
The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides

Abstract
:1. Neurodevelopment
2. Environmental Toxicants and Neurodevelopmental Disorders
2.1. Organochlorines
2.2. Organophosphates
2.3. Pyrethroids
3. Summary
4. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.-S.; McNamara, J.O.; Williams, S.M. Neuronal Migration. In Neuroscience, 5th ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Sanes, D.H.; Reh, T.A.; Harris, W.A. Development of the Nervous System, 3rd ed.; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Arikkath, J.; Reichardt, L.F. Cadherins and catenins at synapses: Roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008, 31, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.; Dirks, A.; Fejtova, A. Bassoon and piccolo regulate ubiquitination and link presynaptic molecular dynamics with activity-regulated gene expression. J. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Purves, D. Neuroscience; Sinauer Associates: Sunderland, MA, USA, 2012. [Google Scholar]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Scheller, R.H. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 2000, 16, 19–49. [Google Scholar] [CrossRef] [PubMed]
- Ropper, A.H.; Samuels, M.A.; Klein, J.P. Chapter 28. Normal development and deviations in development of the nervous system. In Adams and Victor’s Principles of Neurology, 10e; The McGraw-Hill Companies: New York, NY, USA, 2014. [Google Scholar]
- Waxman, S.G. Chapter 2. Development and cellular constituents of the nervous system. In Clinical Neuroanatomy, 27e; The McGraw-Hill Companies: New York, NY, USA, 2013. [Google Scholar]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.; Foldy, C.; Sharma, M.; Tabuchi, K.; Liu, X.; Shamloo, M.; Malenka, R.C.; Sudhof, T.C. Autism-linked neuroligin-3 R451c mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. USA 2011, 108, 13764–13769. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Sudhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Burrows, E.L.; Laskaris, L.; Koyama, L.; Churilov, L.; Bornstein, J.C.; Hill-Yardin, E.L.; Hannan, A.J. A neuroligin-3 mutation implicated in autism causes abnormal aggression and increases repetitive behavior in mice. Mol. Autism 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; Ramanantsoa, N.; Gallego, J.; Ronnenberg, A.; Winter, D.; et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Sudhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M.; Kim, E. The shank family of scaffold proteins. J. Cell Sci. 2000, 113, 1851–1856. [Google Scholar] [PubMed]
- Peykov, S.; Berkel, S.; Schoen, M.; Weiss, K.; Degenhardt, F.; Strohmaier, J.; Weiss, B.; Proepper, C.; Schratt, G.; Nothen, M.M.; et al. Identification and functional characterization of rare shank2 variants in schizophrenia. Mol. Psychiatry 2015, 20, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Cochoy, D.M.; Kolevzon, A.; Kajiwara, Y.; Schoen, M.; Pascual-Lucas, M.; Lurie, S.; Buxbaum, J.D.; Boeckers, T.M.; Schmeisser, M.J. Phenotypic and functional analysis of shank3 stop mutations identified in individuals with asd and/or id. Mol. Autism 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and shank3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of shank mutations in autism spectrum disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ey, E.; Torquet, N.; Le Sourd, A.M.; Leblond, C.S.; Boeckers, T.M.; Faure, P.; Bourgeron, T. The autism prosap1/shank2 mouse model displays quantitative and structural abnormalities in ultrasonic vocalisations. Behav. Brain Res. 2013, 256, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Sungur, A.O.; Vorckel, K.J.; Schwarting, R.K.; Wohr, M. Repetitive behaviors in the shank1 knockout mouse model for autism spectrum disorder: Developmental aspects and effects of social context. J. Neurosci. Methods 2014, 234, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wohr, M. Ultrasonic Vocalizations in Shank Mouse Models for Autism Spectrum Disorders: Detailed Spectrographic Analyses and Developmental Profiles. Neurosci. Biobehav. Rev. 2014, 43, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Kouser, M.; Speed, H.E.; Dewey, C.M.; Reimers, J.M.; Widman, A.J.; Gupta, N.; Liu, S.; Jaramillo, T.C.; Bangash, M.; Xiao, B.; et al. Loss of predominant shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 2013, 33, 18448–18468. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.C.; Speed, H.E.; Xuan, Z.; Reimers, J.M.; Liu, S.; Powell, C.M. Altered striatal synaptic function and abnormal behaviour in shank3 exon4–9 deletion mouse model of autism. Autism Res. 2016, 9, 350–375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Ehlers, M.D. Modeling autism by shank gene mutations in mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Speed, H.E.; Kouser, M.; Xuan, Z.; Reimers, J.M.; Ochoa, C.F.; Gupta, N.; Liu, S.; Powell, C.M. Autism-associated insertion mutation (InsG) of shank3 exon 21 causes impaired synaptic transmission and behavioral deficits. J. Neurosci. 2015, 35, 9648–9665. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two knockdown models of the autism genes syngap1 and shank3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef] [PubMed]
- Uppal, N.; Puri, R.; Yuk, F.; Janssen, W.G.; Bozdagi-Gunal, O.; Harony-Nicolas, H.; Dickstein, D.L.; Buxbaum, J.D.; Hof, P.R. Ultrastructural analyses in the hippocampus CA1 field in shank3-deficient mice. Mol. Autism 2015, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bey, A.L.; Katz, B.M.; Badea, A.; Kim, N.; David, L.K.; Duffney, L.J.; Kumar, S.; Mague, S.D.; Hulbert, S.W.; et al. Altered mglur5-homer scaffolds and corticostriatal connectivity in a shank3 complete knockout model of autism. Nat. Commun. 2016, 7, 11459. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [PubMed]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated SHANK3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman Syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Porton, B.; Wetsel, W.C.; Kao, H.T. Synapsin Iii: Role in neuronal plasticity and disease. Semin. Cell Dev. Biol. 2011, 22, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef] [PubMed]
- Kevenaar, J.T.; Hoogenraad, C.C. The axonal cytoskeleton: From organization to function. Front. Mol. Neurosci. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Benesh, A.E.; Fleming, J.T.; Chiang, C.; Carter, B.D.; Tyska, M.J. Expression and localization of myosin-1d in the developing nervous system. Brain Res. 2012, 1440, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Barr, C.L.; Feng, Y.; Wigg, K.; Bloom, S.; Roberts, W.; Malone, M.; Schachar, R.; Tannock, R.; Kennedy, J.L. Identification of DNA variants in the snap-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol. Psychiatry 2000, 5, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Brophy, K.; Hawi, Z.; Kirley, A.; Fitzgerald, M.; Gill, M. Synaptosomal-associated protein 25 (snap-25) and attention deficit hyperactivity disorder (adhd): Evidence of linkage and association in the Irish population. Mol. Psychiatry 2002, 7, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, V.; Merriman, B.; McGough, J.; McCracken, J.T.; Smalley, S.L.; Nelson, S.F. Biased paternal transmission of snap-25 risk alleles in attention-deficit hyperactivity disorder. Mol. Psychiatry 2003, 8, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Etain, B.; Dumaine, A.; Mathieu, F.; Chevalier, F.; Henry, C.; Kahn, J.P.; Deshommes, J.; Bellivier, F.; Leboyer, M.; Jamain, S. A snap25 promoter variant is associated with early-onset bipolar disorder and a high expression level in brain. Mol. Psychiatry 2010, 15, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Corradini, I.; Fossati, G.; Tomasoni, R.; Menna, E.; Matteoli, M. Snap-25, a known presynaptic protein with emerging postsynaptic functions. Front. Synaptic. Neurosci. 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Littleton, J.T.; Chapman, E.R.; Kreber, R.; Garment, M.B.; Carlson, S.D.; Ganetzky, B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires snare complex assembly and disassembly. Neuron 1998, 21, 401–413. [Google Scholar] [CrossRef]
- Kidokoro, Y. roles of snare proteins and synaptotagmin I in synaptic transmission: Studies at the drosophila neuromuscular synapse. Neurosignals 2003, 12, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Hess, E.J.; Jinnah, H.A.; Kozak, C.A.; Wilson, M.C. Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the snap gene on chromosome 2. J. Neurosci. 1992, 12, 2865–2874. [Google Scholar] [PubMed]
- Ohira, K.; Kobayashi, K.; Toyama, K.; Nakamura, H.K.; Shoji, H.; Takao, K.; Takeuchi, R.; Yamaguchi, S.; Kataoka, M.; Otsuka, S.; et al. Synaptosomal-associated protein 25 mutation induces immaturity of the dentate granule cells of adult mice. Mol. Brain 2013, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, S.C.; Wilson, M.C.; Henriksen, S.J. Coloboma contiguous gene deletion encompassing snap alters hippocampal plasticity. Synapse 1996, 22, 281–289. [Google Scholar] [CrossRef]
- Soderqvist, S.; McNab, F.; Peyrard-Janvid, M.; Matsson, H.; Humphreys, K.; Kere, J.; Klingberg, T. The snap25 gene is linked to working memory capacity and maturation of the posterior cingulate cortex during childhood. Biol. Psychiatry 2010, 68, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Saettini, F.; Panzeri, E.; Arrigoni, F.; Bassi, M.T.; Borgatti, R. A de-novo stxbp1 gene mutation in a patient showing the rett syndrome phenotype. Neuroreport 2015, 26, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Patzke, C.; Han, Y.; Covy, J.; Yi, F.; Maxeiner, S.; Wernig, M.; Sudhof, T.C. Analysis of conditional heterozygous stxbp1 mutations in human neurons. J. Clin. Investig. 2015, 125, 3560–3571. [Google Scholar] [CrossRef] [PubMed]
- Sweet, E.S.; Saunier-Rebori, B.; Yue, Z.; Blitzer, R.D. The parkinson’s disease-associated mutation lrrk2-g2019s impairs synaptic plasticity in mouse hippocampus. J. Neurosci. 2015, 35, 11190–11195. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Buonanno, A.; Passafaro, M.; Sala, C.; Sweet, R.A. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 2013, 126, 165–182. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 2006, 26, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.S.; Bucelli, R.; Jett, D.A.; Bruun, D.; Yang, D.; Lein, P.J. Chlorpyrifos exerts opposing effects on axonal and dendritic growth in primary neuronal cultures. Toxicol. Appl. Pharmacol. 2005, 207, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M. Vulnerability of synapses in the frontal cortex of mice developmentally exposed to an insecticide: Potential contribution to neuropsychiatric disease. Neurotransmitter (Houst.) 2015, 2. [Google Scholar] [CrossRef]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The charge study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Kogut, K.; Huen, K.; Harley, K.G.; Bouchard, M.; Bradman, A.; Boyd-Barr, D.; Johnson, C.; Holland, N. Organophosphate pesticide exposure, pon1, and neurodevelopment in school-age children from the chamacos study. Environ. Res. 2014, 134, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, S.; Liang, D.; Shi, X.; Wang, F.; Liu, W.; Zhang, L.; Chen, L.; Gu, Y.; Tian, Y. Prenatal exposure to organophosphate pesticides and neurobehavioral development of neonates: A birth cohort study in shenyang, China. PLoS ONE 2014, 9, e88491. [Google Scholar] [CrossRef] [PubMed]
- Quiros-Alcala, L.; Mehta, S.; Eskenazi, B. Pyrethroid pesticide exposure and parental report of learning disability and attention deficit/hyperactivity disorder in US. children: Nhanes 1999–2002. Environ. Health Perspect. 2014, 122, 1336–1342. [Google Scholar] [PubMed]
- Wagner-Schuman, M.; Richardson, J.R.; Auinger, P.; Braun, J.M.; Lanphear, B.P.; Epstein, J.N.; Yolton, K.; Froehlich, T.E. Association of pyrethroid pesticide exposure with attention-deficit/hyperactivity disorder in a nationally representative sample of US. children. Environ. Health 2015, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Shafer, T.J.; Meyer, D.A.; Crofton, K.M. Developmental neurotoxicity of pyrethroid insecticides: Critical review and future research needs. Environ. Health Perspect. 2005, 113, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Elwan, M.A.; Richardson, J.R.; Guillot, T.S.; Caudle, W.M.; Miller, G.W. Pyrethroid pesticide-induced alterations in dopamine transporter function. Toxicol. Appl. Pharmacol. 2006, 211, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.W.; Shukla, R.K.; Yadav, R.S.; Seth, K.; Pant, A.B.; Singh, D.; Agrawal, A.K.; Islam, F.; Khanna, V.K. Cholinergic dysfunctions and enhanced oxidative stress in the neurobehavioral toxicity of lambda-cyhalothrin in developing rats. Neurotox. Res. 2012, 22, 292–309. [Google Scholar] [CrossRef] [PubMed]
- De Felice, A.; Scattoni, M.L.; Ricceri, L.; Calamandrei, G. Prenatal exposure to a common organophosphate insecticide delays motor development in a mouse model of idiopathic autism. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Rauh, V.; Arunajadai, S.; Horton, M.; Perera, F.; Hoepner, L.; Barr, D.B.; Whyatt, R. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ. Health Perspect. 2011, 119, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Sealey, L.A.; Hughes, B.W.; Sriskanda, A.N.; Guest, J.R.; Gibson, A.D.; Johnson-Williams, L.; Pace, D.G.; Bagasra, O. Environmental factors in the development of autism spectrum disorders. Environ. Int. 2016, 88, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Hertz-Picciotto, I.; Pessah, I.N. Tipping the balance of autism risk: Potential mechanisms linking pesticides and autism. Environ. Health Perspect. 2012, 120, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Taylor, M.M.; Shalat, S.L.; Guillot, T.S., 3rd; Caudle, W.M.; Hossain, M.M.; Mathews, T.A.; Jones, S.R.; Cory-Slechta, D.A.; Miller, G.W. Developmental pesticide exposure reproduces features of attention deficit hyperactivity disorder. FASEB J. 2015, 29, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.J.; Du, J.C.; Chiou, H.C.; Chung, M.Y.; Yang, W.; Chen, Y.S.; Fuh, M.R.; Chien, L.C.; Hwang, B.; Chen, M.L. Increased risk of attention-deficit/hyperactivity disorder associated with exposure to organophosphate pesticide in Taiwanese children. Andrology 2016, 4, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Kofman, O.; Berger, A.; Massarwa, A.; Friedman, A.; Jaffar, A.A. Motor inhibition and learning impairments in school-aged children following exposure to organophosphate pesticides in infancy. Pediatr. Res. 2006, 60, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Rauh, V.A.; Perera, F.P.; Horton, M.K.; Whyatt, R.M.; Bansal, R.; Hao, X.; Liu, J.; Barr, D.B.; Slotkin, T.A.; Peterson, B.S. Brain anomalies in children exposed prenatally to a common organophosphate pesticide. Proc. Natl. Acad. Sci. USA 2012, 109, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Chen, H.; Weisskopf, M.G.; O’Reilly, E.; McCullough, M.L.; Calle, E.E.; Schwarzschild, M.A.; Thun, M.J. Pesticide exposure and risk for parkinson’s disease. Ann. Neurol. 2006, 60, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.H.; Elias, M.A.; Mankodi, A.K. Acute and reversible parkinsonism due to organophosphate pesticide intoxication: Five cases. Neurology 1999, 52, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Hancock, D.B.; Martin, E.R.; Mayhew, G.M.; Stajich, J.M.; Jewett, R.; Stacy, M.A.; Scott, B.L.; Vance, J.M.; Scott, W.K. Pesticide exposure and risk of parkinson’s disease: A family-based case-control study. BMC Neurol. 2008, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Roy, A.; Shalat, S.L.; von Stein, R.T.; Hossain, M.M.; Buckley, B.; Gearing, M.; Levey, A.I.; German, D.C. Elevated serum pesticide levels and risk for alzheimer disease. JAMA Neurol. 2014, 71, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.R.; Nielsen, J.B.; Grandjean, P. Toxicologic evidence of developmental neurotoxicity of environmental chemicals. Toxicology 2000, 144, 121–127. [Google Scholar] [CrossRef]
- Cole, T.B.; Jampsa, R.L.; Walter, B.J.; Arndt, T.L.; Richter, R.J.; Shih, D.M.; Tward, A.; Lusis, A.J.; Jack, R.M.; Costa, L.G.; et al. Expression of human paraoxonase (Pon1) during development. Pharmacogenetics 2003, 13, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Torres-Sanchez, L.; Schnaas, L.; Rothenberg, S.J.; Cebrian, M.E.; Osorio-Valencia, E.; Hernandez Mdel, C.; Garcia-Hernandez, R.M.; Lopez-Carrillo, L. Prenatal P,P’-Dde exposure and neurodevelopment among children 3.5–5 years of age. Environ. Health Perspect. 2013, 121, 263–268. [Google Scholar] [PubMed]
- Ribas-Fito, N.; Torrent, M.; Carrizo, D.; Munoz-Ortiz, L.; Julvez, J.; Grimalt, J.O.; Sunyer, J. In Utero exposure to background concentrations of ddt and cognitive functioning among preschoolers. Am. J. Epidemiol. 2006, 164, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Torres-Sanchez, L.; Rothenberg, S.J.; Schnaas, L.; Cebrian, M.E.; Osorio, E.; Del Carmen Hernandez, M.; Garcia-Hernandez, R.M.; Del Rio-Garcia, C.; Wolff, M.S.; Lopez-Carrillo, L. In Utero P,P’-Dde Exposure and infant neurodevelopment: A perinatal cohort in mexico. Environ. Health Perspect. 2007, 115, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Rogan, W.J.; Gladen, B.C. Pcbs, Dde, and child development at 18 and 24 months. Ann. Epidemiol. 1991, 1, 407–413. [Google Scholar] [CrossRef]
- Eskenazi, B.; Marks, A.R.; Bradman, A.; Fenster, L.; Johnson, C.; Barr, D.B.; Jewell, N.P. In Utero exposure to dichlorodiphenyltrichloroethane (Ddt) and dichlorodiphenyldichloroethylene (Dde) and neurodevelopment among young Mexican American children. Pediatrics 2006, 118, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Rogan, W.J.; Gladen, B.C.; McKinney, J.D.; Carreras, N.; Hardy, P.; Thullen, J.; Tinglestad, J.; Tully, M. Neonatal effects of transplacental exposure to Pcbs and Dde. J. Pediatr. 1986, 109, 335–341. [Google Scholar] [CrossRef]
- Cartier, C.; Muckle, G.; Jacobson, S.W.; Jacobson, J.L.; Dewailly, E.; Ayotte, P.; Chevrier, C.; Saint-Amour, D. Prenatal and 5-year P,P’-Dde exposures are associated with altered sensory processing in school-aged children in nunavik: A visual evoked potential study. Neurotoxicology 2014, 44, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wendel de Joode, B.; Wesseling, C.; Kromhout, H.; Monge, P.; Garcia, M.; Mergler, D. Chronic nervous-system effects of long-term occupational exposure to Ddt. Lancet 2001, 357, 1014–1016. [Google Scholar] [CrossRef]
- Ross, G.W.; Duda, J.E.; Abbott, R.D.; Pellizzari, E.; Petrovitch, H.; Miller, D.B.; O’Callaghan, J.P.; Tanner, C.M.; Noorigian, J.V.; Masaki, K.; et al. Brain organochlorines and lewy pathology: The honolulu-asia aging study. Mov. Disord. 2012, 27, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Richardson, J.R.; Wang, M.; Miller, G.W. Perinatal heptachlor exposure increases expression of presynaptic dopaminergic markers in mouse striatum. Neurotoxicology 2005, 26, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Caudle, W.M.; Wang, M.; Dean, E.D.; Pennell, K.D.; Miller, G.W. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of parkinson’s disease. FASEB J. 2006, 20, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Bloomquist, J.R.; Barlow, R.L.; Gillette, J.S.; Li, W.; Kirby, M.L. Selective effects of insecticides on nigrostriatal dopaminergic nerve pathways. Neurotoxicology 2002, 23, 537–544. [Google Scholar] [CrossRef]
- Hong, S.; Hwang, J.; Kim, J.Y.; Shin, K.S.; Kang, S.J. Heptachlor induced nigral dopaminergic neuronal loss and parkinsonism-like movement deficits in mice. Exp. Mol. Med. 2014, 46, e80. [Google Scholar] [CrossRef] [PubMed]
- Briz, V.; Molina-Molina, J.M.; Sanchez-Redondo, S.; Fernandez, M.F.; Grimalt, J.O.; Olea, N.; Rodriguez-Farre, E.; Sunol, C. Differential estrogenic effects of the persistent organochlorine pesticides dieldrin, endosulfan, and lindane in primary neuronal cultures. Toxicol. Sci. 2011, 120, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Richardson, J.R.; Guillot, T.S.; McCormack, A.L.; Di Monte, D.A.; Jones, D.P.; Pennell, K.D.; Miller, G.W. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp. Neurol. 2007, 204, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Barlow, R.L.; Bloomquist, J.R. Neurotoxicity of the organochlorine insecticide heptachlor to murine striatal dopaminergic pathways. Toxicol. Sci. 2001, 61, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro, T.; Caride, A.; Romero, A.; Lafuente, A. Effects of in Utero and lactational exposure to endosulfan in prefrontal cortex of male rats. Toxicol. Lett. 2008, 176, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lafuente, A.; Pereiro, N. Neurotoxic effects induced by endosulfan exposure during pregnancy and lactation in female and male rat striatum. Toxicology 2013, 311, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Eriksson, P.; Fredriksson, A.; Buratovic, S.; Viberg, H. Developmental neurotoxic effects of two pesticides: Behavior and neuroprotein studies on endosulfan and cypermethrin. Toxicology 2015, 335, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Caudle, W.M.; Wang, M.Z.; Dean, E.D.; Pennell, K.D.; Miller, G.W. Developmental heptachlor exposure increases susceptibility of dopamine neurons to n-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (Mptp)in a gender-specific manner. Neurotoxicology 2008, 29, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Purkerson-Parker, S.; McDaniel, K.L.; Moser, V.C. Dopamine transporter binding in the rat striatum is increased by gestational, perinatal, and adolescent exposure to heptachlor. Toxicol. Sci. 2001, 64, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.W.; Onyenwe, W.; Bradner, J.M.; Nennig, S.E.; Caudle, W.M. Developmental exposure to the organochlorine insecticide endosulfan alters expression of proteins associated with neurotransmission in the frontal cortex. Synapse 2014, 68, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Briz, V.; Galofre, M.; Sunol, C. Reduction of glutamatergic neurotransmission by prolonged exposure to dieldrin involves nmda receptor internalization and metabotropic glutamate receptor 5 downregulation. Toxicol. Sci. 2010, 113, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Babot, Z.; Vilaro, M.T.; Sunol, C. Long-term exposure to dieldrin reduces gamma-aminobutyric acid type a and N-Methyl-D-Aspartate receptor function in primary cultures of mouse cerebellar granule cells. J. Neurosci. Res. 2007, 85, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Dick, R.B.; Howell, R.J.; Chrislip, D.W.; Hines, C.J.; Reid, T.M.; Lehman, E.; Laber, P.; Krieg, E.F., Jr.; Knott, C. Neurologic function among termiticide applicators exposed to chlorpyrifos. Environ. Health Perspect. 2000, 108, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Seidler, F.J.; Meyer, A.; Thillai, I.; Slotkin, T.A. Serotonergic systems targeted by developmental exposure to chlorpyrifos: Effects during different critical periods. Environ. Health Perspect 2003, 111, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Seidler, F.J.; Slotkin, T.A. Developmental Exposure to chlorpyrifos elicits sex-selective alterations of serotonergic synaptic function in adulthood: Critical periods and regional selectivity for effects on the serotonin transporter, receptor subtypes, and cell signaling. Environ. Health Perspect. 2004, 112, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Raines, K.W.; Seidler, F.J.; Slotkin, T.A. Alterations in serotonin transporter expression in brain regions of rats exposed neonatally to chlorpyrifos. Brain Res. Dev. Brain Res. 2001, 130, 65–72. [Google Scholar] [CrossRef]
- Qiao, D.; Seidler, F.J.; Padilla, S.; Slotkin, T.A. Developmental neurotoxicity of chlorpyrifos: What is the vulnerable period? Environ. Health Perspect. 2002, 110, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Chambers, J.E. Neurochemical effects of repeated gestational exposure to chlorpyrifos in developing rats. Toxicol. Sci. 2004, 77, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Seidler, F.J.; Tate, C.A.; Cousins, M.M.; Slotkin, T.A. Fetal chlorpyrifos exposure: Adverse effects on brain cell development and cholinergic biomarkers emerge postnatally and continue into adolescence and adulthood. Environ. Health Perspect. 2003, 111, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D.; Addy, N.; Baruah, A.; Elias, A.; Christopher, N.C.; Seidler, F.J.; Slotkin, T.A. Prenatal chlorpyrifos exposure in rats causes persistent behavioral alterations. Neurotoxicol. Teratol. 2002, 24, 733–741. [Google Scholar] [CrossRef]
- Sanchez-Santed, F.; Canadas, F.; Flores, P.; Lopez-Grancha, M.; Cardona, D. Long-term functional neurotoxicity of paraoxon and chlorpyrifos: Behavioural and pharmacological evidence. Neurotoxicol. Teratol. 2004, 26, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Eriksson, P.; Fredriksson, A.; Buratovic, S.; Viberg, H. Developmental neurotoxic effects of two pesticides: Behavior and biomolecular studies on chlorpyrifos and Carbaryl. Toxicol. Appl. Pharmacol. 2015, 288, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Guo-Ross, S.X.; Chambers, J.E.; Meek, E.C.; Carr, R.L. Altered muscarinic acetylcholine receptor subtype binding in neonatal rat brain following exposure to chlorpyrifos or methyl parathion. Toxicol. Sci. 2007, 100, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Olivier, K.; Pope, C.N. Comparative neurochemical effects of repeated methyl parathion or chlorpyrifos exposures in neonatal and adult rats. Toxicol Appl. Pharmacol. 1999, 158, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Chambers, J.E. Effects of repeated oral postnatal exposure to chlorpyrifos on cholinergic neurochemistry in developing rats. Toxicol. Sci. 2005, 84, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Carr, R.L.; Chambers, J.E. Changes in rat brain cholinesterase activity and muscarinic receptor density during and after repeated oral exposure to chlorpyrifos in early postnatal development. Toxicol. Sci. 1999, 51, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Pope, C.N. Age-related effects of chlorpyrifos on muscarinic receptor-mediated signaling in rat cortex. Arch. Toxicol. 2002, 75, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Mubarak Hossain, M.; Suzuki, T.; Sato, N.; Sato, I.; Takewaki, T.; Suzuki, K.; Tachikawa, E.; Kobayashi, H. Differential effects of pyrethroid insecticides on extracellular dopamine in the striatum of freely moving rats. Toxicol. Appl. Pharmacol. 2006, 217, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lucki, I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry 1998, 44, 151–162. [Google Scholar] [CrossRef]
- Hossain, M.M.; Suzuki, T.; Unno, T.; Komori, S.; Kobayashi, H. Differential presynaptic actions of pyrethroid insecticides on glutamatergic and gabaergic neurons in the hippocampus. Toxicology 2008, 243, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, J.L. Aldrin and Dieldrin: A review of research on their production, environmental deposition and fate, bioaccumulation, toxicology, and epidemiology in the United States. Environ. Health Perspect. 2001, 109, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. Worldwide trends in ddt levels in human breast milk. Int J. Epidemiol 1999, 28, 179–188. [Google Scholar] [CrossRef] [PubMed]
- LaKind, J.S.; Amina Wilkins, A.; Berlin, C.M., Jr. Environmental Chemicals in Human Milk: A review of levels, infant exposures and health, and guidance for future research. Toxicol. Appl. Pharmacol. 2004, 198, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Main, K.M.; Andersson, A.M.; Damgaard, I.N.; Virtanen, H.E.; Skakkebaek, N.E.; Toppari, J.; Schramm, K.W. Concentrations of persistent organochlorine compounds in human milk and placenta are higher in Denmark Than in Finland. Hum. Reprod. 2008, 23, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Main, K.M.; Virtanen, H.E.; Damggard, I.N.; Haavisto, A.M.; Kaleva, M.; Boisen, K.A.; Schmidt, I.M.; Chellakooty, M.; Skakkebaek, N.E.; et al. From mother to child: Investigation of prenatal and postnatal exposure to persistent bioaccumulating toxicants using breast milk and placenta biomonitoring. Chemosphere 2007, 67, S256–S262. [Google Scholar] [CrossRef] [PubMed]
- Holan, G. New halocyclopropane insecticides and the mode of action of ddt. Nature 1969, 221, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Vijverberg, H.P.; van der Zalm, J.M.; van der Bercken, J. Similar mode of action of pyrethroids and ddt on sodium channel gating in Myelinated Nerves. Nature 1982, 295, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D. Casarett and Doull’s Toxicology: The Basic Science of Poisons, 8th ed.; McGraw-Hill: New York, NY, USA, 2013. [Google Scholar]
- Narahashi, T.; Carter, D.B.; Frey, J.; Ginsburg, K.; Hamilton, B.J.; Nagata, K.; Roy, M.L.; Song, J.H.; Tatebayashi, H. Sodium channels and gabaa receptor-channel complex as targets of environmental toxicants. Toxicol. Lett. 1995, 82–83, 239–245. [Google Scholar] [CrossRef]
- Abalis, I.M.; Eldefrawi, M.E.; Eldefrawi, A.T. Effects of insecticides on gaba-induced chloride influx into rat brain microsacs. J. Toxicol. Environ. Health 1986, 18, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Bloomquist, J.R.; Soderlund, D.M. Neurotoxic insecticides inhibit gaba-dependent chloride uptake by mouse brain vesicles. Biochem. Biophys. Res. Commun. 1985, 133, 37–43. [Google Scholar] [CrossRef]
- Cole, L.M.; Casida, J.E. Polychlorocycloalkane insecticide-induced convulsions in mice in relation to disruption of the gaba-regulated chloride ionophore. Life Sci. 1986, 39, 1855–1862. [Google Scholar] [CrossRef]
- Matsumura, F.; Ghiasuddin, S.M. Evidence for similarities between cyclodiene type insecticides and picrotoxinin in their action mechanisms. J. Environ. Sci. Health B 1983, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gant, D.B.; Eldefrawi, M.E.; Eldefrawi, A.T. Cyclodiene insecticides inhibit gabaa receptor-regulated chloride transport. Toxicol. Appl. Pharmacol. 1987, 88, 313–321. [Google Scholar] [CrossRef]
- Fenster, L.; Eskenazi, B.; Anderson, M.; Bradman, A.; Hubbard, A.; Barr, D.B. In Utero exposure to ddt and performance on the brazelton neonatal behavioral assessment scale. Neurotoxicology 2007, 28, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Ribas-Fito, N.; Cardo, E.; Sala, M.; Eulalia de Muga, M.; Mazon, C.; Verdu, A.; Kogevinas, M.; Grimalt, J.O.; Sunyer, J. Breastfeeding, exposure to organochlorine compounds, and neurodevelopment in infants. Pediatrics 2003, 111, e580–e585. [Google Scholar] [CrossRef] [PubMed]
- Gladen, B.C.; Rogan, W.J.; Hardy, P.; Thullen, J.; Tingelstad, J.; Tully, M. Development after exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene transplacentally and through human milk. J. Pediatr. 1988, 113, 991–995. [Google Scholar] [CrossRef]
- Boucher, O.; Simard, M.N.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Chajes, V.; Dallaire, R.; Monfort, C.; Thome, J.P.; et al. Exposure to an organochlorine pesticide (chlordecone) and development of 18-month-old infants. Neurotoxicology 2013, 35, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.M.; Wetmur, J.; Chen, J.; Zhu, C.; Barr, D.B.; Canfield, R.L.; Wolff, M.S. Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood. Environ. Health Perspect. 2011, 119, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Hoppin, J.A. Association of pesticide exposure with neurologic dysfunction and disease. Environ. Health Perspect. 2004, 112, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Clavel, J.; Rathouz, P.J.; Moisan, F.; Galanaud, J.P.; Delemotte, B.; Alperovitch, A.; Tzourio, C. Professional exposure to pesticides and parkinson disease. Ann. Neurol. 2009, 66, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F. Epidemiology. paths from pesticides to parkinson’s. Science 2013, 341, 722–723. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; Knekt, P.; O’Reilly, E.J.; Lyytinen, J.; Reunanen, A.; Laden, F.; Altshul, L.; Ascherio, A. Persistent organochlorine pesticides in serum and risk of parkinson disease. Neurology 2010, 74, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.L.; Fitzmaurice, A.G.; Cockburn, M.; Bronstein, J.M.; Sinsheimer, J.S.; Ritz, B. Pesticides that inhibit the ubiquitin-proteasome system: Effect measure modification by genetic variation in Skp1 in parkinsons disease. Environ. Res. 2013, 126, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Roy, A.; Shalat, S.L.; Buckley, B.; Winnik, B.; Gearing, M.; Levey, A.I.; Factor, S.A.; O’Suilleabhain, P.; German, D.C. Beta-hexachlorocyclohexane levels in serum and risk of parkinson’s disease. Neurotoxicology 2011, 32, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; McLean, A.J.; Taylor, M.C.; Woodham, B.L.; Board, P.G. Pesticides and parkinson’s disease. Biomed. Pharmacother. 1999, 53, 122–130. [Google Scholar] [CrossRef]
- Priyadarshi, A.; Khuder, S.A.; Schaub, E.A.; Shrivastava, S. A meta-analysis of parkinson’s disease and exposure to pesticides. Neurotoxicology 2000, 21, 435–440. [Google Scholar] [PubMed]
- Semchuk, K.M.; Love, E.J.; Lee, R.G. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology 1992, 42, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Goldman, S.M. Epidemiology of parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Corrigan, F.M.; Murray, L.; Wyatt, C.L.; Shore, R.F. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in parkinson’s disease. Exp. Neurol. 1998, 150, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.M.; Wienburg, C.L.; Shore, R.F.; Daniel, S.E.; Mann, D. Organochlorine insecticides in substantia nigra in parkinson’s disease. J. Toxicol. Environ. Health A 2000, 59, 229–234. [Google Scholar] [PubMed]
- Richardson, J.R.; Shalat, S.L.; Buckley, B.; Winnik, B.; O’Suilleabhain, P.; Diaz-Arrastia, R.; Reisch, J.; German, D.C. Elevated serum pesticide levels and risk of parkinson disease. Arch. Neurol. 2009, 66, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Fleming, L.; Mann, J.B.; Bean, J.; Briggle, T.; Sanchez-Ramos, J.R. Parkinson’s disease and brain levels of organochlorine pesticides. Ann. Neurol. 1994, 36, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.W.; Shapiro, L.P.; Bradner, J.M.; Caudle, W.M. Developmental exposure to the organochlorine insecticide endosulfan damages the nigrostriatal dopamine system in male offspring. Neurotoxicology 2014, 44, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hudson, P.M.; Chen, P.H.; Tilson, H.A.; Hong, J.S. Effects of P,P’-Ddt on the rat brain concentrations of biogenic amine and amino acid neurotransmitters and their association with P,P’-Ddt-induced tremor and hyperthermia. J. Neurochem. 1985, 45, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.F.; Chevrier, J.; Harley, K.G.; Kogut, K.; Vedar, M.; Calderon, N.; Trujillo, C.; Johnson, C.; Bradman, A.; Barr, D.B.; et al. Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children. Environ. Health Perspect. 2011, 119, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rasoul, G.M.; Abou Salem, M.E.; Mechael, A.A.; Hendy, O.M.; Rohlman, D.S.; Ismail, A.A. Effects of occupational pesticide exposure on children applying pesticides. Neurotoxicology 2008, 29, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Eckerman, D.A.; Gimenes, L.S.; de Souza, R.C.; Galvao, P.R.; Sarcinelli, P.N.; Chrisman, J.R. Age related effects of pesticide exposure on neurobehavioral performance of adolescent farm workers in Brazil. Neurotoxicol. Teratol. 2007, 29, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Marks, A.R.; Bradman, A.; Harley, K.; Barr, D.B.; Johnson, C.; Morga, N.; Jewell, N.P. Organophosphate pesticide exposure and neurodevelopment in young Mexican-American children. Environ. Health Perspect. 2007, 115, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Harari, R.; Barr, D.B.; Debes, F. Pesticide exposure and stunting as independent predictors of neurobehavioral deficits in ecuadorian school children. Pediatrics 2006, 117, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guodong, D.; Pei, W.; Ying, T.; Jun, Z.; Yu, G.; Xiaojin, W.; Rong, S.; Guoquan, W.; Xiaoming, S. Organophosphate pesticide exposure and neurodevelopment in young Shanghai children. Environ. Sci. Technol. 2012, 46, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Handal, A.J.; Harlow, S.D.; Breilh, J.; Lozoff, B. Occupational exposure to pesticides during pregnancy and neurobehavioral development of infants and toddlers. Epidemiology 2008, 19, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Handal, A.J.; Lozoff, B.; Breilh, J.; Harlow, S.D. Effect of community of residence on neurobehavioral development in infants and young children in a flower-growing region of Ecuador. Environ. Health Perspect. 2007, 115, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Harari, R.; Julvez, J.; Murata, K.; Barr, D.; Bellinger, D.C.; Debes, F.; Grandjean, P. Neurobehavioral deficits and increased blood pressure in school-age children prenatally exposed to pesticides. Environ. Health Perspect. 2010, 118, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizardi, P.S.; O’Rourke, M.K.; Morris, R.J. The effects of organophosphate pesticide exposure on hispanic children’s cognitive and behavioral functioning. J. Pediatr. Psychol. 2008, 33, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Rohlman, D.S.; Arcury, T.A.; Quandt, S.A.; Lasarev, M.; Rothlein, J.; Travers, R.; Tamulinas, A.; Scherer, J.; Early, J.; Marin, A.; et al. Neurobehavioral performance in preschool children from agricultural and non-agricultural communities in Oregon and North Carolina. Neurotoxicology 2005, 26, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rohlman, D.S.; Lasarev, M.; Anger, W.K.; Scherer, J.; Stupfel, J.; McCauley, L. Neurobehavioral performance of adult and adolescent agricultural workers. Neurotoxicology 2007, 28, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Ruckart, P.Z.; Kakolewski, K.; Bove, F.J.; Kaye, W.E. Long-term neurobehavioral health effects of Methyl Parathion exposure in children in Mississippi and Ohio. Environ. Health Perspect. 2004, 112, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.F.; Bellinger, D.C.; Wright, R.O.; Weisskopf, M.G. Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides. Pediatrics 2010, 125, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R.; Harley, K.; Bradman, A.; Kogut, K.; Barr, D.B.; Johnson, C.; Calderon, N.; Eskenazi, B. Organophosphate pesticide exposure and attention in young mexican-american children: The chamacos study. Environ. Health Perspect. 2010, 118, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.M.; Berkowitz, G.S.; Barr, D.B.; Teitelbaum, S.L.; Siskind, J.; Meisel, S.J.; Wetmur, J.G.; Wolff, M.S. Prenatal organophosphate metabolite and organochlorine levels and performance on the brazelton neonatal behavioral assessment scale in a Multiethnic Pregnancy Cohort. Am. J. Epidemiol. 2007, 165, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, J.G.; Takhar, H.S.; Ruffalo, C.A.; Zwass, M. Health effects of diazinon on a family. J. Toxicol. Clin. Toxicol. 2004, 42, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Young, J.G.; Eskenazi, B.; Gladstone, E.A.; Bradman, A.; Pedersen, L.; Johnson, C.; Barr, D.B.; Furlong, C.E.; Holland, N.T. Association between in Utero organophosphate pesticide exposure and abnormal reflexes in Neonates. Neurotoxicology 2005, 26, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Sheets, L.P. A consideration of age-dependent differences in susceptibility to organophosphorus and pyrethroid insecticides. Neurotoxicology 2000, 21, 57–63. [Google Scholar] [PubMed]
- Holland, N.; Furlong, C.; Bastaki, M.; Richter, R.; Bradman, A.; Huen, K.; Beckman, K.; Eskenazi, B. Paraoxonase polymorphisms, haplotypes, and enzyme activity in latino mothers and newborns. Environ. Health Perspect. 2006, 114, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Furlong, M.A.; Engel, S.M.; Barr, D.B.; Wolff, M.S. Prenatal exposure to organophosphate pesticides and reciprocal social behavior in childhood. Environ. Int. 2014, 70, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Brix, K.A. Review of health consequences from high-, intermediate- and low-level exposure to organophosphorus nerve agents. J. Appl. Toxicol. 1998, 18, 393–408. [Google Scholar] [CrossRef]
- Jamal, G.A.; Hansen, S.; Pilkington, A.; Buchanan, D.; Gillham, R.A.; Abdel-Azis, M.; Julu, P.O.; Al-Rawas, S.F.; Hurley, F.; Ballantyne, J.P. A clinical neurological, neurophysiological, and neuropsychological study of sheep farmers and dippers exposed to organophosphate pesticides. Occup Environ. Med. 2002, 59, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, L.; Keifer, M.; Daniell, W.E.; McConnell, R.; Claypoole, K. Chronic central nervous system effects of acute organophosphate pesticide intoxication. the pesticide health effects study group. Lancet 1991, 338, 223–227. [Google Scholar] [CrossRef]
- Steenland, K. Chronic neurological effects of organophosphate pesticides. BMJ 1996, 312, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Speed, H.E.; Blaiss, C.A.; Kim, A.; Haws, M.E.; Melvin, N.R.; Jennings, M.; Eisch, A.J.; Powell, C.M. Delayed reduction of hippocampal synaptic transmission and spines following exposure to repeated subclinical doses of organophosphorus pesticide in adult mice. Toxicol. Sci. 2012, 125, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, T.K.; Farrar, J.D.; Pope, C.N. Comparative neurochemical and neurobehavioral effects of repeated chlorpyrifos exposures in young and adult rats. Pharmacol. Biochem. Behav. 1993, 46, 219–224. [Google Scholar] [CrossRef]
- Chanda, S.M.; Harp, P.; Liu, J.; Pope, C.N. Comparative developmental and maternal neurotoxicity following acute gestational exposure to chlorpyrifos in rats. J. Toxicol. Environ. Health 1995, 44, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Pennell, K.D.; Miller, G.W. Parkinson’s disease and pesticides: A toxicological perspective. Trends Pharmacol. Sci. 2008, 29, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Panuwet, P.; Prapamontol, T.; Chantara, S.; Barr, D.B. Urinary pesticide metabolites in school students from Northern Thailand. Int. J. Hyg. Environ. Health 2009, 212, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Rohitrattana, J.; Siriwong, W.; Robson, M.; Panuwet, P.; Barr, D.B.; Fiedler, N. Pyrethroid insecticide exposure in school-aged children living in rice and aquacultural farming regions of Thailand. Risk Manag. Healthc. Policy 2014, 7, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Singleton, S.T.; Lein, P.J.; Farahat, F.M.; Farahat, T.; Bonner, M.R.; Knaak, J.B.; Olson, J.R. Characterization of alpha-cypermethrin exposure in Egyptian agricultural workers. Int. J. Hyg. Environ. Health 2014, 217, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Ostrea, E.M., Jr.; Villanueva-Uy, E.; Bielawski, D.; Birn, S.; Janisse, J.J. Trends in Long term exposure to propoxur and pyrethroids in young children in the Philippines. Environ. Res. 2014, 131, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zheng, M.; Wu, C.; Wang, G.; Feng, C.; Zhou, Z. urinary pyrethroid metabolites among pregnant women in an agricultural area of the province of Jiangsu, China. Int. J. Hyg. Environ. Health 2012, 215, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Babina, K.; Dollard, M.; Pilotto, L.; Edwards, J.W. Environmental exposure to organophosphorus and pyrethroid pesticides in South Australian preschool children: A cross sectional study. Environ. Int. 2012, 48, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Burr, S.A.; Ray, D.E. Structure-activity and interaction effects of 14 different pyrethroids on Voltage-Gated chloride ion channels. Toxicol. Sci. 2004, 77, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Symington, S.B. Neurotoxic implications of the agonistic action of cs-syndrome pyrethroids on the N-type ca(v)2.2 calcium channel. Pest. Manag. Sci. 2008, 64, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.S.; Kim, K.B.; Padilla, S.; Muralidhara, S.; Kim, H.J.; Fisher, J.W.; Bruckner, J.V. Ontogeny of hepatic and plasma metabolism of deltamethrin in vitro: Role in Age-Dependent Acute Neurotoxicity. Drug. Metab. Dispos. 2006, 34, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Horton, M.K.; Rundle, A.; Camann, D.E.; Boyd Barr, D.; Rauh, V.A.; Whyatt, R.M. Impact of prenatal exposure to piperonyl butoxide and permethrin on 36-month neurodevelopment. Pediatrics 2011, 127, e699–e706. [Google Scholar] [CrossRef] [PubMed]
- Oulhote, Y.; Bouchard, M.F. Urinary metabolites of organophosphate and pyrethroid pesticides and behavioral problems in Canadian children. Environ. Health Perspect. 2013, 121, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Viel, J.F.; Warembourg, C.; Le Maner-Idrissi, G.; Lacroix, A.; Limon, G.; Rouget, F.; Monfort, C.; Durand, G.; Cordier, S.; Chevrier, C. Pyrethroid insecticide exposure and cognitive developmental disabilities in children: The pelagie mother-child cohort. Environ. Int. 2015, 82, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, N.; Rohitrattana, J.; Siriwong, W.; Suttiwan, P.; Ohman Strickland, P.; Ryan, P.B.; Rohlman, D.S.; Panuwet, P.; Barr, D.B.; Robson, M.G. Neurobehavioral effects of exposure to organophosphates and pyrethroid pesticides among Thai children. Neurotoxicology 2015, 48, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Gillette, J.S.; Bloomquist, J.R. Differential up-regulation of striatal dopamine transporter and alpha-synuclein by the pyrethroid insecticide permethrin. Toxicol Appl. Pharmacol. 2003, 192, 287–293. [Google Scholar] [CrossRef]
- Collotta, M.; Bertazzi, P.A.; Bollati, V. Epigenetics and pesticides. Toxicology 2013, 307, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kanthasamy, A.; Anantharam, V.; Sun, F.; Kanthasamy, A.G. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, L.; Nasuti, C.; Mirto, M.; Caradonna, F.; Gabbianelli, R. Intergenerational effect of early life exposure to permethrin: Changes in global dna methylation and in nurr1 gene expression. Toxics 2015, 3, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004, 23, 588s–595s. [Google Scholar] [CrossRef] [PubMed]
- Tartaglione, A.M.; Venerosi, A.; Calamandrei, G. Early-life toxic insults and onset of sporadic neurodegenerative diseases-an overview of experimental studies. Curr. Top. Behav. Neurosci. 2015, 29, 231–264. [Google Scholar]
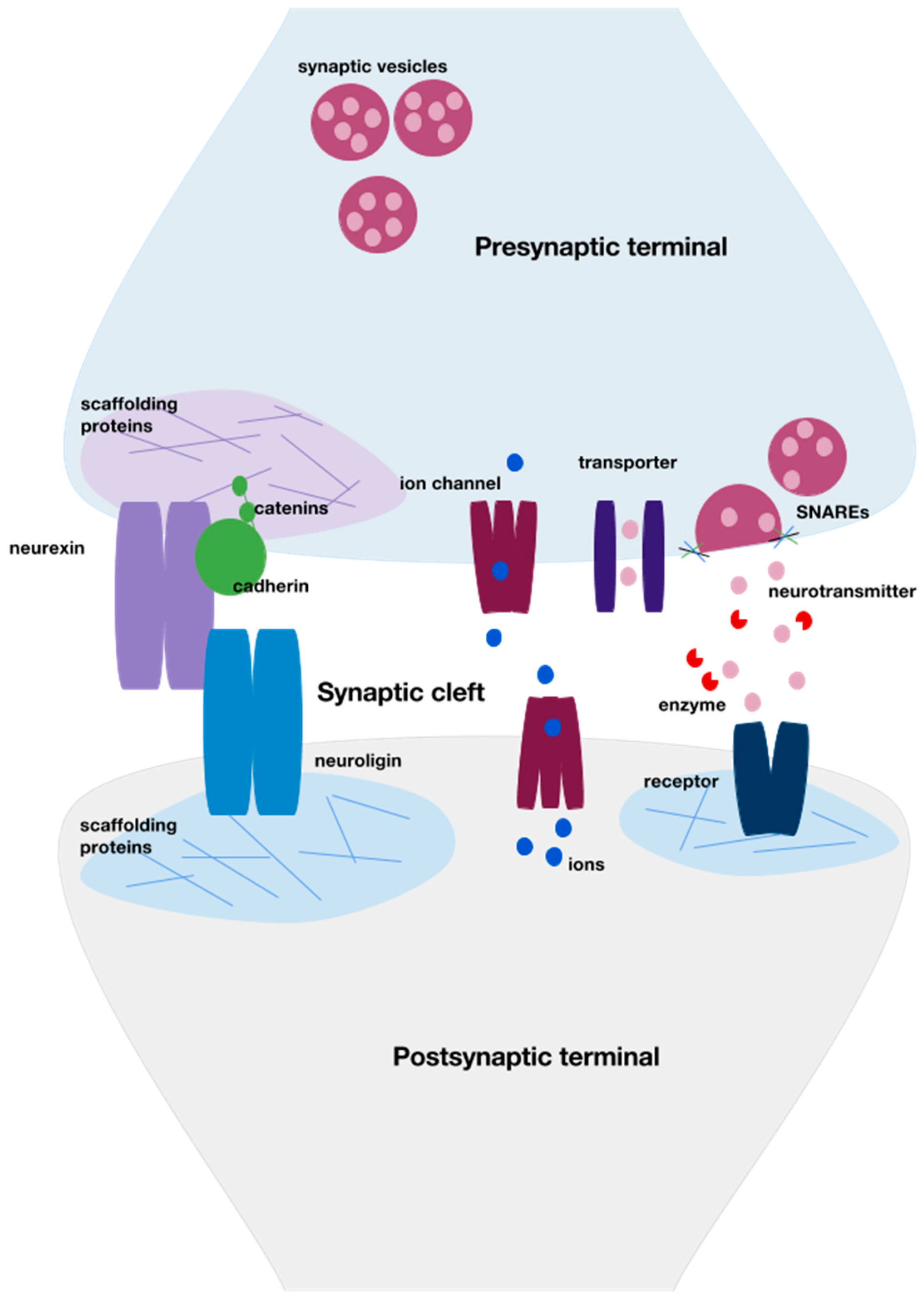

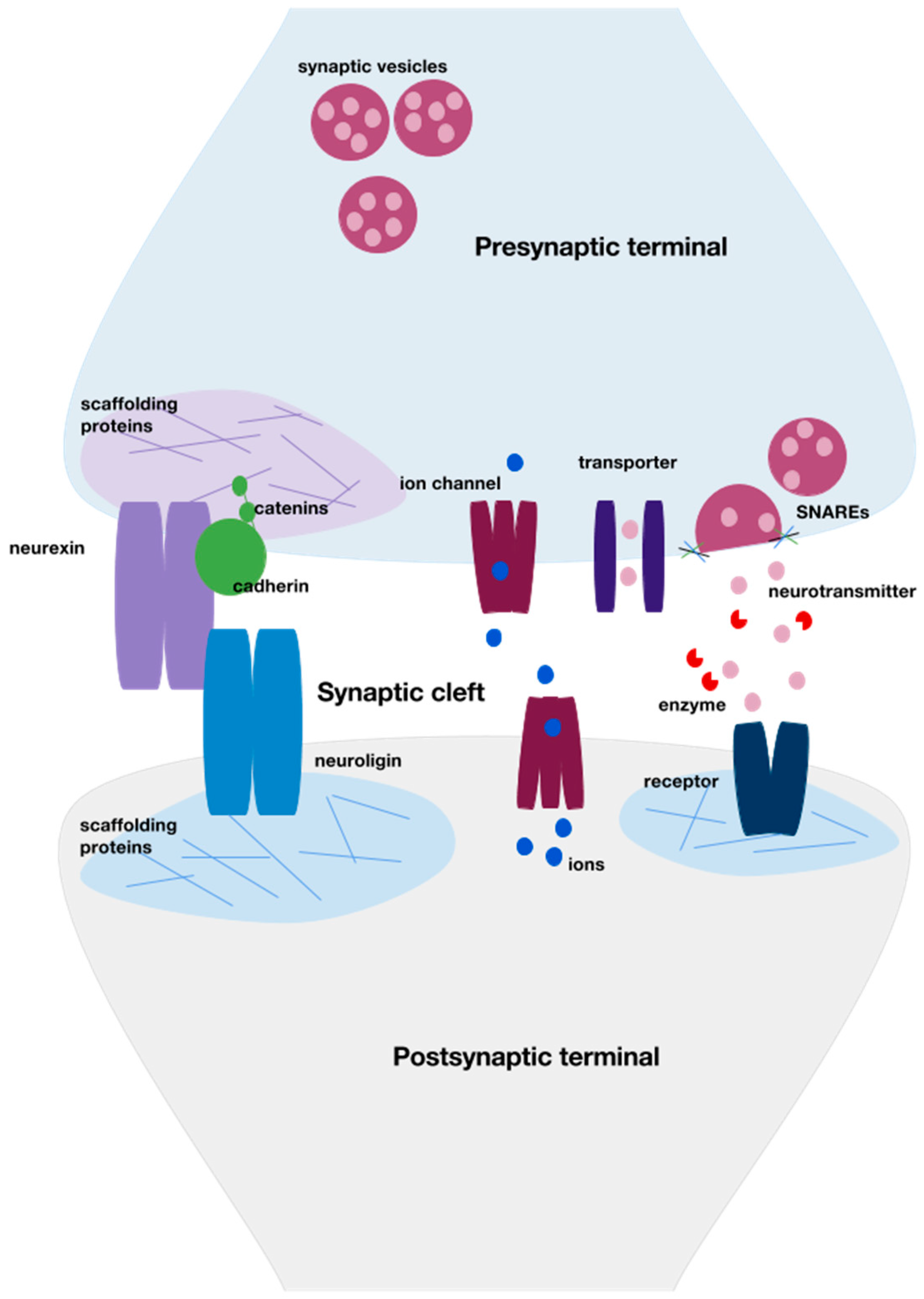{kind=link}
| Subject | Findings | Reference(s) |
|---|---|---|
| Organochlorines | ||
| Human (Children) | Decr. cognitive, quantitative, verbal, sensory, memory functions; hyporeflexia | [81,82,83,84,85,86,87] |
| Human (Adults) | Decr. neurobehavioral performance | [88] |
| Incr. serum and brain DDE levels associated with AD | [78] | |
| Presence of heptachlor assoc. with Lewy body pathology | [89] | |
| Mice | Incr. DAT, increased DA uptake, VMAT2, TH in striatum | [90,91,92] |
| DA neuron loss in substantia nigra, gliosis in ventral midbrain | [93,94,95,96] | |
| Incr. NE in hippocampus, brainstem | [97] | |
| Incr. 5HT in frontal cortex, decr. 5HT in striatum | [98] | |
| Altered GABAA, GluN2B, D2 receptors in frontal cortex | [58] | |
| Altered CAMKII, GluR1, tau in hippocampus and frontal cortex | [99] | |
| Parkinsonism-like movement | [93] | |
| Motor, cognitive behavioral deficits | [99] | |
| Developmental exposure potentiates MPTP toxicity | [91,100] | |
| Rats | Incr. DAT binding | [101] |
| Mouse cortex primary culture | Decr. synaptic puncta, neurite outgrowth, synaptogenesis | [56,102] |
| Altered MAPK, PI3K/Akt, estrogen receptor pathways | [94] | |
| Incr. NMDA receptor internalization and decr. mGLUR5 levels | [103] | |
| Mouse cerebellum primary culture | Decr. GABAA and NMDA receptors | [104] |
| Organophosphates | ||
| Human (Children) | Decr. IQ, working memory index, cognition | [68,72] |
| Incr. risk of ADHD | [68] | |
| Human (Adults) | Self-reported memory, fatigue, muscle strength issues | [105] |
| Mice | Altered 5HT transporter, 5HT receptors in forebrain, brainstem | [106,107,108] |
| Reduced ChAT, vAchT | [109,110,111] | |
| Learning, memory defects, incr. locomotion | [112,113,114] | |
| Decr. CAMKII in hippocampus, synaptophysin in frontal cortex | [114] | |
| Rats | Decr. muscarinic receptors, decr. AChE activity, ChAT, vAchT | [115,116,117,118,119] |
| Pyrethroids | ||
| Mice | ADHD-like behaviors: working memory and attention deficits, hyperactivity, impulse-like behaviors | [71] |
| Incr. striatal DA uptake mediated by DAT, incr. D1 receptor | [71,120] | |
| Altered GluR1 in hippocampus, altered tau in frontal cortex | [99] | |
| Rats | Striatal administration causes altered extracellular 5HT release | [121] |
| Incr. extracellular glutamate, decr. GABA | [122] | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vester, A.; Caudle, W.M. The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides. Toxics 2016, 4, 18. https://doi.org/10.3390/toxics4030018
Vester A, Caudle WM. The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides. Toxics. 2016; 4(3):18. https://doi.org/10.3390/toxics4030018
Chicago/Turabian StyleVester, Aimee, and W. Michael Caudle. 2016. "The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides" Toxics 4, no. 3: 18. https://doi.org/10.3390/toxics4030018