2 and canine lymphocytes. Vet. Immunol. Immunopathol. 1992, 33, 1–16. [Google Scholar] [CrossRef]
- Helfand, S.C.; Soergel, S.A.; MacWilliams, P.S.; Hank, J.A.; Sondel, P.M. Clinical and immunological effects of human recombinant interleukin-2 given by repetitive weekly infusion to normal dogs. Cancer Immunol. Immunother. 1994, 39, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Helfand, S.C.; Soergel, S.A.; Modiano, J.F.; Hank, J.A.; Sondel, P.M. Induction of lymphokine-activated killer (LAK) activity in canine lymphocytes with low dose human recombinant interleukin-2 in vitro. Cancer Biother. 1994, 9, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Anderson, P.M.; Hasz, D.E.; Katsanis, E.; Neville, M.; Klausner, J.S. Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases. Cancer 1997, 79, 1409–1421. [Google Scholar] [CrossRef]
- Khanna, C.; Hasz, D.E.; Klausner, J.S.; Anderson, P.M. Aerosol delivery of interleukin 2 liposomes is nontoxic and biologically effective: Canine studies. Clin. Cancer Res. 1996, 2, 721–734. [Google Scholar] [PubMed]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Guth, A.M.; Hafeman, S.D.; Dow, S.W. Depletion of phagocytic myeloid cells triggers spontaneous T cell- and NK cell-dependent antitumor activity. Oncoimmunology 2012, 1, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Hafeman, S.; London, C.; Elmslie, R.; Dow, S. Evaluation of liposomal clodronate for treatment of malignant histiocytosis in dogs. Cancer Immunol. Immunother. 2010, 59, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Guth, A.M.; Hafeman, S.D.; Elmslie, R.E.; Dow, S.W. Liposomal clodronate treatment for tumour macrophage depletion in dogs with soft-tissue sarcoma. Vet. Comp. Oncol. 2013, 11, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T. Rational combinations of immunotherapeutics that target discrete pathways. J. Immunother. Cancer 2013, 1. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Regan, D.; Dow, S. Manipulation of Innate Immunity for Cancer Therapy in Dogs. Vet. Sci. 2015, 2, 423-439. https://doi.org/10.3390/vetsci2040423
AMA Style
Regan D, Dow S. Manipulation of Innate Immunity for Cancer Therapy in Dogs. Veterinary Sciences. 2015; 2(4):423-439. https://doi.org/10.3390/vetsci2040423
Chicago/Turabian StyleRegan, Daniel, and Steven Dow. 2015. "Manipulation of Innate Immunity for Cancer Therapy in Dogs" Veterinary Sciences 2, no. 4: 423-439. https://doi.org/10.3390/vetsci2040423