Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration

FNRS—ULB, Unité d'Etude du Mouvement, 808 Route de Lennik, 1070 Bruxelles, Belgium
Toxics 2014, 2(2), 327-345; https://doi.org/10.3390/toxics2020327
Submission received: 6 May 2014
/
Revised: 4 June 2014
/
Accepted: 5 June 2014
/
Published: 18 June 2014
(This article belongs to the Collection Heavy Metals Toxicology)
Abstract
:As a cofactor of proteins and enzymes involved in critical molecular pathways in mammals and low eukaryotes, copper is a transition metal essential for life. The intra-cellular and extra-cellular metabolism of copper is under tight control, in order to maintain free copper concentrations at very low levels. Copper is a critical element for major neuronal functions, and the central nervous system is a major target of disorders of copper metabolism. Both the accumulation of copper and copper deficiency are associated with brain dysfunction. The redox capacities of free copper, its ability to trigger the production of reactive oxygen species and the close relationships with the regulation of iron and zinc are remarkable features. Major advances in our understanding of the relationships between copper, neuronal functions and neurodegeneration have occurred these last two decades. The metabolism of copper and the current knowledge on the consequences of copper dysregulation on brain disorders are reviewed, with a focus on neurodegenerative diseases, such as Wilson’s disease, Alzheimer’s disease and Parkinson’s disease. In vitro studies, in vivo experiments and evidence from clinical observations of the neurotoxic effects of copper provide the basis for future therapies targeting copper homeostasis.
1. Introduction
Copper is a cofactor of proteins and enzymes (called cuproenzymes) involved in fundamental mechanisms, such as energy generation, oxygen transportation, hematopoiesis, cellular metabolism and signal transduction [1]. It is therefore essential for living cells. There is a noticeable evolutionary conservation of regulating mechanisms [2]. Its metabolism is linked to the metabolism of iron and zinc, two other essential transition metals [3,4]. The bulk of evidence that copper is involved in the pathogenesis of neurodegenerative disorders is now huge. The recent literature is reviewed, focusing on the impacts of copper dyshomeostasis on neurodegeneration.
2. Metabolism of Copper
In the human body, most of the copper, Cu, is present as Cu+ (cuprous) and oxidized Cu2+ (cupric) compounds. Copper is therefore an intermediary for electron transfer in redox reactions. The oxidation states, Cu3+ and Cu4+, are uncommon [5]. The average daily intake of copper is between 0.5 and 1.5 mg, coming mainly from seeds, grains, shellfish, nuts, beans and liver [6]. The current recommended dietary intake in the USA is 0.9 mg/day [7]. Copper is mainly absorbed in the duodenum and proximal jejunum, with a little bit of absorption occurring in the stomach and the distal portion of the small intestine [8]. The human copper transport protein 1 (hCTR1), located at the level of enterocytes, transfers the ion following the reduction of dietary Cu2+ into Cu+. In hepatocytes, copper binds to metallothioneins (MTs), to reduced glutathione (GSH) or to one of the copper chaperones regulating the traffic of intracellular copper (CCS: chaperone for superoxide dismutase 1 SOD1, which is the sole cytosolic cuproenzyme; COX17: chaperone for cytochrome C oxygenase; ATOX1 antioxidant-1: chaperone for the ATPases, ATP7A and ATP7B). The group of transmembrane copper transporters includes CTR1, ATP7A and ATP7B. ATP7A (expressed in the placenta, gut and nervous system) and ATP7B (expressed in the hepatocytes, where it exports copper into the bile and provides copper to nascent ceruloplasmin, and in the nervous system) are linked to the enzyme, tyrosinase, and the ceruloplasmin, respectively. In blood, about 65%–90% of the copper Cu2+ is bound to ceruloplasmin. The remaining 10–35 percent participate in exchanges with albumin, transcuprein, alpha 2 macroglobulin, and low-molecular-weight compounds [1].
In terms of storage, the total amount of copper in an adult is estimated to be about 90–110 mg. The organs with the highest concentrations are the liver, brain, kidney and heart [9]. Bones and skeletal muscles contain about 47% and 27% of the copper, respectively [10]. Both the liver and brain contain about 8%–11% of the total body copper. Figure 1 illustrates the metabolism of copper in the brain. Brain concentrations range from 3.1 to 5.1 mg/g wet weight [11,12]. Within the brain, the distribution of copper is heterogeneous. Concentrations are higher in the hippocampus, substantia nigra and locus coeruleus [5,13]. Glial cells are enriched in copper as compared to neurons [5]. The concentrations of copper in the synaptic cleft range from 0.2 to 1.7 µM. Some studies have shown concentrations higher than 200 µM [14]. Concentrations in cerebrospinal fluid (CSF) are 100-fold lower as compared to plasmatic levels, and intra-neuronal concentrations are kept very low [15]. Choroid plexus could serve as a storage compartment in the vicinity of the brain.
Copper is excreted endogenously in the saliva, the gastric fluids and intestinal liquids. Copper is excreted from the body either in a non-absorbed form or via the bile. The estimated amount of copper in feces is 1–1.5 mg/day. The quantities lost in urine, saliva and perspiration are even smaller.
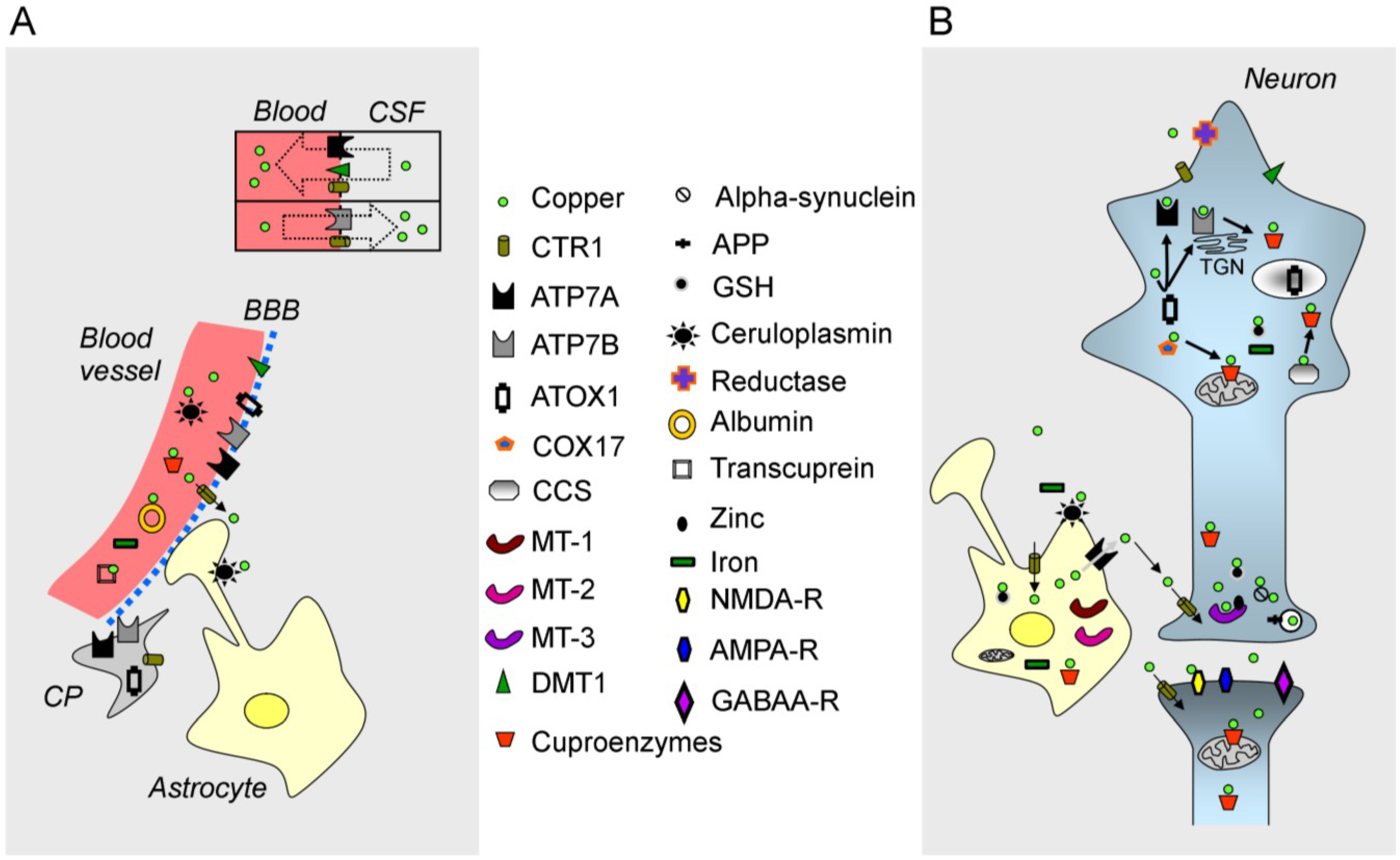
Figure 1.
Illustration of copper metabolism in the central nervous system (CNS). (A) A blood vessel (in red on the left), the blood-brain barrier (BBB), the blood-CSF barrier (shown by a rectangle with two compartments), a choroid plexus (CP) and an astrocyte are illustrated; (B) A neuron, a synapse and an astrocyte are shown. Copper is represented by a green circle. At the blood-CSF barrier, CTR1 (copper transporter 1), DMT1 (divalent metal transporter) and the ATPase, ATP7A, transport copper towards the blood, whereas the ATPase, ATP7B, and CTR1 transport copper in the opposite direction. Organelles and proteins involved in the cellular regulation of copper are represented. CTR1 is the main transporter for transferring copper within cells. Chaperones (ATOX1, COX17, CCS) deliver copper to the ATPases, ATP7A/ATP7B, and to cuproenzymes, including in the mitochondria. Metallothioneins (MT-1, MT-2, MT-3) exert a function as a buffer. Copper is also found in secretory granules. Abbreviations: APP, amyloid precursor protein; GSH, glutathione; NMDA-R, NMDA receptor; AMPA-R, AMPA receptor; GABAA-R, GABAA receptor; TGN, trans-Golgi network.
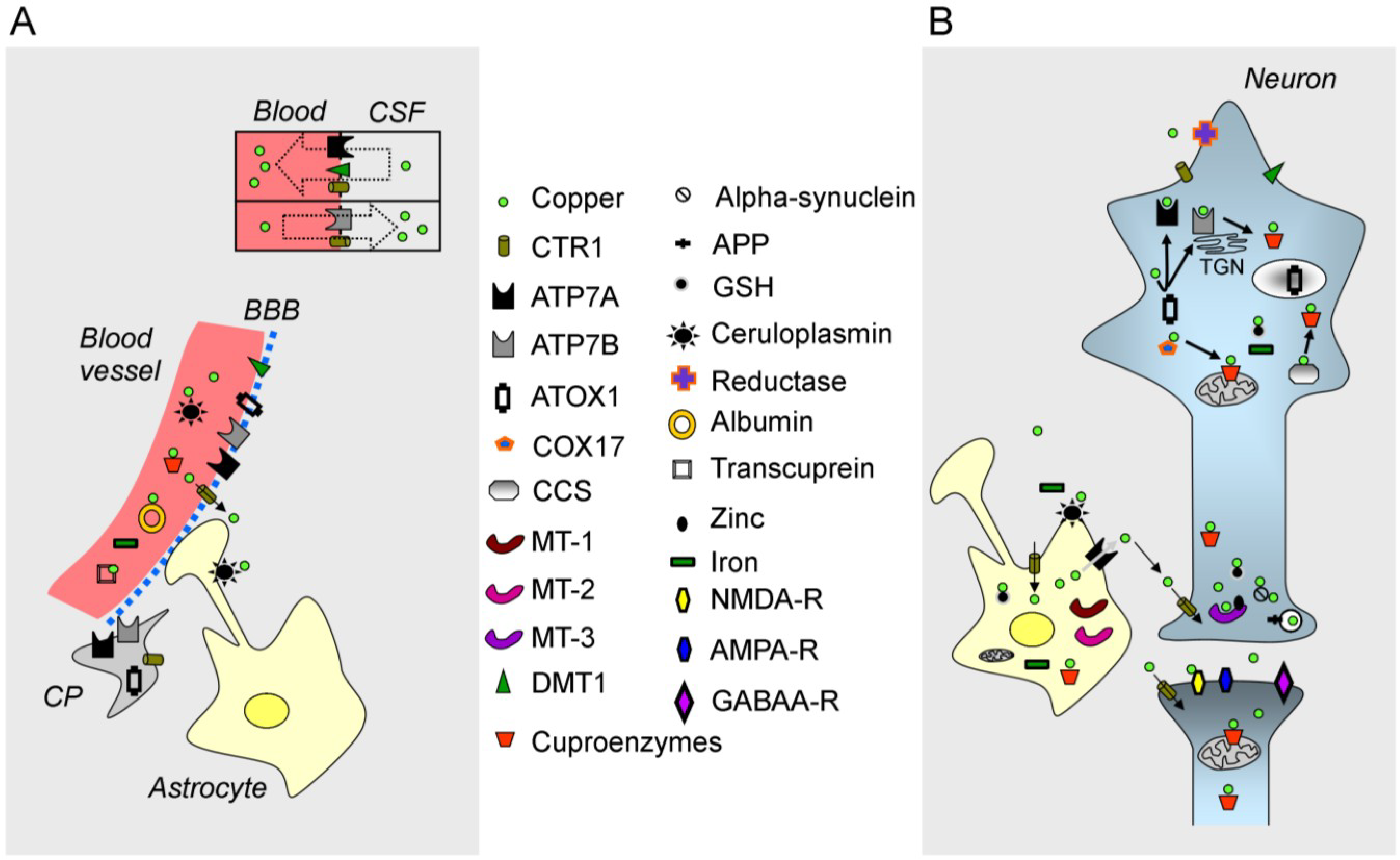
Figure 1.
Illustration of copper metabolism in the central nervous system (CNS). (A) A blood vessel (in red on the left), the blood-brain barrier (BBB), the blood-CSF barrier (shown by a rectangle with two compartments), a choroid plexus (CP) and an astrocyte are illustrated; (B) A neuron, a synapse and an astrocyte are shown. Copper is represented by a green circle. At the blood-CSF barrier, CTR1 (copper transporter 1), DMT1 (divalent metal transporter) and the ATPase, ATP7A, transport copper towards the blood, whereas the ATPase, ATP7B, and CTR1 transport copper in the opposite direction. Organelles and proteins involved in the cellular regulation of copper are represented. CTR1 is the main transporter for transferring copper within cells. Chaperones (ATOX1, COX17, CCS) deliver copper to the ATPases, ATP7A/ATP7B, and to cuproenzymes, including in the mitochondria. Metallothioneins (MT-1, MT-2, MT-3) exert a function as a buffer. Copper is also found in secretory granules. Abbreviations: APP, amyloid precursor protein; GSH, glutathione; NMDA-R, NMDA receptor; AMPA-R, AMPA receptor; GABAA-R, GABAA receptor; TGN, trans-Golgi network.
2.1. Free Copper
The fact that cells contain one free copper ion or less highlights the tight regulation [16]. The redox capacities of free copper and its ability to initiate the production of free radicals are two important features. Free copper is a catalyst of the Fenton reaction (Cu+ and H2O2 generate Cu2+ + OH− + OH). The hydrogen peroxide is transformed into the very reactive hydroxyl radical, which combines with nucleic acids, proteins and lipids. An increase in free copper is potentially harmful for brain circuits, despite the presence of MTs and chaperones (see below).
2.2. Ceruloplasmin
Ceruloplasmin is a multicopper-containing protein mainly synthesized by the liver. Although the rate of synthesis is not influenced by copper intake, ceruloplasmin lacking bound copper has a shorter half-life [17]. Ceruloplasmin plays important functions, acting as an iron oxidase, an amine oxidase, an antioxidant and a glutathione peroxidase [18,19]. As a multicopper oxidase, ceruloplasmin reduces dioxygen, O2, to two water molecules. Ceruloplasmin is a scavenger of reactive oxygen species (ROS). It can be considered that the genuine link between copper metabolism and iron metabolism is mediated by ceruloplasmin. Its soluble form controls the oxidation of iron to be included into transferrin. A deficit in copper reduces the ferroxidase activity of ceruloplasmin (Fe2+ to Fe3+). Dietary and recycled iron are in the Fe2+ oxidation state, but iron is transported in serum by transferrin only as Fe3+ after its export by ferroportin [4]. Iron itself contributes to the formation of ROS. A deficit of dietary copper leads not only to an accumulation of iron in the liver, but also to an impaired distribution within the spinal cord (see zinc-induced myeloneuropathy in the next section). In the central nervous system (CNS), a glycosylphosphatidylinositol-linked ceruloplasmin bound to the cell membranes is the major isoform of this protein [20]. Astrocytes can synthesize their own ceruloplasmin. This glial ceruloplasmin controls also the process of iron oxidation, which allows the clearance of iron from the CNS [21]. Overall, functional ceruloplasmin promotes the synthesis of proteins involved in iron efflux. The maintenance of the iron balance in the brain is thus closely linked to the metabolism of copper.
2.3. The ATPases ATP7A/ATP7B, CTR1 and Chaperones
The ATPases, ATP7A and ATP7B (belonging to the family of P1B-type ATPases), play an important role in the metabolism of copper. They depend on ATP hydrolysis to move heavy metals across cellular membranes. The ATP7A, a highly conserved ion-motive ATPase [22,23], is an intra-cellular pump transferring copper into the Golgi apparatus. This is required to incorporate the ion into cuproenzymes, such as the dopamine-beta-hydroxylase (see Table 1 for the main cuproenzymes). ATP7A is critical to deliver copper from endothelial cells in the direction of the brain [24]. ATP7A is essential not only for the CNS, but also for peripheral nerves [25]. ATP7B is also involved in the translocation of copper across membranes, being located mainly at the apical membrane. Both ATPases can be redistributed to post-Golgi vesicles and cellular membranes if the concentration of copper rises, in order to regulate the export of the ion [26].
CTR1, a protein of 190 amino acids, transports dietary copper within the cells and is the key-regulator of copper influx [27]. CTR1 is particularly expressed in the intestinal cells, in the endothelial cells of brain capillaries, in choroid plexus and brain parenchyma [28]. CTR1 is a transmembrane carrier, which may shift easily between plasma membrane and intra-cellular vesicles [29]. It is estimated that the uptake of at least 80% of the copper is controlled by the carrier [30]. CTR1 is encoded by SLC31A1. The transcription factor, SP1 (specificity protein 1), responds to changes in concentrations of copper [27]. When the extra-cellular concentration of copper rises, CTR1 is internalized. CTR1 regulates also the transport of chemotherapeutic drugs, such as cisplatin. The degree of expression of the carrier is correlated with the prognosis in cancer [31]. CTR2 is a homologous carrier, whose expression profile remains poorly defined [27]. CTR2 probably plays a role in the recycling of copper in internal compartments of the cell [32]. CTR1, the nonspecific divalent metal transporter 1 (DMT1) and ATP7A transport copper from CSF to blood, whereas ATP7B and also CTR1 control the transport in the opposite direction [33]. CTR1 is considered as the main transporter of copper in the direction of the brain. Deletion of CTR1 is lethal, suggesting that it plays a major role in transporting copper in the developing embryo [34].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Function | Physiological Roles |
|---|---|---|
| Superoxide dismutase 1 (SOD1) and 3 (SOD3) * | Converts superoxide to hydrogen peroxide | Anti-oxidative defense |
| Dopamine-beta-hydroxylase | Catecholamine production | Regulation of autonomic nervous system |
| Monoamine oxidase | Pigment and neurotransmitter metabolism | Oxidation of monoamines |
| Cytochrome C oxidase COX (COX, complex IV of the respiratory chain) | Converts molecular oxygen to water | Energy metabolism |
| Tyrosinase | Production of melanin; conversion of tyrosine to L-DOPA | Protection of skin |
| Catalase | Conversion of hydrogen peroxide to water and oxygen | Prevents oxidative-induced damage in the heart |
| Glutathione peroxidase | Converts hydroperoxide and hydrogen peroxide | Antioxidative defense |
| Hephaestin (homolog of ceruloplasmin) | Ferroxidase activity Involved in intestinal iron absorption | Control of iron efflux |
| Cartilage matrix glycoprotein (homolog of ceruloplasmin) | Ferroxidase activity Oxidase activity | Synthesis of the extracellular matrix |
| Lysyl oxidase | Cross-linking of elastin and oxygen | Stabilization of connective tissues |
# Ceruloplasmin is a member of the multi-copper oxidase family of enzymes; * SOD1 and SOD3 contain catalytic copper and structural zinc ions in the active sites. SOD2 contains manganese as a metal cofactor.
Copper chaperones are proteins that play the function of a shuttle. They regulate the traffic of copper. Without chaperones, copper would be directly scavenged by MTs, GSH (copper-GSH complexes are the main contributor for the exchangeable cytosolic copper pool) and mitochondria. ATOX1 is a cytosolic and nuclear copper chaperone protein, which is mainly expressed in the choroid plexus, cerebral cortex, hippocampus, brainstem nuclei, olfactory bulbs and cerebellar cortex [35]. It delivers copper to ATP7A and ATP7B. The high levels of ATOX1 in subsets of neurons sequestering metals indicates that ATOX1 likely contributes to the functionality of metals requiring enzymes [35]. A chaperone for CuZn-SOD (CCS) and another for cytochrome oxidase (COX17) are expressed in the brain [5]. The chaperone for SOD is involved in the maturation of the enzyme.
2.4. Metallothioneins (MTs)
These low-molecular weight proteins are characterized by a high number of cysteine residues. This explains why they bind to metals, such as copper and zinc. Type I and type II are found in almost all organs. They are particularly expressed in astrocytes. Type III is constitutively expressed in neurons. It is currently presumed that MTs exert a neuroprotective role and participate in the compensatory mechanisms facing oxidative stress [36,37]. At the level of glutamatergic synapses, they might work as a buffer for free copper. An increase in brain copper levels is associated with an increase in the levels of MTs, likely as a rapid regulatory response [5,38].
2.5. The Blood-Brain Barrier (BBB)
Free copper crosses the blood-brain barrier [39]. CTR1 and ATP7A contribute to the net influx of copper towards the CNS. As mentioned earlier, this copper is potentially toxic, being available for the Haber–Weiss and Fenton reactions. Nevertheless, copper is rapidly sequestrated by GSH, a mechanism of protection against the toxic effects of free copper [5].
2.6. Copper and Synapses in the CNS
There is evidence that copper is enriched in synaptic levels and could even play a signaling role [40]. Copper is found in secretary granules containing SOD3, in constitutive vesicles and in endosomes. Copper in the secretory pathway is released in a calcium-dependent manner. Copper interacts with glutamatergic and GABAergic synapses and modulates voltage-gated calcium channels [40]. Stimulation of the NMDA receptor (NMDA-R) evokes the release of copper in hippocampal neurons and is associated with a repositioning of the ATP7A transporter towards the hyperactive sites [41]. Copper interacts closely with the NMDA-R and may inhibit currents related to NMDA-R activation [42]. Copper acts on the S-nitrosylation of the NMDA receptor [43]. Copper also interacts with AMPA-R, but the IC50 for inhibition is higher (4.5 µM) as compared to the NMDA-R (0.27 µM) [44]. The transition metal modulates also GABAA-R, with an impact on Cl− currents [45]. The IC50 is 2.4 µM. It affects also the extra-synaptic GABA receptors [46]. Overall, the acute effects of copper on AMPA-R and GABAA-R is inhibitory [5].
3. Copper Deficiency Syndromes (CDS): Menkes’ Disease (MD), Occipital Horn Syndrome, ATP7A-Related Isolated Distal Motor Neuropathy and Zinc-Induced Myeloneuropathy
This group of disorders is characterized by a large phenotypic variation. Mutations in the gene encoding the ATP7A transporter are implicated in distinct phenotypes [47,48]. Menkes’ disease (MD) is an X-linked disease due to a mutation of the ATP7A gene. The large majority of patients are males [49]. Most ATP7A mutations are intragenic mutations or partial gene deletions [49]. Female carriers are mosaics of wild-type and mutant cells due to the random X inactivation [50]. In affected females, clinical symptoms are milder compared to boys having the same mutations. MD is characterized by mental retardation, seizures, abnormal lightly pigmented hair, bone fragility and aortic aneurysms. MD presents in infants between six weeks and one year. Patients present a state of copper deficiency, because copper accumulates in the intestine and cannot be absorbed in blood. A similar process occurs at the level of the BBB, causing a lack of ions within the CNS. The typical biochemical findings are low serum copper and ceruloplasmin, abnormal plasma and CSF neurochemicals and increased concentrations of β-2-microglobulin in urine samples [47]. SOD1 immunoreactivity is reduced, unlike the expression of SOD2 [48]. Therapy for MD is based on copper replacement. Parenteral administration is used. Gene therapy with adeno-associated viral (AAV) vectors is promising [51].
Occipital horn syndrome (OHS) is allelic to MD and is considered as a milder variant. Symptoms start in the first decade. The association of coarse hair and joint hyperlaxity is very suggestive. Occipital bone exostoses and hammer-shaped clavicular heads are typical. Some patients exhibit a dysautonomia, which is related to the reduced activity of the dopamine-β-hydroxylase, whereas the connective tissue abnormalities are attributed to the deficit of lysyl oxidase. Serum copper and ceruloplasmin levels are normal or decreased.
The ATP7A-related isolated distal motor neuropathy is a rare disorder affecting the peripheral motor nerves. The phenotypic presentation resembles Charcot-Marie-Tooth disease type 2 [52]. The mechanisms of the peripheral neuropathy remain to be elucidated. Two unique ATP7A missense mutations (T994I and P1386S) cause subtle defects in ATP7A intra-cellular trafficking, resulting in a preferential accumulation at the plasma membrane [53]. An abnormal interaction between ATP7A and adaptor protein complexes is presumed to be the molecular mechanism [53].
Zinc-induced myeloneuropathy is also caused by a deficit in copper [54]. The syndrome is the human equivalent to the copper deficiency myelopathy occurring in ruminants (“enzootic ataxia”) [55]. The disorder occurs mainly after sustained zinc exposure [56]. Regular use of zinc-containing dental fixatives has been identified as a triggering factor [57]. Patients undergoing gastrojejunal bypass surgery are also at risk [58]. The first disturbances are sensory symptoms in the feet. After a few years, the disorder mimics the subacute combined degeneration of the spinal cord, a disorder due to a deficit in vitamin B12. MRI of the spinal cord demonstrates signal changes in the dorsal columns. Chronic zinc ingestion may also cause bone marrow suppression with anemia, thrombocytopenia and neutropenia. By contrast to Wilson’s disease (WD, see the next section), urinary copper levels are typically decreased [59]. Although copper replacement may revert cytopenias, neurological deficits are irreversible in 40% of cases, highlighting the importance of detecting the hypocupremic state as soon as possible.
Figure 2.
Brain MRI (axial flair images) showing zones of impaired signal at the level of mesencephalon (A) and thalami (B) in a 21-year-old patient presenting Wilson’s disease. In the left panel, note the relative sparing of red nuclei and part of pars reticulata (giving a typical aspect called the “Panda sign”). In the right panel, the simultaneous hyposignals of basal ganglia (blue arrows) and the hypersignals in thalamic nuclei and internal capsule (red arrows) are very suggestive.
Figure 2.
Brain MRI (axial flair images) showing zones of impaired signal at the level of mesencephalon (A) and thalami (B) in a 21-year-old patient presenting Wilson’s disease. In the left panel, note the relative sparing of red nuclei and part of pars reticulata (giving a typical aspect called the “Panda sign”). In the right panel, the simultaneous hyposignals of basal ganglia (blue arrows) and the hypersignals in thalamic nuclei and internal capsule (red arrows) are very suggestive.
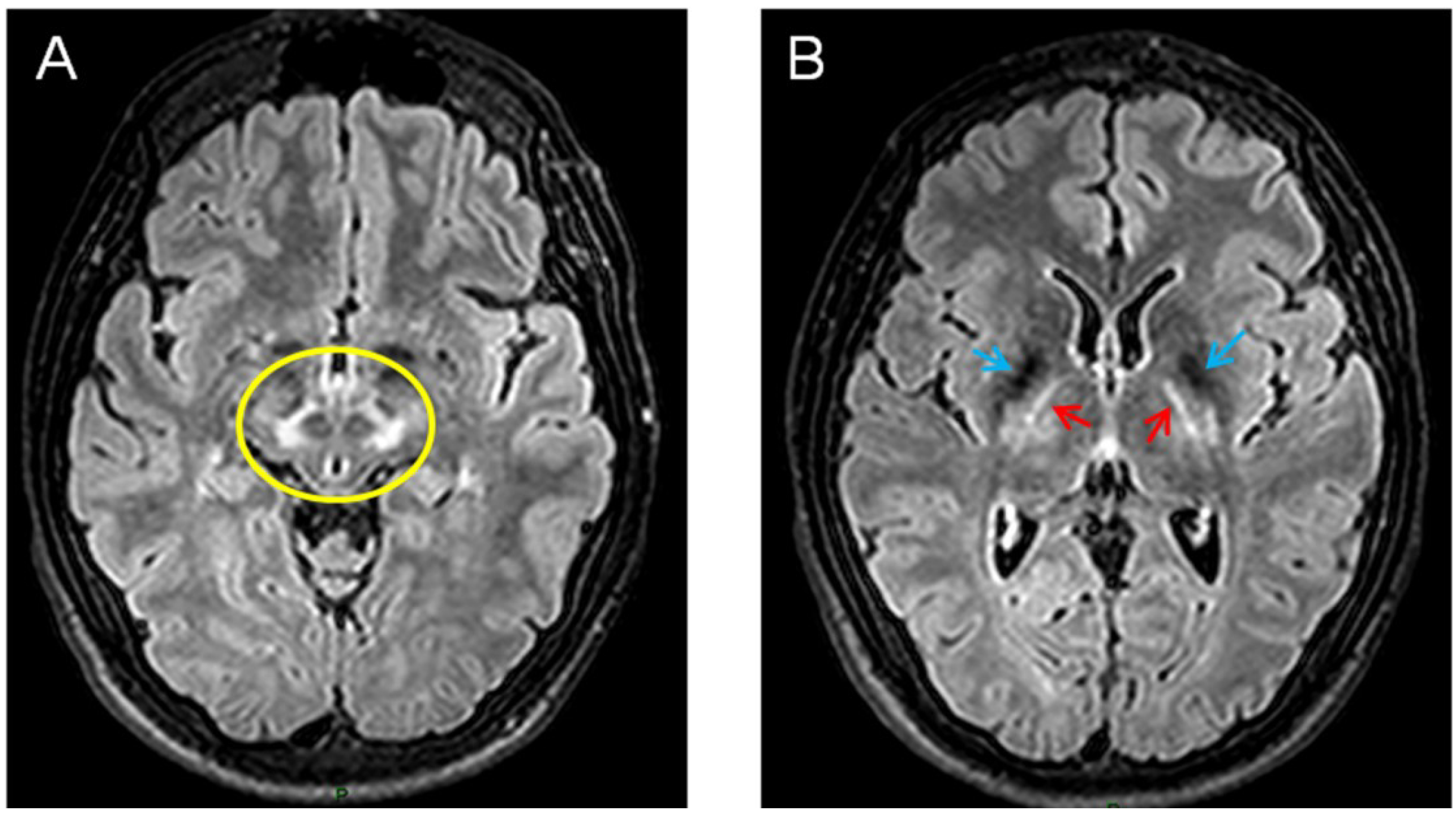
4. Wilson’s Disease (WD)
Wilson’s disease (WD) is an autosomal recessive disease involving the ATP7B gene. This causes deficits not only for the excretion of copper into the bile, but also in terms of the binding of copper to ceruloplasmin. Patients exhibit, in particular, deficits of the nervous system (with both neurological and psychiatric signs) and the liver. The Kayser–Fleischer ring (brown discoloration of the cornea) is very suggestive. Whereas the juvenile form is mainly associated with liver symptoms, the adult presentation tends to manifest mainly with neurological deficits [60]. Brain MRI shows areas of increased signals (Figure 2).
In WD, the concentrations of copper in liver are very high, contrasting with very low levels in blood. Levels of urinary copper are typically increased and are used as a biomarker of the disease. In an affected brain, the contents of copper are between two and eight times greater compared to a normal brain [61].
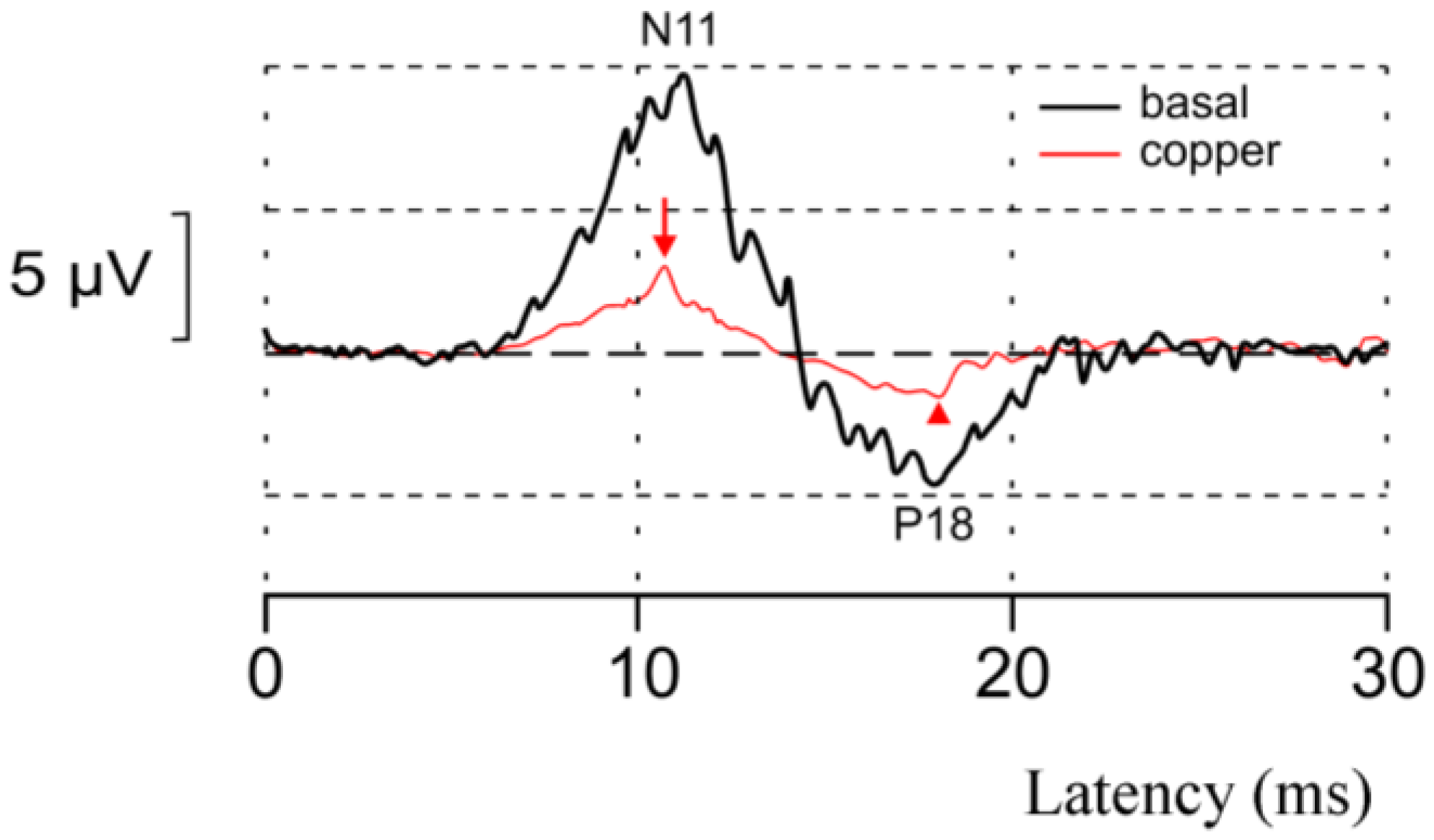
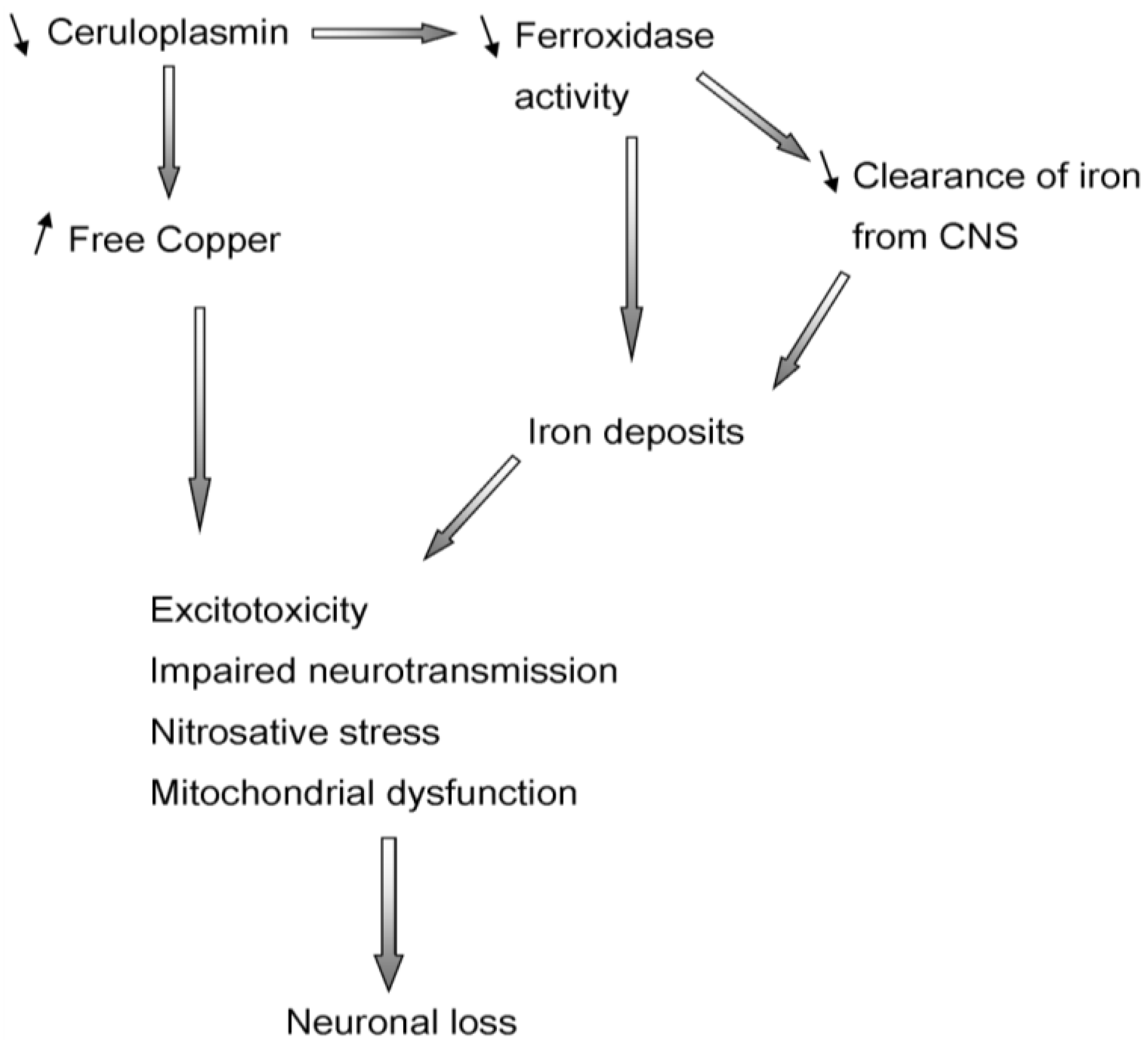
In vitro, the administration of copper markedly decreases neuronal survival [62]. The ion impairs the NMDA-mediated regulation of glutamate, triggering an excitotoxic cascade, and increases the production of nitric oxide (NO) by stimulating the transcription of nitric oxide synthesis (NOS1-3). NMDA antagonists partially protect the neurons. The in vivo acute administration of copper in the cerebellum markedly impairs the spinocerebellar-evoked potentials (Figure 3). A copper-related oxidative damage has been demonstrated in models of WD. Copper overload causes a mitochondrial dysfunction, lipid peroxidation and the synthesis of 4-hydroxy nonenal, which inhibits pyruvate dehydrogenase and alpha-ceto-glutarate dehydrogenase [63,64,65]. Neuropathological studies in WD show abnormal astrocytes (Opalski cells and Alzheimer cells) [66]. Figure 4 illustrates the mechanisms of neuronal loss associated with increased concentrations of free copper following a decrease in ceruloplasmin levels.
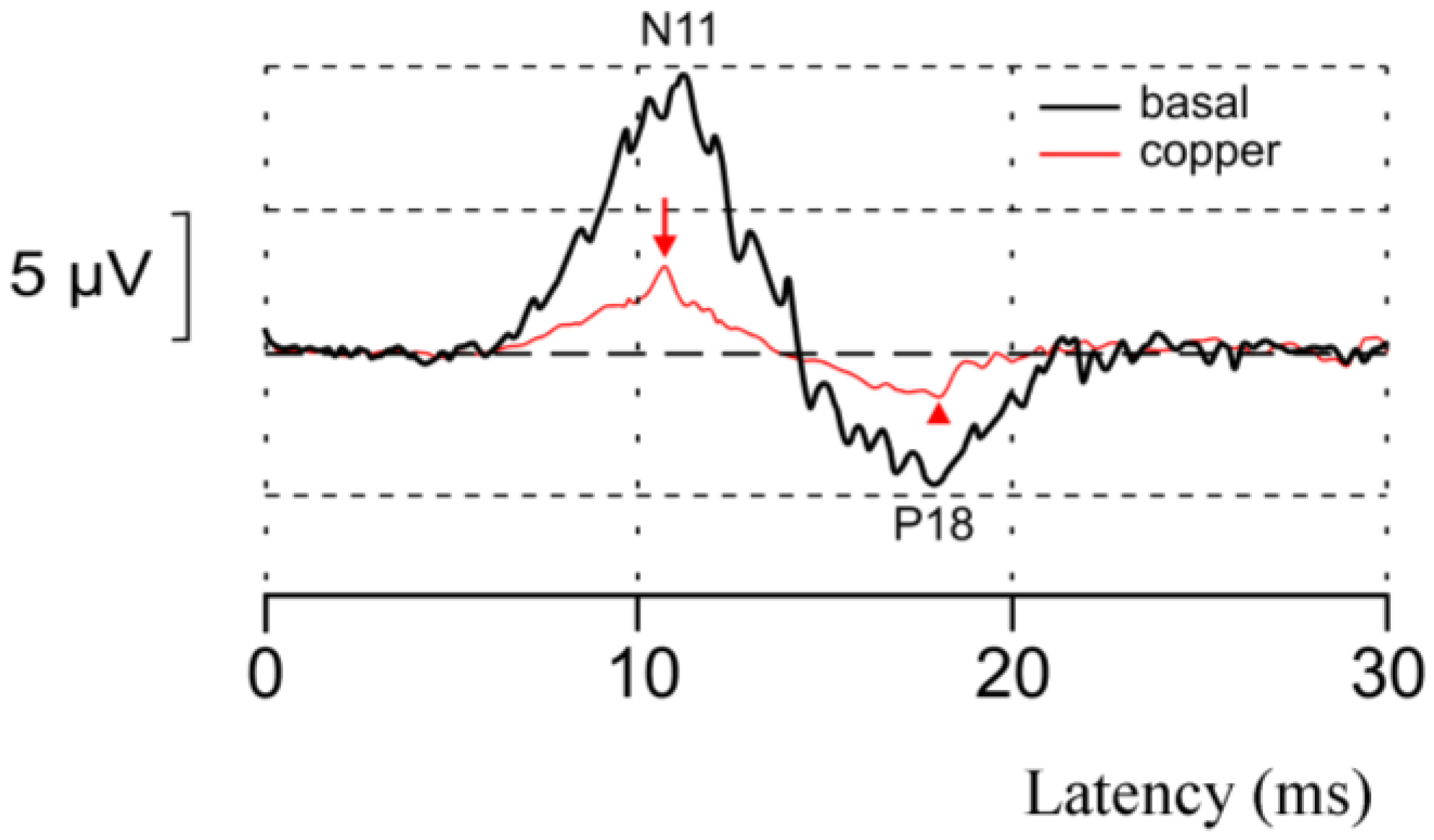
Figure 3.
An example of the acute neurotoxic effect of copper in vivo. The spinocerebellar N11-P18 complex (spinocerebellar evoked potentials (SCEP)) at baseline (black trace) and 20 min after the administration of copper (at a concentration of 71.2 µM) in left interpositus nucleus (cerebellum) in an adult Wistar rat maintained under general anesthesia with chloral hydrate. Recordings performed over left cerebellar hemisphere with electrical peripheral stimuli applied on the left tibial nerve. Note the marked decrease of the N11 wave (red arrow) and the P18 wave (red arrowhead), indicating a major depressive effect of copper on cerebellar afferent signals mediated by mossy fibers.
Figure 3.
An example of the acute neurotoxic effect of copper in vivo. The spinocerebellar N11-P18 complex (spinocerebellar evoked potentials (SCEP)) at baseline (black trace) and 20 min after the administration of copper (at a concentration of 71.2 µM) in left interpositus nucleus (cerebellum) in an adult Wistar rat maintained under general anesthesia with chloral hydrate. Recordings performed over left cerebellar hemisphere with electrical peripheral stimuli applied on the left tibial nerve. Note the marked decrease of the N11 wave (red arrow) and the P18 wave (red arrowhead), indicating a major depressive effect of copper on cerebellar afferent signals mediated by mossy fibers.
The therapy includes the administration of chelating agents (penicillamine, tetrathiomolybdate, trientine) and zinc (sulfate or acetate). The latter not only competes with intestinal uptake of copper, but induces also the intestinal MT-T, which blocks the transfer of copper to the blood. A diet with low levels of copper is recommended.
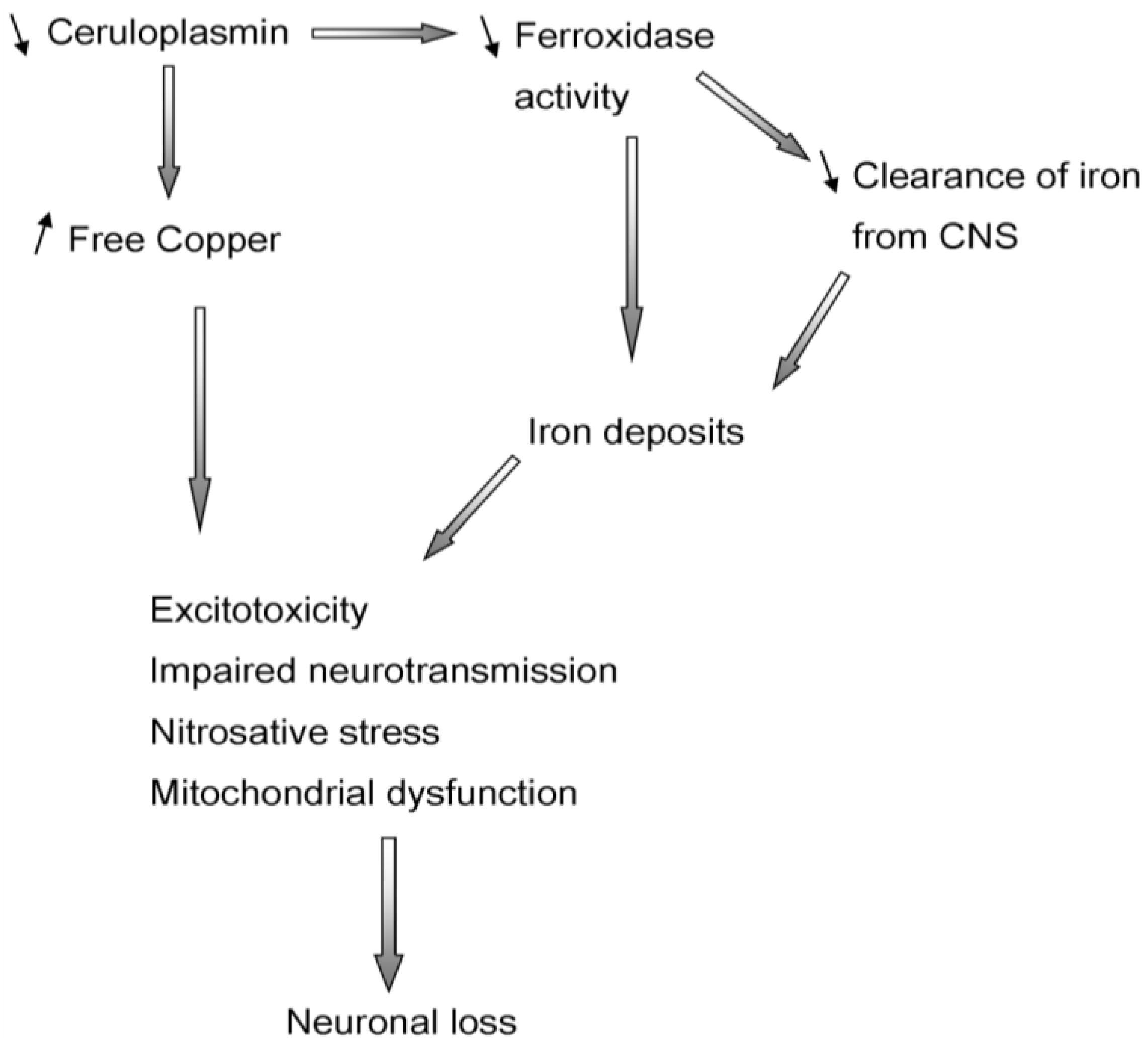
Figure 4.
Cascade of the events triggered by decreased levels of ceruloplasmin. Levels of free copper rise and iron deposits occur, leading to excitotoxicity, enhanced nitrosative/oxidative stress and damage to mitochondria.
Figure 4.
Cascade of the events triggered by decreased levels of ceruloplasmin. Levels of free copper rise and iron deposits occur, leading to excitotoxicity, enhanced nitrosative/oxidative stress and damage to mitochondria.
5. Aceruloplasminemia
Aceruloplasminemia is an autosomal recessive disease resulting in the absence of ceruloplasmin in blood. This causes the accumulation of iron in the CNS, especially in basal ganglia, in retina, liver and pancreas. Clinically, patients exhibit diabetes mellitus, retinal degeneration and a progressive neurological syndrome combining extrapyramidal signs, cerebellar ataxia and dementia, usually between the age of 25 and 60 years [67,68,69]. From a biochemical standpoint, ceruloplasmin knockout mice are characterized by enhanced lipid peroxidation caused by iron-mediated cellular radical injury [67]. Blood studies in aceruloplasminemia show low serum concentrations of copper and iron, a microcytic anemia, which contrasts with a high serum ferritin concentration. Hepatic concentrations of iron are increased. Brain MRI shows low intensities due to iron accumulation, mainly in striatum, thalamus and cerebellum. MRI of the liver is also suggestive. Iron chelators, such as desferrioxamine, are recommended [68]. Fresh-frozen human plasma (FFP) decreases iron contents in the liver and may improve neurologic deficits. Antioxidants, such as vitamin E, and the oral administration of zinc might prevent tissue damage [69].
6. Copper Toxicosis
The symptoms of acute copper intoxication include nausea, vomiting, diarrhea, abdominal pain, tachycardia and hemolytic anemia. The intoxication usually follows the ingestion of contaminated water or food [70].
The daily intake of copper determines the occurrence of toxicosis. High levels of copper in drinking water and food can cause a subacute to chronic syndrome of copper toxicosis [3]. The current tolerable upper intake level is 10 mg/day [7]. An oral supplement of copper 10 mg/day (supplementation with plain tap water between meals, consisting of two 5-mg copper gelatin capsules as copper sulfate) during two months induces a transient, but significant, elevation of aminotransferases [71]. Drinking water from wells via copper pipes has been incriminated in copper toxicosis [72]. The syndrome affects mainly young children. Their parents may be clinically healthy [72]. The disorder can lead to death. In India, a micronodular cirrhosis has been reported [73]. Copper toxicosis may be idiopathic, but these cases are very rare.
Rats exposed to copper in drinking water develop liver damage due to increased hepatocellular stores, with a significant increase of concentrations of the metal in the CNS and biochemical evidence of oxidative stress [74]. The activity of SOD decreases, and the concentrations of malondialdehyde rise.
7. Alzheimer’s Disease (AD)
The major histopathological features of Alzheimer’s disease (AD), the commonest form of dementia worldwide, are senile plaques and neurofibrillary tangles. AD is due to an abnormal processing of the APP (amyloid precursor protein) by β- and γ-secretases (Table 2A summarizes the main pathogenic mechanisms). The epsilon 4 allele of the apolipoprotein E (APOE) gene augments the risk of AD by three-fold and decreases age onset [75]. Patients with two epsilon 4 alleles have a 15-fold increase in AD risk as compared with the APOE epsilon 3 [76]. The regions of the brain that are particularly involved by atrophy at the beginning of the disease are the entorhinal cortex, the hippocampus and the amygdala.
Table 2.
Pathogenic mechanisms of Alzheimer’s disease (AD, left) and mechanisms of protein aggregation in Parkinson’s disease (PD, right).
| A. Alzheimer’s disease (AD) | B. Mechanisms of protein aggregation in Parkinson’s disease (PD) |
|---|---|
| Aggregation of β-Amyloid (Aβ) Peptide and Tau Proteins (Amyloid Cascade) * | Defect of the Ubiquitin-Proteasome System |
| Oxidative stress * | Overproduction of free radicals * |
| Inflammation * | Mitochondrial dysfunction * |
| Impaired energy metabolism * | Inflammation * |
| Impaired neurotransmission * | Impaired homeostasy of biometals * |
| Exposure to environmental pollutants * |
* Evidence that copper participates in the pathogenesis.
In addition to the hypothesis of the amyloid cascade per se, the role of biometals in the pathogenesis of AD is a subject of growing interest. Indeed, free plasma levels of copper increase with ageing [77]. Metal dyshomeostasis might impact not only on AD onset, but also on its progression [1]. There is a negative correlation between free copper in blood and cognitive status in AD [78]. Although the total levels of copper are similar in healthy subjects and in AD, the ratios of plasma/serum copper levels are significantly higher in AD [79]. Copper not bound to ceruloplasmin, rather than absolute serum copper levels, is a key-concept for the understanding of the pathogenesis of AD [80]. Both in vitro and in vivo studies have shown local and systemic defects in copper metabolism in AD. Very high concentrations of copper have been found in senile plaques [8]. There is a very appealing relationship between the levels of free copper in serum and the levels of free copper in the more severely affected areas of the brain, arguing strongly in favor of a systemic dyshomeostasy of copper [81]. The impaired homeostasis of copper is presumed to participate in oxidative stress, promoting free radicals-mediated pathways. The hypermetallation of the Aβ peptide might be at the origin of redox cycles of oxidative stress and H2O2 production, Aβ oligomer formation and precipitation [1]. Whereas Cu2+ can bind to nitrogen donors or oxygen donors, such as glutamate, Cu+ preferentially binds to free thiols of cysteine/methionine. This causes a cross-linking between proteins. It should be emphasized that copper-related oxidative stress is also associated with states of copper deficiency, indicating that the fine regulation of copper concentrations within margins is critical. From the genetic standpoint, some loci in the ATP7B gene are associated with an increased risk of developing AD and Parkinson’s disease (PD) [1]. ATP7B loss-of-function variants in transmembrane domains increase disease risk. Patients with some genetic background might be at greater risk of AD in the case of chronic copper exposure. Interestingly, the possibility of environmental contamination has been raised. Indeed, one recent hypothesis is that the ingestion of inorganic copper in drinking-water (contamination from copper pipe-lines) could increase the risk of AD [82].
The role of copper in the initiation and propagation of an inflammatory cascade within the aging brain has been suggested [83]. Copper may independently initiate inflammatory events and could interact with aluminum to increase the levels of APP [84]. In vitro studies have demonstrated that copper triggers a pro-inflammatory state by modulating the production of molecules, such as IL-1alpha or IL-12 [85].
Overall, the hypothesis of a genuine copper-related phenotype in AD is now solid. Critical outcomes in terms of prevention and active therapies might emerge for a devastating disorder whose prevalence is now a major public health issue. Clinical trials with metal modulators are in progress, in order to assess the effects of therapies redistributing copper amongst the different compartments [86]. Chelating agents improve cognitive symptoms in animal models of AD [87].
8. Parkinson’s Disease (PD)
Parkinson’s disease (PD) is the second most common neurodegenerative disease. Patients exhibit various combinations of bradykinesia, rigidity, tremor and impaired postural capacities with a tendency to fall. The pathological hallmark of the disease is the loss of dopamine-producing neurons in the substantia nigra (pars compacta). Aggregates of misfolded proteins, including alpha-synuclein, form the intra-cellular Lewy bodies. Alpha-synuclein is a copper-binding protein (with 2 sites for binding) enriched at the presynaptic terminals of neurons and catabolized by the ubiquitin-proteasome pathway [88]. Bound to copper, alpha-synuclein promotes the contribution of iron in biosynthesis of free radicals by exerting a ferrireductase effect. Triplication of the gene encoding the protein causes a familial PD [89]. The phenomenon of protein aggregation is a subject of intense investigation. Several mechanisms have been proposed for the pathogenesis of PD (Table 2B). The hypothesis of an impaired metabolism of metals has gained interest these last few years. One of the key findings comes from post-mortem studies demonstrating decreased copper levels contrasting with iron accumulation [90]. Copper might be missing in neurons of the striatum, being no longer available for the CuZn-SOD, which is a major contributor of the antioxidant system in striatal neurons. In addition, decreased copper in striatal neurons would result in iron accumulation. Iron deposits would occur in the substantia nigra before the beginning of motor symptoms. The accumulation of iron can be quantified indirectly by the measurement of the echogenicity of substantia nigra with transcranial ultrasound [91]. A positive correlation has been observed between serum ceruloplasmin levels and the age of onset of PD, and a negative correlation between iron accumulation in the brain and the copper-dependent ferroxidase plasma activity has been demonstrated [92,93]. Still, further studies are required to elucidate whether a deficiency of copper in the striatum is really the first step leading to iron deposits.
The concentrations of free copper in CSF are higher in PD and have even been suggested as a biochemical marker of the disease [94]. Environmental studies have shown that long-term exposure to copper and manganese increase the risk of PD [95]. As stated above, the activity of ferroxidase (which is maintained via ceruloplasmin) is reduced in PD [96]. Copper ions enhance the oxidation of dopamine, leading to DNA damage [97]. Intranigral administration of copper promotes the apoptosis of dopaminergic neurons [98]. Experimentally, the overexpression of CuZn-SOD protects against the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and ceruloplasmin knockout mice show a parkinsonism, which is rescued by iron chelation [96,99]. Furthermore, mice overexpressing MTs are more protected against peroxynitrite (ONOO-)-releasing agents and MPTP, suggesting that MT gene induction might provide neuroprotection [100]. Ceruloplasmin itself is now considered as a potential therapeutic agent in terms of neuroprotection [101].
9. Conclusions
Copper is an essential transition metal. It participates in critical cuproenzymes preventing neurodegeneration and regulating neurotransmission. Via the ferroxidase activity of ceruloplasmin, copper is a metabolic regulator of the contents of iron in the CNS. An excess of free copper is directly involved to neurodegeneration. WD, AD and PD are major neurodegenerative disorders associated with copper dyshomeostasis. Acting on copper metabolism represents a therapeutic approach for these severe disorders of the CNS. Gene therapy, gene induction, metal modulators promoting the redistribution between compartments and administration of ceruloplasmin are promising possibilities that deserve specific studies.
Acknowledgments
Mario Manto is supported by the National Reasearch Funds (FNRS), Belgium. The author has received a research grant from Pfizer (USA) to investigate the effects of pregabalin in a model of copper-induced neurotoxicity. He has been funded by the NIH, the European Commission (EC) and the National Research Fund (FNRS), Belgium.
Conflicts of Interest
The author declares no conflict of interest.
References
- Squitti, R.; Polimanti, R. Copper phenotype in Alzheimer’s disease: Dissecting the pathway. Am. J. Neurodegener. Dis. 2013, 2, 46–56. [Google Scholar]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper in medicine. Curr. Opin. Chem. Biol. 2003, 7, 207–212. [Google Scholar] [CrossRef]
- Eid, C.; Hémadi, M.; Ha-Duong, N.T.; El Hage Chahine, J.M. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta 2014, 1840, 1771–1781. [Google Scholar] [CrossRef]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116C, 33–57. [Google Scholar]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed. Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Chambers, A.; Krewski, D.; Birkett, N.; Plunkett, L.; Hertzberg, R.; Danzeisen, R.; Aggett, P.J.; Starr, T.B.; Baker, S.; Dourson, M.; et al. An exposure-response curve for copper excess and deficiency. J. Toxicol. Environ. Health B 2010, 13, 546–578. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Popiołek, L.; Kocot, J. The many “faces” of copper in medicine and treatment. Biometals 2014. [Google Scholar] [CrossRef]
- Thiele, D.J. Integrating trace element metabolism from the cell to the whole organism. J. Nutr. 2003, 133, 1579S–1580S. [Google Scholar]
- Linder, M.C. Biochemistry of Copper; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.H.; Bush, A.I.; La Fontaine, S. Links between copper and cholesterol in Alzheimer’s disease. Front. Physiol. 2013, 4, 111. [Google Scholar]
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Kretzschmar, H.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef]
- Huffman, D.L.; O’Halloran, T.V. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 2001, 70, 677–701. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Vashchenko, G.; MacGillivray, R.T. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef]
- Beaumont, C. Molecular mechanisms of iron homeostasis. Med. Sci. 2004, 20, 68–72. [Google Scholar]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [CrossRef]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13. [Google Scholar] [CrossRef]
- Chelly, J.; Tümer, Z.; Tønnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19. [Google Scholar] [CrossRef]
- Qian, Y.; Tiffany-Castiglioni, E.; Welsh, J.; Harris, E.D. Copper efflux from murine microvascular cells requires expression of the menkes disease Cu-ATPase. J. Nutr. 1998, 128, 1276–1282. [Google Scholar]
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352. [Google Scholar] [CrossRef]
- Petris, M.J.; Mercer, J.F.; Culvenor, J.G.; Lockhart, P.; Gleeson, P.A.; Camakaris, J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996, 15, 6084–6095. [Google Scholar]
- Wee, N.K.; Weinstein, D.C.; Fraser, S.T.; Assinder, S.J. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int. J. Biochem. Cell Biol. 2013, 45, 960–963. [Google Scholar] [CrossRef]
- Choi, B.S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar]
- Van den Berghe, P.V.; Klomp, L.W. Posttranslational regulation of copper transporters. J. Biol. Inorg. Chem. 2010, 15, 37–46. [Google Scholar] [CrossRef]
- Larson, C.A.; Adams, P.L.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the methionines and histidines in the transmembrane domain of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Mol. Pharmacol. 2010, 78, 333–339. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar]
- Bertinato, J.; Swist, E.; Plouffe, L.J.; Brooks, S.P.; L’abbé, M.R. Ctr2 is partially localized to the plasma membrane and stimulates copper uptake in COS-7 cells. Biochem. J. 2008, 409, 731–740. [Google Scholar] [CrossRef]
- Zheng, G.; Chen, J.; Zheng, W. Relative contribution of CTR1 and DMT1 in copper transport by the blood-CSF barrier: Implication in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 285–293. [Google Scholar] [CrossRef]
- Lee, J.; Prohaska, J.R.; Thiele, D.J. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl. Acad. Sci. USA 2001, 98, 6842–6847. [Google Scholar] [CrossRef]
- Naeve, G.S.; Vana, A.M.; Eggold, J.R.; Kelner, G.S.; Maki, R.; Desouza, E.B.; Foster, A.C. Expression profile of the copper homeostasis gene, rAtox1, in the rat brain. Neuroscience 1999, 93, 1179–1187. [Google Scholar] [CrossRef]
- Penkowa, M.; Tio, L.; Giralt, M.; Quintana, A.; Molinero, A.; Atrian, S.; Vasák, M.; Hidalgo, J. Specificity and divergence in the neurobiologic effects of different metallothioneins after brain injury. J. Neurosci. Res. 2006, 83, 974–984. [Google Scholar] [CrossRef]
- Michael, G.J.; Esmailzadeh, S.; Moran, L.B.; Christian, L.; Pearce, R.K.; Graeber, M.B. Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 2011, 12, 295–305. [Google Scholar] [CrossRef]
- Haywood, S.; Vaillant, C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative diseases. J. Comp. Pathol. 2014, 150, 216–224. [Google Scholar] [CrossRef]
- Squitti, R. Copper dysfunction in Alzheimer’s disease: From meta-analysis of biochemical studies to new insight into genetics. J. Trace Elem. Med. Biol. 2012, 26, 93–96. [Google Scholar]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar]
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246. [Google Scholar] [CrossRef]
- Vlachová, V.; Zemková, H.; Vyklický, L., Jr. Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur. J. Neurosci. 1996, 8, 2257–2264. [Google Scholar] [CrossRef]
- Schlief, M.L.; Gitlin, J.D. Copper homeostasis in the CNS: A novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 2006, 33, 81–90. [Google Scholar] [CrossRef]
- Weiser, T.; Wienrich, M. The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res. 1996, 742, 211–218. [Google Scholar] [CrossRef]
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- McGee, T.P.; Houston, C.M.; Brickley, S.G. Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J. Neurosci. 2013, 33, 13431–13435. [Google Scholar] [CrossRef]
- Kaler, S.G. Translational research investigations on ATP7A: An important human copper ATPase. Ann. N. Y. Acad. Sci. 2014, 1314, 64–68. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Umahara, T.; Kawanami, T.; Asayama, K. Cerebellar superoxide dismutase expression in Menkes’ kinky hair disease: An immunohistochemical investigation. Acta Neuropathol. 1995, 90, 198–202. [Google Scholar] [CrossRef]
- Tümer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mutat. 2013, 34, 417–429. [Google Scholar] [CrossRef]
- Møller, L.B.; Lenartowicz, M.; Zabot, M.T.; Josiane, A.; Burglen, L.; Bennett, C.; Riconda, D.; Fisher, R.; Janssens, S.; Mohammed, S.; et al. Clinical expression of Menkes disease in females with normal karyotype. Orphanet J. Rare Dis. 2012, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Haddad, M.R.; Choi, E.Y.; Kaler, S.G. AAVrh10 ATP7A administration to the cerebrospinal fluid, in combination with subcutaneous copper, normalizes neurological outcomes in a mouse model of Menkes disease. Mol. Ther. 2014, 22 (Suppl.). in press. [Google Scholar]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Yi, L.; Kaler, S. ATP7A trafficking and mechanisms underlying the distal motor neuropathy induced by mutations in ATP7A. Ann. N.Y. Acad. Sci. 2014, 1314, 49–54. [Google Scholar] [CrossRef]
- Schleper, B.; Stuerenburg, H.J. Copper deficiency-associated myelopathy in a 46-year-old woman. J. Neurol. 2001, 248, 705–706. [Google Scholar] [CrossRef]
- Bennetts, H.W.; Chapman, F.E. Copper deficiency in sheep in Western Australia: A preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust. Vet. J. 1937, 13, 138–149. [Google Scholar] [CrossRef]
- Lanska, D.J.; Remler, B. Myelopathy among zinc-smelter workers in Upper Silesia during the late 19th century. Neurology 2014, 82, 1175–1179. [Google Scholar] [CrossRef]
- Gabreyes, A.A.; Abbasi, H.N.; Forbes, K.P.; McQuaker, G.; Duncan, A.; Morrison, I. Hypocupremia associated cytopenia and myelopathy: A national retrospective review. Eur. J. Haematol. 2013, 90, 1–9. [Google Scholar]
- Choi, E.H.; Strum, W. Hypocupremia-related myeloneuropathy following gastrojejunal bypass surgery. Ann. Nutr. MeTable 2010, 57, 190–192. [Google Scholar] [CrossRef]
- Jaiser, S.R.; Winston, G.P. Copper deficiency myelopathy. J. Neurol. 2010, 257, 869–881. [Google Scholar] [CrossRef]
- Lorincz, M.T. Neurologic Wilson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef]
- Faa, G.; Lisci, M.; Caria, M.P.; Ambu, R.; Sciot, R.; Nurchi, V.M.; Silvagni, R.; Diaz, A.; Crisponi, G. Brain copper, iron, magnesium, zinc, calcium, sulfur and phosphorus storage in Wilson’s disease. J. Trace Elem. Med. Biol. 2001, 15, 155–160. [Google Scholar] [CrossRef]
- Marmolino, D.; Manto, M. Pregabalin antagonizes copper-induced toxicity in the brain: In vitro and in vivo studies. Neurosignals 2010, 18, 210–222. [Google Scholar] [CrossRef]
- Sokol, R.J.; Devereaux, M.W.; O’Brien, K.; Khandwala, R.A.; Loehr, J.P. Abnormal hepatic mitochondrial respiration and cytochrome C oxidase activity in rats with long-term copper overload. Gastroenterology 1993, 105, 178–187. [Google Scholar]
- Sheline, C.T.; Choi, D.W. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann. Neurol. 2004, 55, 645–653. [Google Scholar] [CrossRef]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar]
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T.; Blaauwgers, H.G.; Czlonkowska, A.; Dymecki, J. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 2001, 39, 73–79. [Google Scholar]
- Kono, S. Aceruloplasminemia. Curr. Drug Targets 2012, 13, 1190–1199. [Google Scholar] [CrossRef]
- Pan, P.L.; Tang, H.H.; Chen, Q.; Song, W.; Shang, H.F. Desferrioxamine treatment of aceruloplasminemia: Long-term follow-up. Mov. Disord. 2011, 26, 2142–2144. [Google Scholar] [CrossRef]
- Miyajima, H. Aceruloplasminemia. In GeneReviews; Pagon, R.A., Adam, M.P., Ardinger, H.H., Bird, T.D., Dolan, C.R., Fong, C.T., Smith, R.J.H., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 2003; pp. 1993–2014. [Google Scholar]
- Puig, S.; Thiele, D. Molecular mechanism of copper uptake and distribution. Curr. Opin. Chem. Biol. 2002, 6, 171–180. [Google Scholar] [CrossRef]
- Araya, M.; Olivares, M.; Pizarro, F.; Méndez, M.A.; González, M.; Uauy, R. Supplementing copper at the upper level of the adult dietary recommended intake induces detectable but transient changes in healthy adults. J. Nutr. 2005, 135, 2367–2371. [Google Scholar]
- Schramel, P.; Müller-Höcker, J.; Meyer, U.; Weiss, M.; Eife, R. Nutritional copper intoxication in three German infants with severe liver cell damage (features of Indian childhood cirrhosis). J. Trace Elem. Electrolytes Health Dis. 1988, 2, 85–89. [Google Scholar]
- Baker, A.; Gormally, S.; Saxena, R.; Baldwin, D.; Drumm, B.; Bonham, J.; Portmann, B.; Mowat, A.P. Copper-associated liver disease in childhood. J. Hepatol. 1995, 23, 538–543. [Google Scholar] [CrossRef]
- Ozcelik, D.; Uzun, H. Copper intoxication, antioxidant defenses and oxidative damage in rat brain. Biol. Trace Elem. Res. 2009, 127, 45–52. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Bettens, K.; Sleegers, K.; van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Noda, Y.; Asada, M.; Kubota, M.; Maesako, M.; Watanabe, K.; Uemura, M.; Kihara, T.; Shimohama, S.; Takahashi, R.; Kinoshita, A.; et al. Copper enhances APP dimerization and promotes Ab production. Neurosci. Lett. 2013, 547, 10–15. [Google Scholar] [CrossRef]
- Arnal, N.; Morel, G.R.; de Alaniz, M.J.T.; Castillo, O.; Marra, C.A. Role of copper and cholesterol association in the neurodegenerative process. Int. J. Alzheimer’s Dis. 2013, 10, 1–15. [Google Scholar]
- Ventriglia, M.; Bucossi, S.; Panetta, V.; Squitti, R. Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimer’s Dis. 2012, 30, 981–984. [Google Scholar]
- Squitti, R.; Pasqualetti, P.; dal Forno, G.; Moffa, F.; Cassetta, E.; Lupoi, D.; Vernieri, F.; Rossi, L.; Baldassini, M.; Rossini, P.M. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 2005, 64, 1040–1046. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper toxicity in Alzheimer’s disease: Cognitive loss from ingestion of inorganic copper. J. Trace Elem. Med. Biol. 2012, 26, 89–92. [Google Scholar] [CrossRef]
- Campbell, A. The role of aluminum and copper on neuroinflammation and Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 165–172. [Google Scholar]
- Becaria, A.; Lahiri, D.K.; Bondy, S.C.; Chen, D.; Hamadeh, A.; Li, H.; Taylor, R.; Campbell, A. Aluminum and copper in drinking water enhance inflammatory or oxidative events specifically in the brain. J. Neuroimmunol. 2006, 176, 16–23. [Google Scholar] [CrossRef]
- Spisni, E.; Valerii, M.C.; Manerba, M.; Strillacci, A.; Polazzi, E.; Mattia, T.; Griffoni, C.; Tomasi, V. Effect of copper on extracellular levels of key pro-inflammatory molecules in hypothalamic GN11 and primary neurons. Neurotoxicology 2009, 30, 605–612. [Google Scholar] [CrossRef]
- Crouch, P.J.; Barnham, K.J. Therapeutic redistribution of metal ions to treat Alzheimer’s disease. Acc. Chem. Res. 2012, 45, 1604–1611. [Google Scholar] [CrossRef]
- Ceccom, J.; Cosledan, F.; Halley, H.; Frances, B.; Lassalle, J.M.; Meunier, B. Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLoS One 2012, 7, e43105. [Google Scholar]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Insights into the bioinorganic chemistry of Parkinson’s disease. Structural characterization of copper(II) binding to alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Berg, D.; Siefker, C.; Becker, G. Echogenicity of the substantia nigra in Parkinson’s disease and its relation to clinical findings. J. Neurol. 2001, 248, 684–689. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Montes, S.; Higuera-Calleja, J.; Yescas, P.; Boll, M.C.; Diaz-Ruiz, A.; Rios, C. Plasma ceruloplasmin ferroxidase activity correlates with the nigral sonographic area in Parkinson’s disease patients: A pilot study. Neurochem. Res. 2011, 36, 2111–2115. [Google Scholar] [CrossRef]
- Bharucha, K.J.; Friedman, J.K.; Vincent, A.S.; Ross, E.D. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson’s disease. J. Neurol. 2008, 255, 1957–1962. [Google Scholar] [CrossRef]
- Boll, M.C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Spencer, W.A.; Jeyabalan, J.; Kichambre, S.; Gupta, R.C. Oxidatively generated DNA damage after Cu(II) catalysis of dopamine and related catecholamine neurotransmitters and neurotoxins: Role of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 139–147. [Google Scholar]
- Yu, W.R.; Jiang, H.; Wang, J.; Xie, J.X. Copper (Cu2+) induces degeneration of dopaminergic neurons in the nigrostriatal system of rats. Neurosci. Bull. 2008, 24, 73–78. [Google Scholar] [CrossRef]
- Przedborski, S.; Kostic, V.; Jackson-Lewis, V.; Naini, A.B.; Simonetti, S.; Fahn, S.; Carlson, E.; Epstein, C.J.; Cadet, J.L. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J. Neurosci. 1992, 12, 1658–1667. [Google Scholar]
- Ebadi, M.; Sharma, S. Metallothioneins 1 and 2 attenuate peroxynitrite-induced oxidative stress in Parkinson disease. Exp. Biol. Med. 2006, 231, 1576–1583. [Google Scholar]
- Ayton, S.; Zhang, M.; Roberts, B.R.; Lam, L.Q.; Lind, M.; McLean, C.; Bush, A.I.; Frugier, T.; Crack, P.J.; Duce, J.A. Ceruloplasmin and β-amyloid precursor protein confer neuroprotection in traumatic brain injury and lower neuronal iron. Free Radic. Biol. Med. 2014, 69, 331–337. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Manto, M. Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics 2014, 2, 327-345. https://doi.org/10.3390/toxics2020327
AMA Style
Manto M. Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics. 2014; 2(2):327-345. https://doi.org/10.3390/toxics2020327
Chicago/Turabian StyleManto, Mario. 2014. "Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration" Toxics 2, no. 2: 327-345. https://doi.org/10.3390/toxics2020327