The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats


Abstract
:
1. Introduction
2. Material and Methods
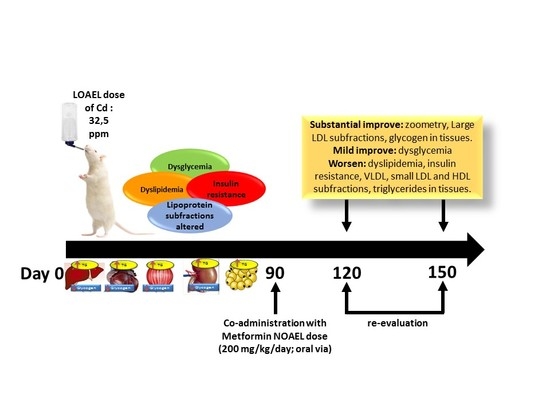
2.1. Animals and Treatments
2.2. Animal Zoometry
2.3. Biochemical Assays in Serum
2.4. Insulin Resistance Analysis
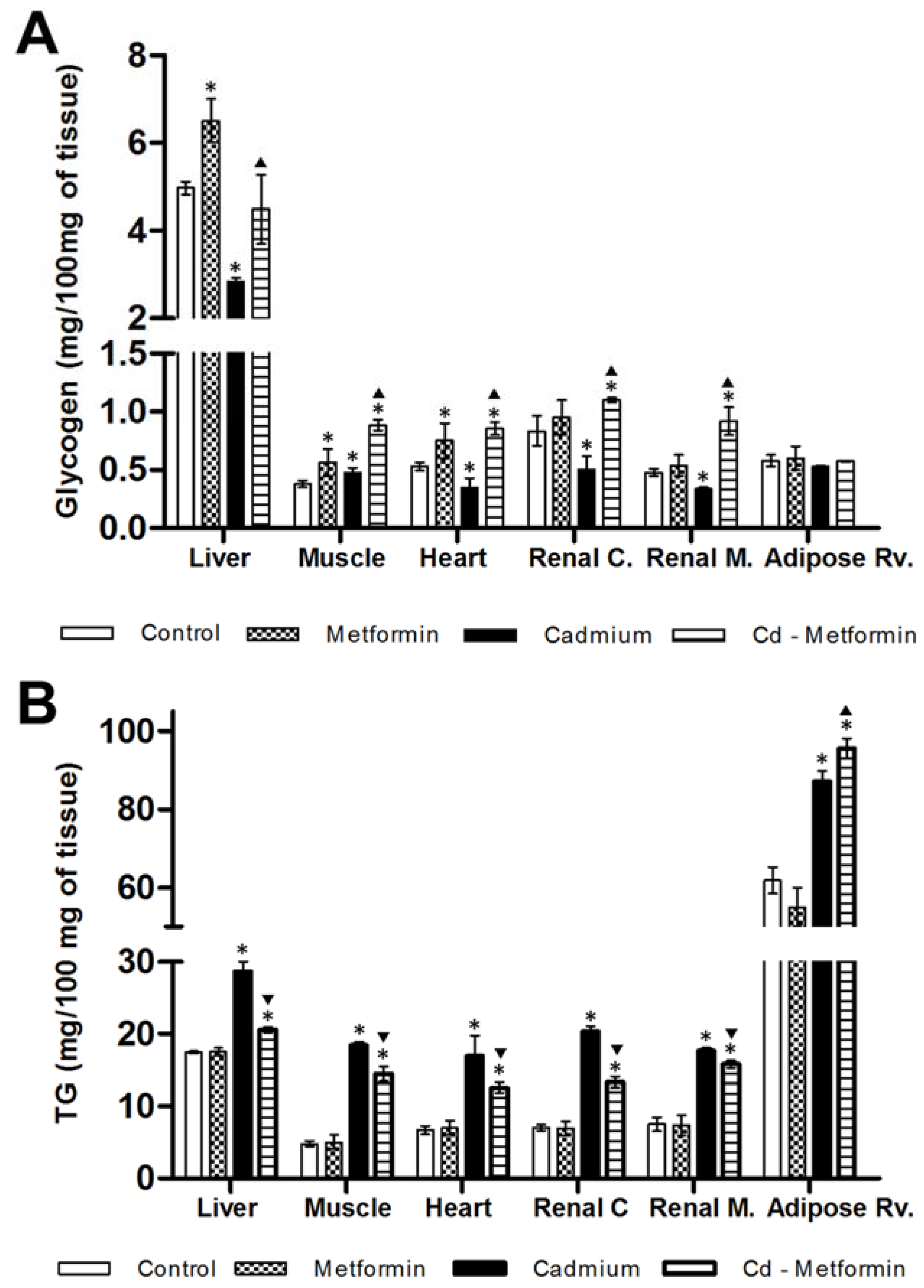
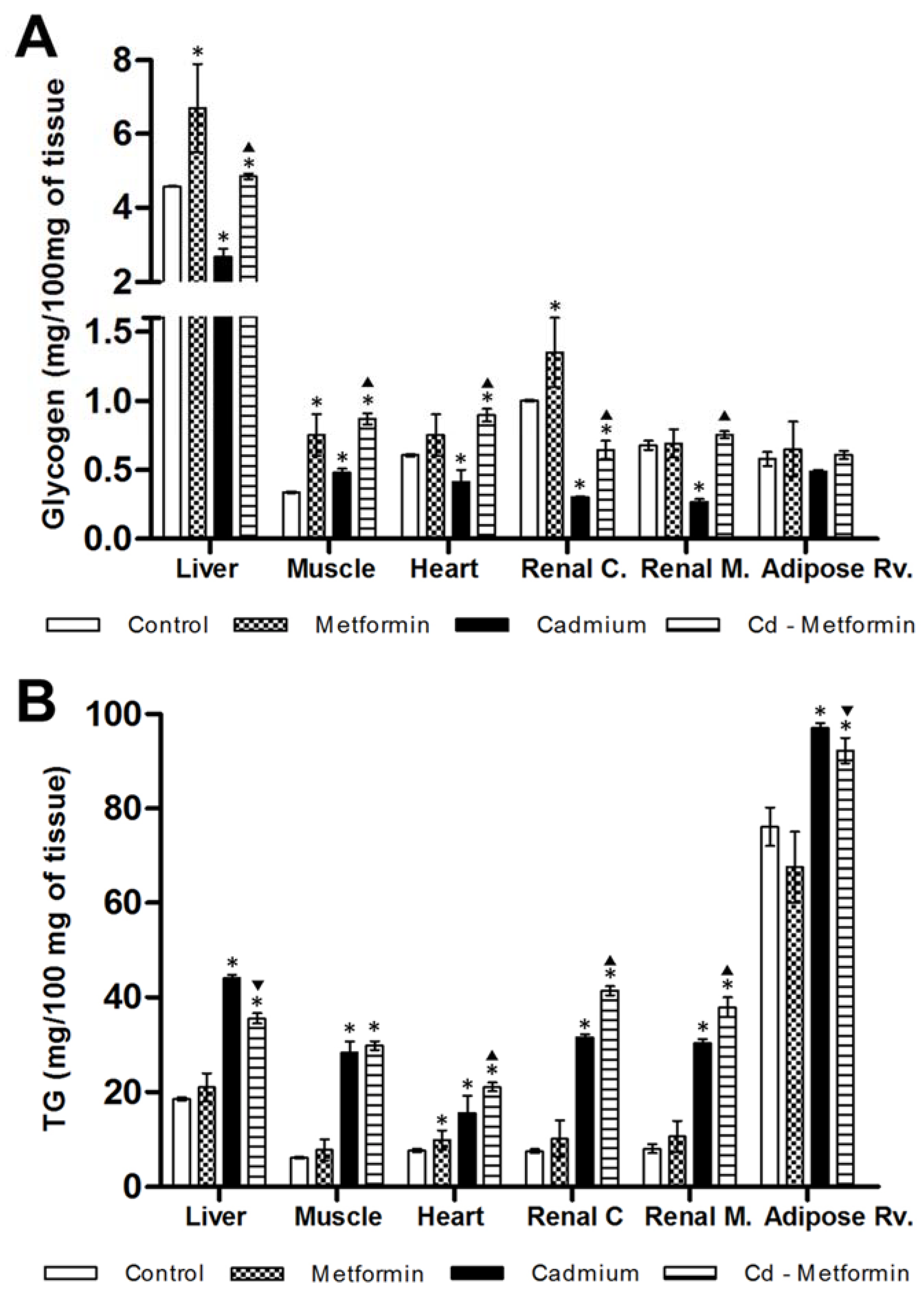
2.5. Glycogen and Triglycerides Content in Tissues
2.6. Statistical Analysis
3. Results
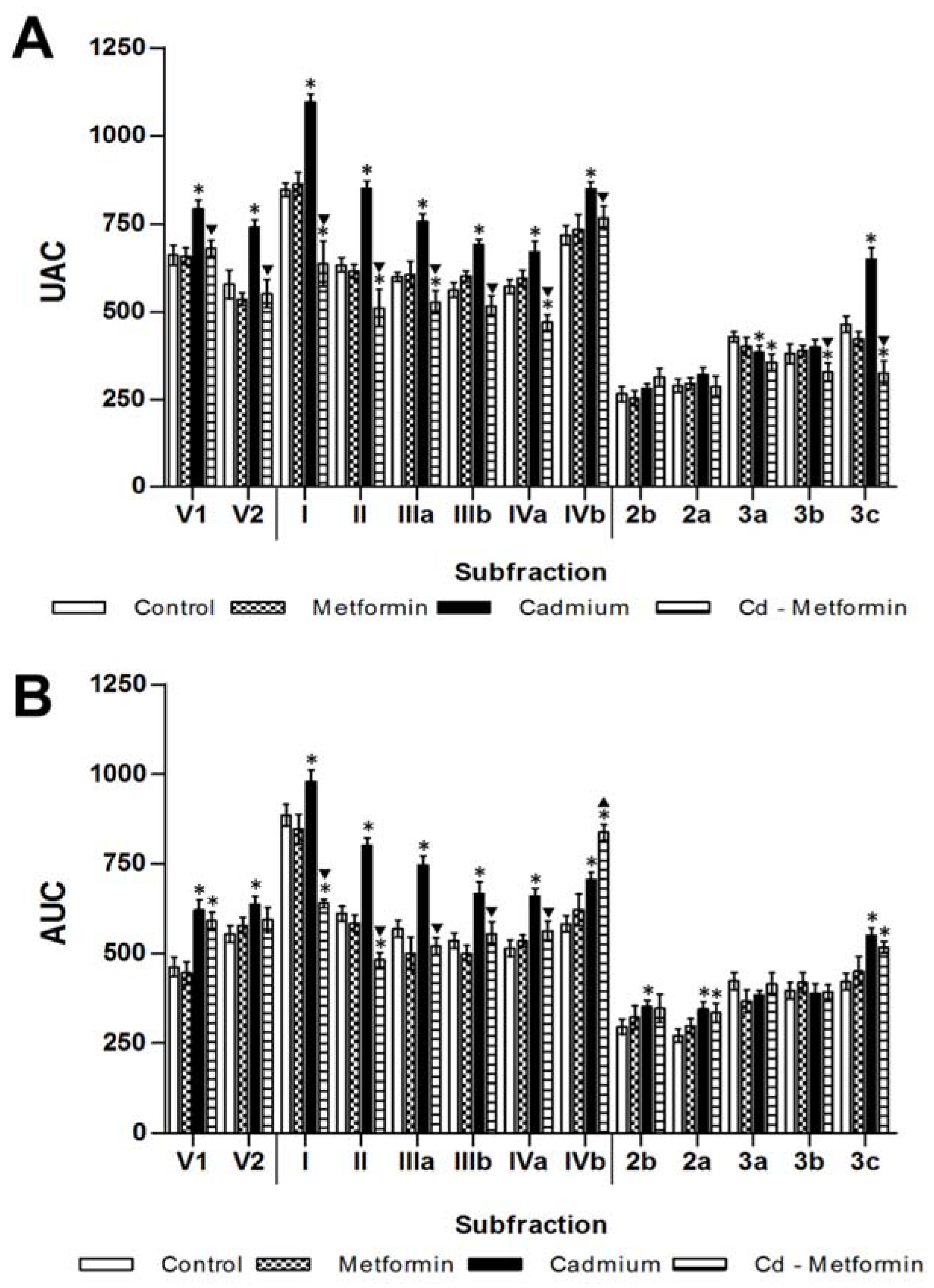
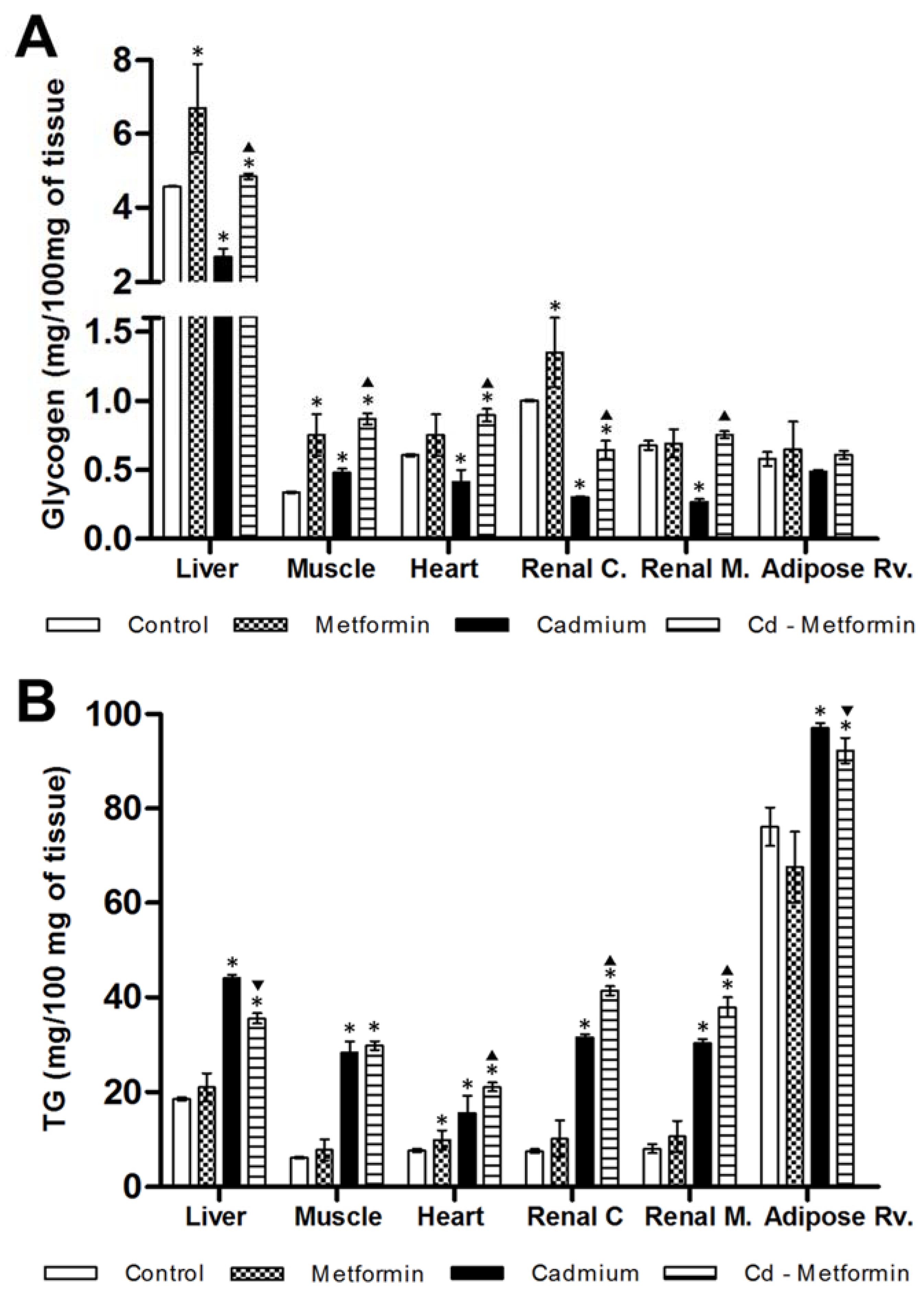
3.1. Morphometry, Lipids and Carbohydrates in Serum and Tissues after 3 Months of Cd Exposure
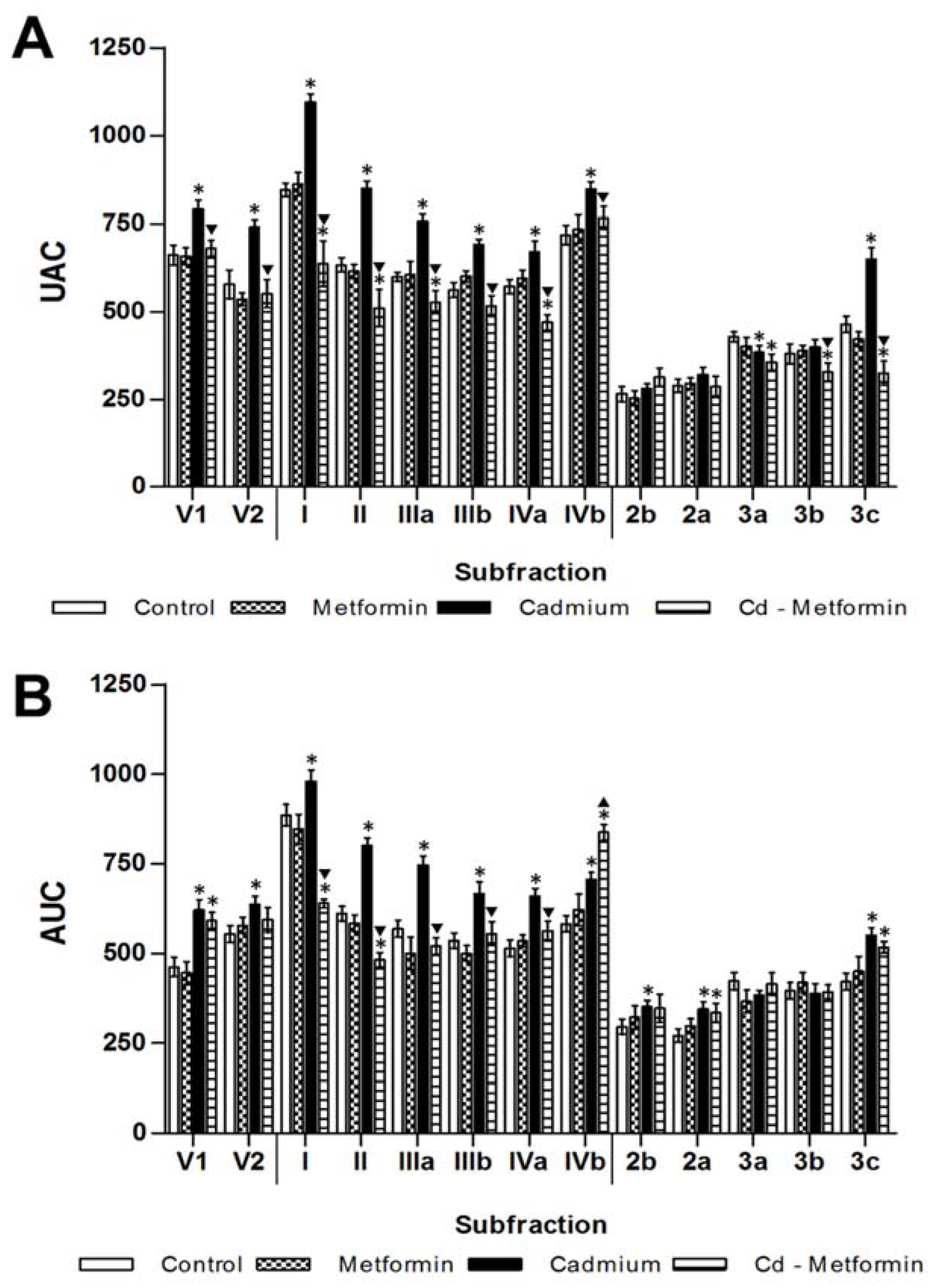
3.2. Metformin Treatment on the Metabolic Disruption Caused by Cd Exposure
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- ATSDR, Agency for Toxic Substance and Disease Registry, U.S. Toxicological Profile for Cadmium. 2012. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp.asp?id=48&tid=15 (accessed on 15 May 2018).
- Bernhoft, R.A. Cadmium Toxicity and Treatment. Sci. World J. 2013, 2013, 394652. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, B.; Hay, E.B.; Nebert, D.W. Discovery of ZIP transporters that participate in cadmium damage to testis and kidney. Toxicol. Appl. Pharmacol. 2009, 238, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordberg, G.F. Historical perspectives on cadmium toxicology. Toxicol. Appl. Pharmacol. 2009, 238, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 2003, 186, 163–188. [Google Scholar] [CrossRef]
- Klaassen, D.C.; Liu, J.; Diwan, B.A. Metallothionein Protection of Cadmium Toxicity. Toxicol. Appl. Pharmacol. 2009, 238, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Habeebu, S.S.; Liu, J.; Liu, Y.; Klaassen, C.D. Metallothionein-null mice are more sensitive than wild-type mice to liver injury induced by repeated exposure to cadmium. Toxicol. Sci. 2000, 55, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F.; Ciarimboli, G.; Leistner, M.; Wolff, N.A.; Lee, W.K.; Schatz, I.; Keller, T.; AlMonajjed, R.; Gorboulev, V.; Koepsell, H. Substrate- and cell contact-dependent inhibitor affinity of human organic cation transporter 2: Studies with two classical organic cation substrates and the novel substrate Cd2+. Mol. Pharm. 2013, 10, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F. Nephrotoxicity and the proximal tubule: Insights from Cadmium. Nephron Physiol. 2003, 93, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 743–1786. [Google Scholar] [CrossRef] [PubMed]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Moulis, J.M. Cellular mechanisms of cadmium toxicity related to the homeostasis of essential metals. Biometals 2010, 23, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Moulis, J.M.; Thévenod, F. New perspectives in cadmium toxicity: An introduction. Biometals 2010, 23, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, M.; Yoshihara, S.I. New aspects of cadmium as an endocrine disruptor. Environ. Sci. 2005, 13, 107–116. [Google Scholar]
- Silva, N.; Peiris-John, R.; Wickremasinghe, R.; Senanayake, H.; Sathiakumar, N. Cadmium a metalloestrogen: Are we convinced? J. Appl. Toxicol. 2012, 35, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Ortega, V.; Cano Barquilla, P.; Fernández-Mateos, P.; Cardinali, D.P.; Esquifino, A.I. Cadmium as an endocrine disruptor: Correlation with anterior pituitary redox and circadian clock mechanisms and prevention by melatonin. Free Radic. Biol. Med. 2012, 53, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.D.; Cortizo, A.M.; Sedlinsky, C. Metformin revisited: Does this regulator of AMP-activated protein kinase secondarily affect bone metabolism and prevent diabetic osteopathy. World J. Diabetes 2016, 7, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Kim, Y. Blood cadmium, mercury, and lead and metabolic syndrome in South Korea: 2005–2010. Am. J. Ind. Med. 2012, 56, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Moore, M.R. Emerging roles of cadmium and heme oxygenase in type-2 diabetes and cancer susceptibility. Tohoku J. Exp. Med. 2012, 228, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yang, C.Y.; Huang, C.F.; Hung, D.Z.; Leung, Y.M.; Liu, S.H. Heavy metals, islet function, and diabetes development. Islets 2009, 1, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hruby, A.; Hu, F.B. The epidemiology of obesity: A big picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, F.; Bener, A.; Eliyan, A.; Delghan, H.; Nofal, E.; Shalabi, L.; Wadi, N. Projection of Diabetes Burden through 2025 and Contributing Risk Factors of Changing Disease Prevalence: An Emerging Public Health Problem. J. Diabetes Metab. 2014, 5, 341. [Google Scholar]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus present and future perspectives. Nat. Rev. Endocrinol. 2011, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B. Epidemiology of type 2 diabetes and cardiovascular disease: Translation from population to prevention: The Kelly West award lecture 2009. Diabetes Care 2010, 33, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Sánchez-Lara, E.; Sarmiento-Ortega, V.E.; Sánchez-Lombardo, I.; Flores-Hernández, J.A.; Pérez-Benítez, A.; Brambila-Colombres, E.; González-Vergara, E. Hypoglycemic, lipid-lowering and metabolic regulation activities of metforminium decavanadate (H2Metf)3 [V10O28]·8H2O using hypercaloric-induced carbohydrate and lipid deregulation in Wistar rats as a biological model. J. Inorg. Biochem. 2015, 147, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Desroches, S.; Lamarche, B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Shaw, J.E.; Zimmet, P.Z. The metabolic syndrome: Prevalence in worldwide populations. Endocrinol. Metab. Clin. N. Am. 2004, 33, 351–375. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Fleming, G.A.; Chenc, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metab. Clin. Exp. 2016, 65, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. Endocrinol. (Paris) 2013, 74, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, B.; Guigas, B.; Sanz-Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. (Lond.) 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summary of Revisions: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 4 (Suppl. 1), S4–S6. [CrossRef]
- Chamberlain, J.J.; Herman, W.H.; Leal, S.; Rhinehart, A.S.; Shubrook, J.H.; Skolnik, N.; Kalyani, R.R. Pharmacologic Therapy for Type 2 Diabetes: Synopsis of the 2017 American Diabetes Association Standards of Medical Care in Diabetes. Ann. Intern. Med. 2017, 166, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Quaile, M.P.; Melich, D.H.; Jordan, H.L.; Nold, J.B.; Chism, J.P.; Polli, J.W.; Smith, G.A.; Rhodes, M.C. Toxicity and toxicokinetics of metformin in rats. Toxicol. Appl. Pharmacol. 2010, 243, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, P. AMP-activated protein kinase: Structure and regulation. Curr. Protein Pept. Sci. 2008, 9, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Luong, D.Q.; Oster, R.; Ashraf, A.P. Metformin treatment improves weight and dyslipidemia in children with metabolic syndrome. J. Pediatr. Endocrinol. Metab. 2015, 28, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Waalkes, M.P.; Flores-Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Warnick, G.R.; Knopp, R.H.; Fitzpatrick, V.; Branson, L. Estimating low-density lipoprotein cholesterol by the Friedewald equation is adequate for classifying patients on the basis of the nationally recommended cut point. Clin. Chem. 1990, 36, 15–19. [Google Scholar] [PubMed]
- Brunk, S.D.; Swanson, J.R. Colorimetric method for free fatty acids in serum validated by comparison with gas chromatography. Clin. Chem. 1981, 27, 924–926. [Google Scholar] [PubMed]
- Rainwater, D.L.; Moore, P.H., Jr.; Gamboa, I.O. Improved method for making nondenaturing composite gradient gels for the electrophoretic separation of lipoproteins. J. Lipid Res. 2004, 45, 773–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, L.W.; Keirs, R.W.; Peebles, E.D.; Gerard, P.D. Methodologies of tissue preservation and analysis of the glycogen content of the broiler chick liver. Poult. Sci. 2007, 86, 2653–2665. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, G.F.; Nogawa, K.; Nordberg, M.; Friberg, L. Handbook of the Toxicology of Metals, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Haswell-Elkins, M.; Mcgrath, V.; Moore, M.; Satarug, S.; Walmby, M.; Ng, J. Exploring potential dietary contributions including traditional seafood and other determinants of urinary cadmium levels among indigenous women of a Torres Strait Island (Australia). J. Expo. Sci. Environ. Epidemiol. 2007, 17, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalnaya, M.G.; Tinkov, A.A.; Demidov, V.A.; Serebryansky, E.P.; Nikonorov, A.A.; Skalny, A.V. Hair toxic element content in adult men and women in relation to body mass index. Biol. Trace Elem. Res. 2014, 161, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Nishiyama, K.; Tanaka, J.I.; Kadota, Y.; Sato, M.; Suzuki, S. Changes in macrophage migration and adipokine gene expression induced by cadmium in the white adipose tissue of metallothionein-null mice. J. Toxicol. Sci. 2012, 37 (Suppl. II), AP-46. [Google Scholar]
- Kawakami, T.; Sugimoto, H.; Furuichi, R.; Kadota, Y.; Inoue, M.; Setsu, K.; Suzuki, S.; Sato, M. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology 2010, 267, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Il’yasova, D.; Ivanova, A. Urinary cadmium, impaired fasting glucose, and diabetes in the NHANES III. Diabetes Care 2003, 26, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.R.; Puga, M.P.; Wrobel, K.; Sevilla, M.E.G.; Wrobel, K. Trace elements status in diabetes mellitus type 2: Possible role of the interaction between molybdenum and copper in the progress of typical complications. Diabetes Res. Clin. Pract. 2011, 91, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Afridi, H.I.; Kazi, T.G.; Kazi, N.G.; Jamali, M.K.; Arain, M.B.; Sirajuddin Baig, J.A.; Kandhro, G.A.; Wadhwa, S.K.; Shah, A.Q. Evaluation of cadmium, lead, nickel and zinc status in biological samples of smokers and non-smokers hypertensive patients. J. Hum. Hypertens. 2010, 24, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jin, Y.; Zeng, Z.; Liu, Z.; Fu, Z. Subchronic exposure of mice to cadmium perturbs their hepatic energy metabolism and gut microbiome. Chem. Res. Toxicol. 2015, 28, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Chapatwala, K.D.; Rajanna, B.; Desaiah, D. Cadmium-induced changes in gluconeogenic enzymes in rat kidney and liver. Drug Chem. Toxicol. 1980, 3, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Chapatwala, K.D.; Hobson, M.; Desaiah, D.; Rajanna, B. Effect of cadmium on hepatic and renal gluconeogenic enzymes in female rats. Toxicol. Lett. 1982, 12, 27–34. [Google Scholar] [CrossRef]
- Han, J.C.; Park, S.Y.; Hah, B.G.; Choi, G.H.; Kim, Y.K.; Kwon, T.H.; Kim, E.K.; Lachaal, M.; Jung, C.Y.; Lee, W. Cadmium induces impaired glucose tolerance in the rat by down-regulating GLUT4 expression in adipocytes. Arch. Biochem. Biophys. 2003, 413, 213–220. [Google Scholar] [CrossRef]
- Shanbaky, I.O.; Borowitz, J.L.; Kessler, W.V. Mechanisms of cadmium- and barium-induced adrenal catecholamine release. Toxicol. Appl. Pharmacol. 1978, 44, 99–105. [Google Scholar] [CrossRef]
- Edwards, J.R.; Prozialeck, W.C. Cadmium, diabetes and chronic kidney disease. Toxicol. Appl. Pharmacol. 2009, 238, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hectors, T.L.M.; Vanparys, C.; van der Ven, K.; Martens, G.A.; Jorens, P.G.; Van Gaal, L.F.; Covaci, A.; De Coen, W.; Blust, R. Environmental pollutants and type 2 diabetes: A review of mechanisms that can disrupt beta cell function. Diabetologia 2011, 54, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Larregle, E.V.; Varas, S.M.; Oliveros, L.B.; Martínez, L.D.; Anton, R.; Marchevsky, E.; Gimenez, M.S. Lipid metabolism in liver of rat exposed to cadmium. Food Chem. Toxicol. 2008, 46, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, P.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, J.D.; Sparks, C.E.; Adeli, K. Selective Hepatic Insulin Resistance, VLDL Overproduction, and Hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2104–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, B.; Bernhard, D. Cadmium and cardiovascular diseases: Cell biology, pathophysiology, and epidemiological relevance. Biometals 2010, 23, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Messner, B.; Knoflach, M.; Seubert, A.; Ritsch, A.; Pfaller, K.; Henderson, B.; Shen, Y.H.; Zeller, I.; Willeit, J.; Laufer, G.; et al. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Plaza, M.; Jones, M.R.; Dominguez-Lucas, A.; Guallar, E.; Navas-Acien, A. Cadmium Exposure and Clinical Cardiovascular Disease: A Systematic Review. Curr. Atheroscler. Rep. 2013, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Takamura, M.; Kojima, S. Some heavy metals affecting the lecithin-cholesterol acyltransferase reaction in human plasma. J. Biochem. 1977, 81, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Rosso, L.; Lhomme, M.; Meroño, T.; Dellepiane, A.; Sorroche, P.; Hedjazi, L.; Zakiev, E.; Sukhorukov, V.; Orekhov, A.; Gasparri, J.; et al. Poor glycemic control in type 2 diabetes enhances functional and compositional alterations of small, dense HDL3c. Biochim. Biophys. Acta 2017, 1862, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pari, L.; Ramakrishnan, L. Protective effect of Tetrahydrocurcumin on plasma lipids and lipoproteins in cadmium intoxicated rats. Int. J. Toxicol. Appl. Pharmacol. 2013, 3, 26–32. [Google Scholar]
- Yang, H.; Guo, D.; Shu, Y. Cadmium Ion Upregulates the activity of Human Organic Transporter 2. FASEB J. 2017, 31, 819.10. [Google Scholar]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanovski, J.A.; Krakoff, J.; Salaita, C.G.; McDuffie, J.R.; Kozlosky, M.; Sebring, N.G.; Reynolds, J.C.; Brady, S.M.; Calis, K.A. Effects of Metformin on Body Weight and Body Composition in Obese Insulin-Resistant Children: A Randomized Clinical Trial. Diabetes 2011, 60, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current knowledge. J. Res. Med. Sci. 2014, 19, 658–664. [Google Scholar] [PubMed]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signaling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Lomako, L.; Lomako, W.M.; Whelan, W.G. The Biogenesis of Muscle Glycogen: Regulation of the Activity of the Autocatalytic Primer Protein. BioFactors 1990, 2, 251–254. [Google Scholar] [PubMed]
- Vytla, V.S.; Ochs, R.S. Metformin Increases Mitochondrial Energy Formation in L6 Muscle Cell Cultures. J. Biol. Chem. 2013, 288, 20369–20377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Mithieux, G.; Guignot, L.; Bordet, J.C.; Wiernsperger, J.C. Intrahepatic Mechanisms Underlying the Effect of Metformin in Decreasing Basal Glucose Production in Rats Fed a High-Fat Diet. Diabetes 2002, 51, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithieux, G.; Rajas, F.; Zitoun, C. Glucose Utilization Is Suppressed in the Gut of Insulin-Resistant High Fat-Fed Rats and Is Restored by Metformin. Biochem. Pharmacol. 2006, 72, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, J.; Al-Mass, A.; Nolan, C.J.; Corkey, B.E.; Madiraju, S.R.M.; Joly, E.; Prentki, M. Identification of the signals for glucose-induced insulin secretion in INS1 (832/13) β-cells using metformin-induced metabolic deceleration as a model. J. Biol. Chem. 2017, 292, 19458–19468. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Robitaille, K.; Faubert, B.; Reeks, C.; Dai, X.-Q.; Hardy, A.B.; Sankar, K.S.; Ogrel, S.; Al-Dirbashi, O.Y.; Rocheleau, J.V.; et al. LKB1 couples glucose metabolism to insulin secretion in mice. Diabetologia 2015, 58, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugabo, Y.; Zhao, S.; Lamontagne, J.; Al-Mass, A.; Peyot, M.-L.; Corkey, B.E.; Joly, E.; Madiraju, S.R.M.; Prentki, M. Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic β-cells. J. Biol. Chem. 2017, 292, 7407–7422. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurements | Control | Cadmium | Metabolite mg/100 mg of Tissue | Control | Cadmium |
|---|---|---|---|---|---|
| n = 30 | n = 50 | n = 10 | n = 10 | ||
| Morphometric panel: | Triglycerides: | ||||
| Weight (g) | 341 ± 3.4 | 439.2 ± 8.1 * | Liver | 16.2 ± 0.7 | 22.5 ± 1 * |
| Abdominal perimeter (cm) | 18.4 ± 0.1 | 22.4 ± 0.2 * | Muscle | 3.36 ± 0.2 | 10.0 ± 0.3 * |
| Body mass index | 0.93 ± 0.02 | 1.35 ± 0.05 * | Heart | 5.06 ± 0.3 | 9 ± 0.2 * |
| % Body fat | 35.9 ± 0.1 | 41.3 ± 0.5 * | Renal cortex | 5.98 ± 0.4 | 12.7 ± 0.6 * |
| Lipidic panel (mg/dL): | Renal medulla | 7.46 ± 0.3 | 11.5 ± 0.7 * | ||
| Total lipids | 184 ± 12 | 243 ± 5.1 * | Rv Adipose | 51.2 ± 1.4 | 64.8 ± 1.6 * |
| FFA | 2.18 ± 0.03 | 5.18 ± 1.1 * | |||
| Triglycerides | 64.4 ± 2.5 | 106.8 ± 3.1 * | Glycogen: | ||
| Total Cholesterol | 103.1 ± 7.5 | 103.4 ± 6 | Liver | 4.2 ± 0.4 | 2.9 ± 0.3 * |
| Cholesterol fraction: | Muscle | 0.3 ± 0.03 | 0.62 ± 0.1 * | ||
| VLDL | 13.5 ± 1.5 | 19.2 ± 1.2 * | Heart | 0.9 ± 0.05 | 0.57 ± 0.8 * |
| LDL | 24.2 ± 4 | 47.9 ± 3.1 * | Renal cortex | 1.2 ± 0.2 | 0.57 ± 0.11 * |
| HDL | 65.4 ± 2 | 36.3 ± 1.5 * | Renal medulla | 0.56 ± 0.2 | 0.51 ± 0.6 |
| Carbohydrate panel: | Rv. Adipose | 0.55 ± 0.03 | 0.48 ± 0.01 | ||
| Lactate (mmol/L) | 7.3 ± 0.7 | 9.4 ± 0.5 * | Insulin resistance panel: | ||
| Fasting glucose (mg/dL) | 80 ± 3.2 | 165 ± 5 * | Insulin (µUI/mL) | 12 ± 3.1 | 21 ± 4.5 * |
| ‡ Glucose 30 min (mg/dL) | 107.1 ± 2.9 | 241 ± 8.1 * | HOMA-IR | 0.44 ± 0.05 | 1.39 ± 0.19 * |
| ‡ Glucose 60 min (mg/dL) | 90.6 ± 3.4 | 265 ± 7 * | IDA-IR | 0.07 ± 0.03 | 0.46 ± 0.15 * |
| ‡ Glucose 90 min (mg/dL) | 81.2 ± 3 | 251.5 ± 4.2 * | HIS | 18.8 ± 3.5 | 5.2 ± 2.2 * |
| Measurements | Control | Metformin | Cadmium | Cd + Metformin |
|---|---|---|---|---|
| n = 10 | n = 10 | n = 10 | n = 10 | |
| Morphometric panel: | ||||
| Weight (g) | 401.6 ± 7.9 | 380.2 ±15.3 | 470 ± 6.3 * | 418 ± 10 ▼ |
| Abdominal perimeter (cm) | 20.8 ± 0.1 | 19.4 ± 0.6 | 24.2 ± 0.7 * | 21.1 ± 0.4 ▼ |
| Body mass index | 1.1 ± 0.04 | 1.0 ± 0.02 | 1.4 ± 0.01 * | 1.1 ± 0.03 ▼ |
| % Body fat | 37.8 ± 0.3 | 35.7 ± 1.6 | 42.1 ± 0.1 * | 40.0 ± 0.3 *▼ |
| Lipid panel (mg/dL): | ||||
| Total lipids | 187.2 ± 9.6 | 180.1 ± 3.3 | 253.6 ± 4.5 * | 232 ± 6.1 *▼ |
| FFA | 2.85 ± 0.2 | 3.01 ± 0.2 | 6.49 ± 0.1 * | 5.1 ± 0.1 *▼ |
| Triglycerides | 56 ± 6 | 51 ± 2 | 112.3 ± 4.5 * | 93.1 ± 3.5 *▼ |
| Total Cholesterol | 111.9 ± 5.6 | 107.9 ± 4.9 | 108.1 ± 6.7 | 84.9 ± 5.7 * ▼ |
| Cholesterol fraction | ||||
| VLDL | 16 ± 1.6 | 18 ± 0.9 | 21.2 ± 0.9 * | 18.2 ± 1 *▼ |
| LDL | 36 ± 3.5 | 40 ± 6.1 | 54.8 ± 1.7 * | 26 ± 2.1 *▼ |
| HDL | 59.9 ± 2.6 | 49.9 ± 7.2 | 32.1 ± 2.5 * | 40.7 ± 3.1 *▲ |
| Carbohydrate panel: | ||||
| Lactate (mmol/L) | 7.3 ± 0.7 | 7.5 ± 0.2 | 8.55 ± 0.3 * | 8.2 ± 0.1 ▼ |
| Fasting glucose (mg/dL) | 80.0 ± 6.3 | 76.0 ± 4.8 | 135 ± 4.1 * | 79.7 ± 4.3 ▼ |
| Glucose 30 min (mg/dL) | 107.1 ± 3.8 | 97.7 ± 5.2 | 238 ± 6.7 * | 154.4 ± 5.4 *▼ |
| Glucose 60 min (mg/dL) | 90.6 ± 4.3 | 88.4 ± 7.1 | 250 ± 9.4 * | 160.4 ± 3.8 *▼ |
| Glucose 90 min (mg/dL) | 81.2 ± 5.0 | 73.9 ± 9.4 | 235 ± 8.8 * | 142.2 ± 6.1 *▼ |
| Insulin resistance panel: | ||||
| Insulin (µUI/mL) | 10 ± 3.4 | 9.8 ± 4.8 | 19 ± 2.4 * | 27 ± 4.9 * |
| HOMA-IR | 0.44 ± 0.03 | 0.40 ± 0.08 | 1.42 ± 0.19 * | 0.88 ± 0.21 *▼ |
| IDA-IR | 0.02 ± 0.02 | 0.02 ± 0.01 | 0.54 ± 0.12 * | 0.35 ± 0.11 * |
| HIS | 22.5 ± 5.19 | 20.1 ± 3.3 | 7.01 ± 3.8 * | 8.3 ± 2.2 * |
| Measurements | Control | Metformin | Cadmium | Cd + Metformin |
|---|---|---|---|---|
| n = 10 | n = 10 | n = 10 | n = 10 | |
| Morphometric panel: | ||||
| Weight (g) | 434.3 ± 6.2 | 420.9 ± 12.8 | 502.6 ± 9.5 * | 442.6 ± 10.5 ▼ |
| Abdominal perimeter (cm) | 22.1 ± 0.6 | 20.7 ± 0.9 | 26.2 ± 0.5 * | 22.9 ± 0.6 ▼ |
| Body mass index | 1.1 ± 0.04 | 1.0 ± 0.08 | 1.5 ± 0.03 * | 1.2 ± 0.03 ▼ |
| % Body fat | 38.8 ± 0.3 | 36.2 ± 1.1 | 43.1 ± 0.1 * | 40.8 ± 0.3 *▼ |
| Lipid panel (mg/dL): | ||||
| Total lipids | 180 ± 13 | 178 ± 9 | 270 ± 6.1 * | 259 ± 4.5 * |
| FFA | 3.49 ± 0.12 | 3.98 ± 0.6 | 6.8 ± 0.16 * | 5.9 ± 0.2 *▼ |
| Triglycerides | 50.5 ± 5.8 | 47.7 ± 5.1 | 121.7 ± 2.1 * | 113.6 ± 3.1 *▼ |
| Total Cholesterol | 102.2 ± 10.8 | 98.5 ± 6.7 | 112.9 ± 5.2 | 102.6 ± 3.2 |
| Cholesterol fraction | ||||
| VLDL | 10.1 ± 1.8 | 10.1 ± 1.8 | 24.9 ± 1.2 * | 24.3 ± 1.2 * |
| LDL | 39 ± 7 | 41 ± 3 | 59.5 ± 2.7 * | 43.5 ± 4.7 ▼ |
| HDL | 53.1 ± 2 | 50.9 ± 3 | 28.5 ± 2.4 * | 34.8 ± 1.4 *▲ |
| Carbohydrate panel: | ||||
| Lactate (mmol/L) | 8.2 ± 0.5 | 9.1 ± 1.3 | 11.3 ± 0.4 * | 10.1 ± 0.3 *▼ |
| Fasting glucose (mg/dL) | 72.9 ± 4 | 67.7 ± 7 | 139.5 ± 4.2 * | 85 ± 5.9 *▼ |
| Glucose 30 min (mg/dL) | 93.1 ± 0.6 | 85.3 ± 4.9 | 157.4 ± 14.4 * | 108.3 ± 6.0 *▼ |
| Glucose 60 min (mg/dL) | 98.2 ± 3.0 | 104.4 ± 5.5 | 171.3 ± 8.7 * | 117.0 ± 5.3 *▼ |
| Glucose 90 min (mg/dL) | 90.0 ± 5.0 | 98.0 ± 7.3 | 164 ± 6.3 * | 116.2 ± 4.8 *▼ |
| Insulin resistance panel: | ||||
| Insulin (µUI/mL) | 11 ± 1.8 | 14 ± 2.9 | 25 ± 3.2 * | 34.7 ± 4.1 *▲ |
| HOMA-IR | 0.47 ± 0.03 | 0.50 ± 0.1 | 1.69 ± 0.2 * | 1.21 ± 0.23 * |
| IDA-IR | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.63 ± 0.17 * | 0.51 ± 0.2 * |
| HIS | 22.4 ± 4.7 | 24.7 ± 5.1 | 6.02 ± 1.9 * | 6.10 ± 2.1 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmiento-Ortega, V.E.; Brambila, E.; Flores-Hernández, J.Á.; Díaz, A.; Peña-Rosas, U.; Moroni-González, D.; Aburto-Luna, V.; Treviño, S. The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats. Toxics 2018, 6, 55. https://doi.org/10.3390/toxics6030055
Sarmiento-Ortega VE, Brambila E, Flores-Hernández JÁ, Díaz A, Peña-Rosas U, Moroni-González D, Aburto-Luna V, Treviño S. The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats. Toxics. 2018; 6(3):55. https://doi.org/10.3390/toxics6030055
Chicago/Turabian StyleSarmiento-Ortega, Victor Enrique, Eduardo Brambila, José Ángel Flores-Hernández, Alfonso Díaz, Ulises Peña-Rosas, Diana Moroni-González, Violeta Aburto-Luna, and Samuel Treviño. 2018. "The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats" Toxics 6, no. 3: 55. https://doi.org/10.3390/toxics6030055