Investigation of the Spin Crossover Properties of Three Dinulear Fe(II) Triple Helicates by Variation of the Steric Nature of the Ligand Type


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Syntheses
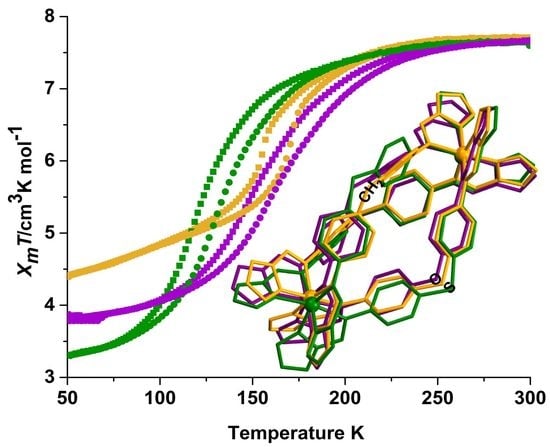
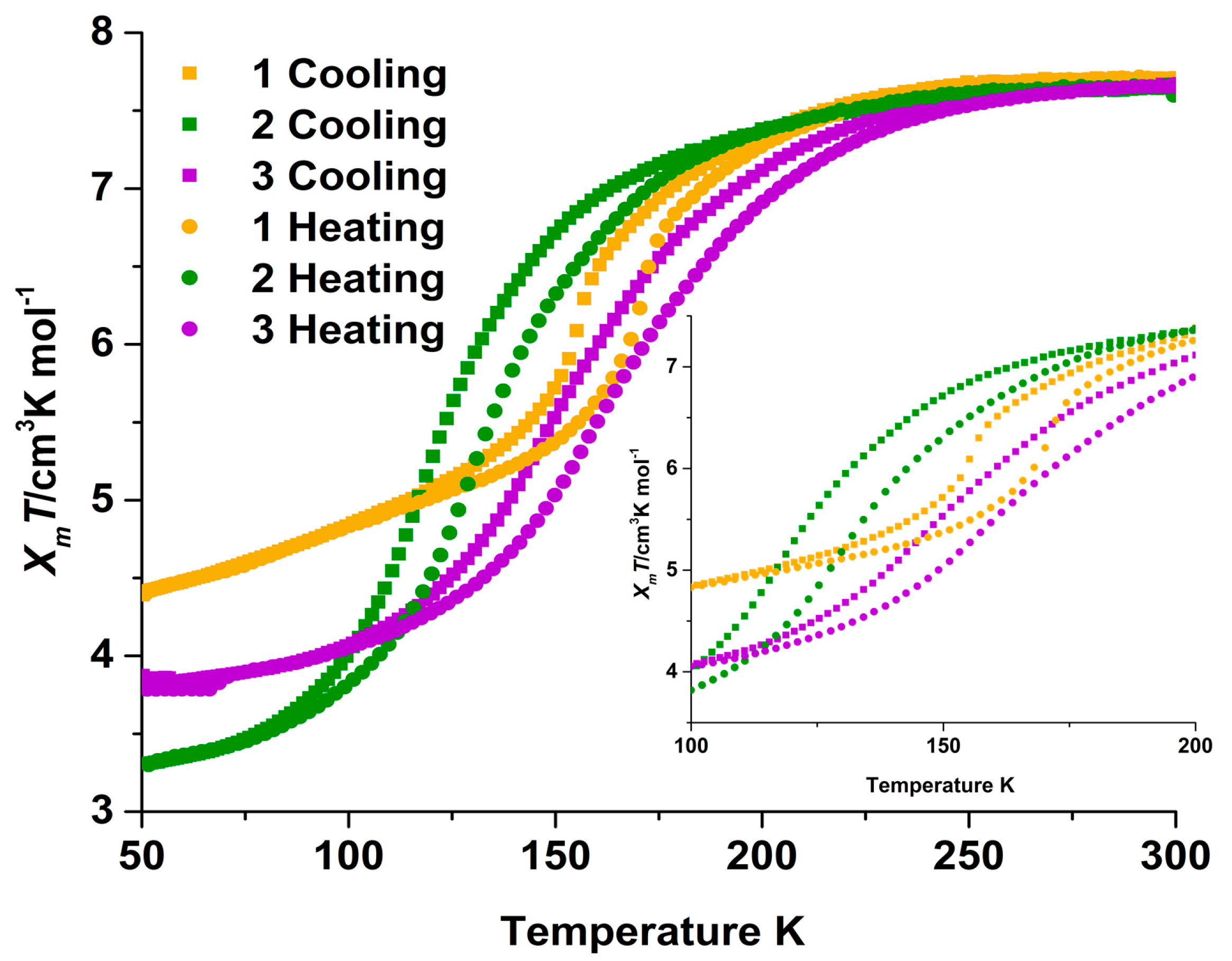
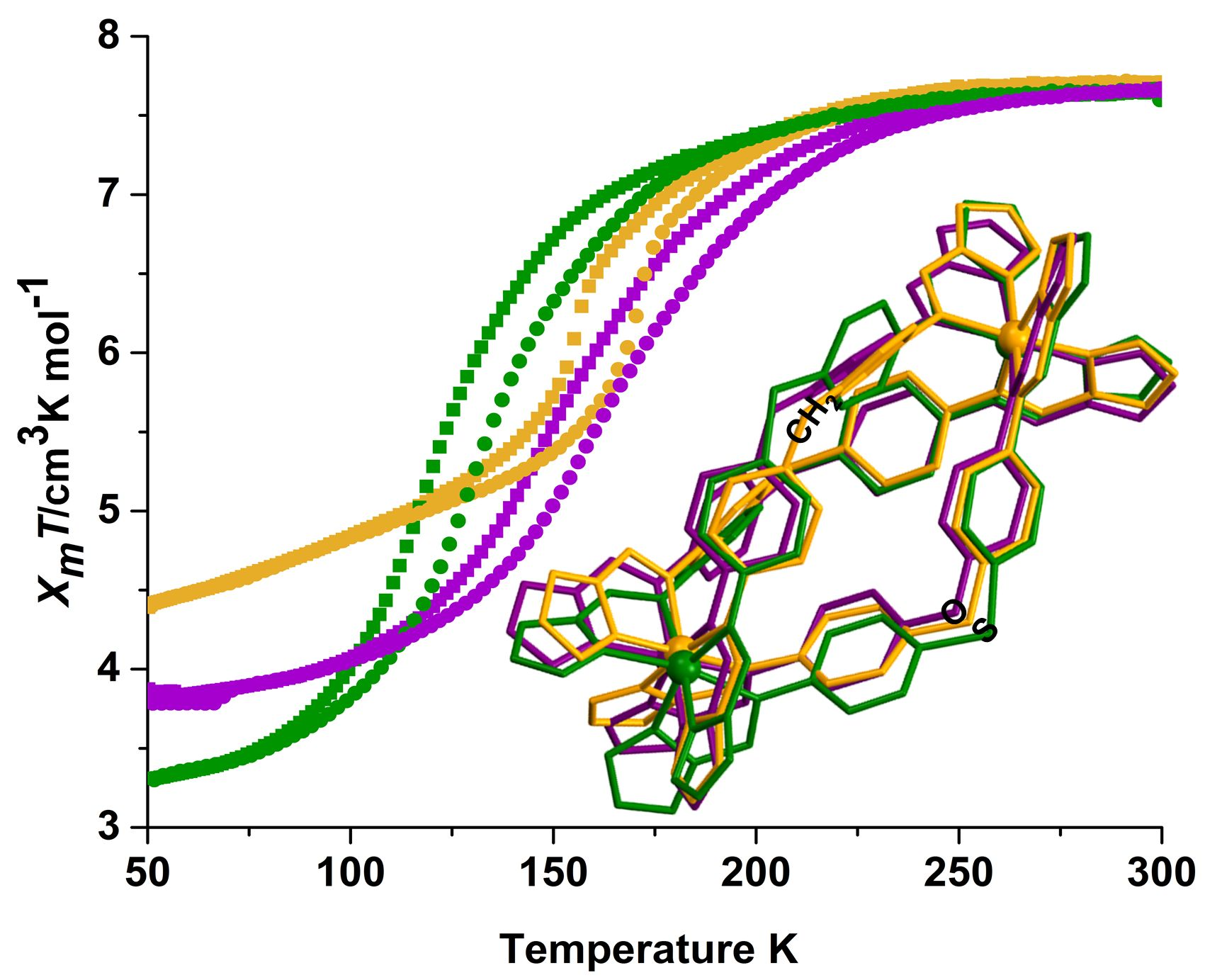
2.2. Magnetic Susceptibility Measurements
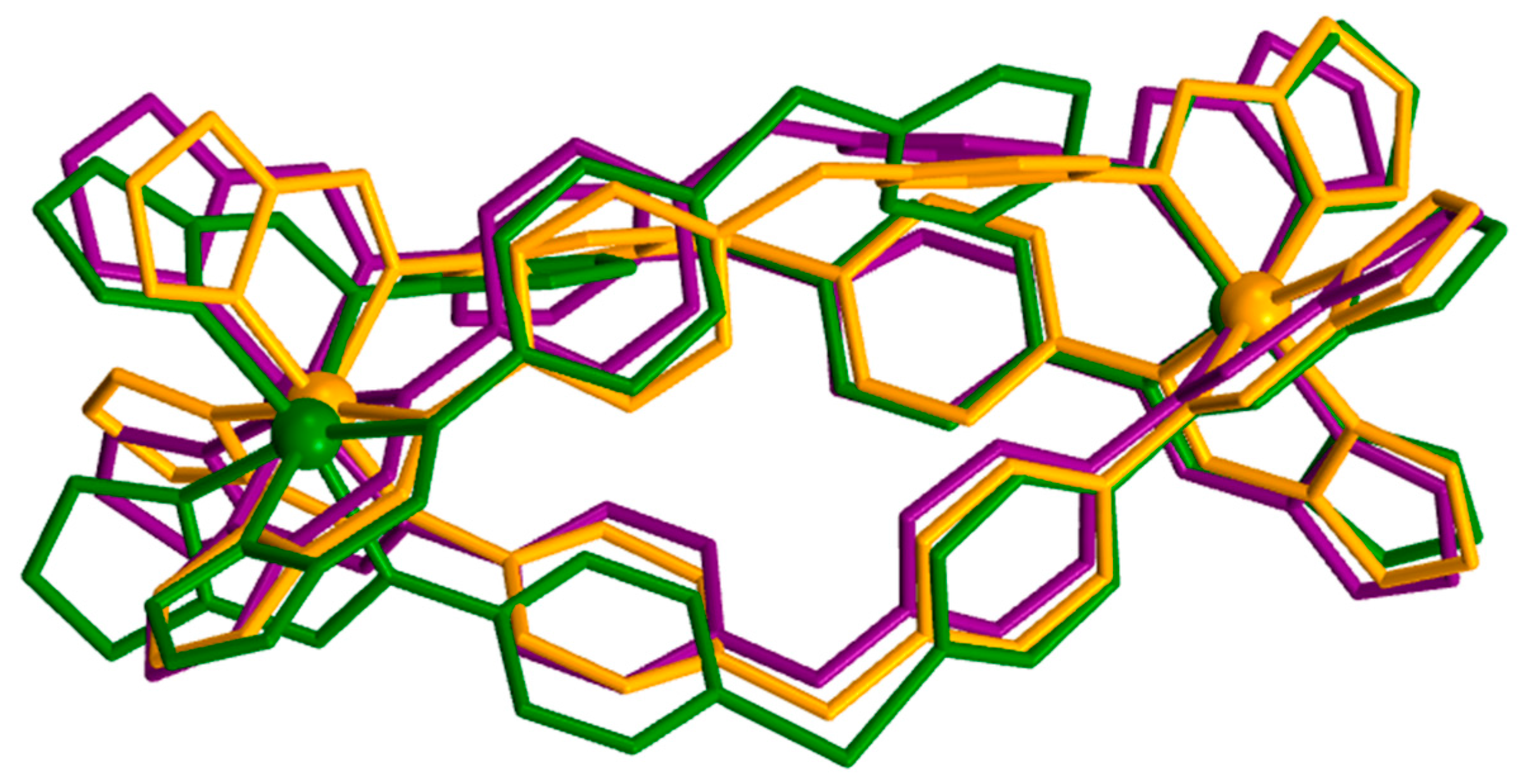
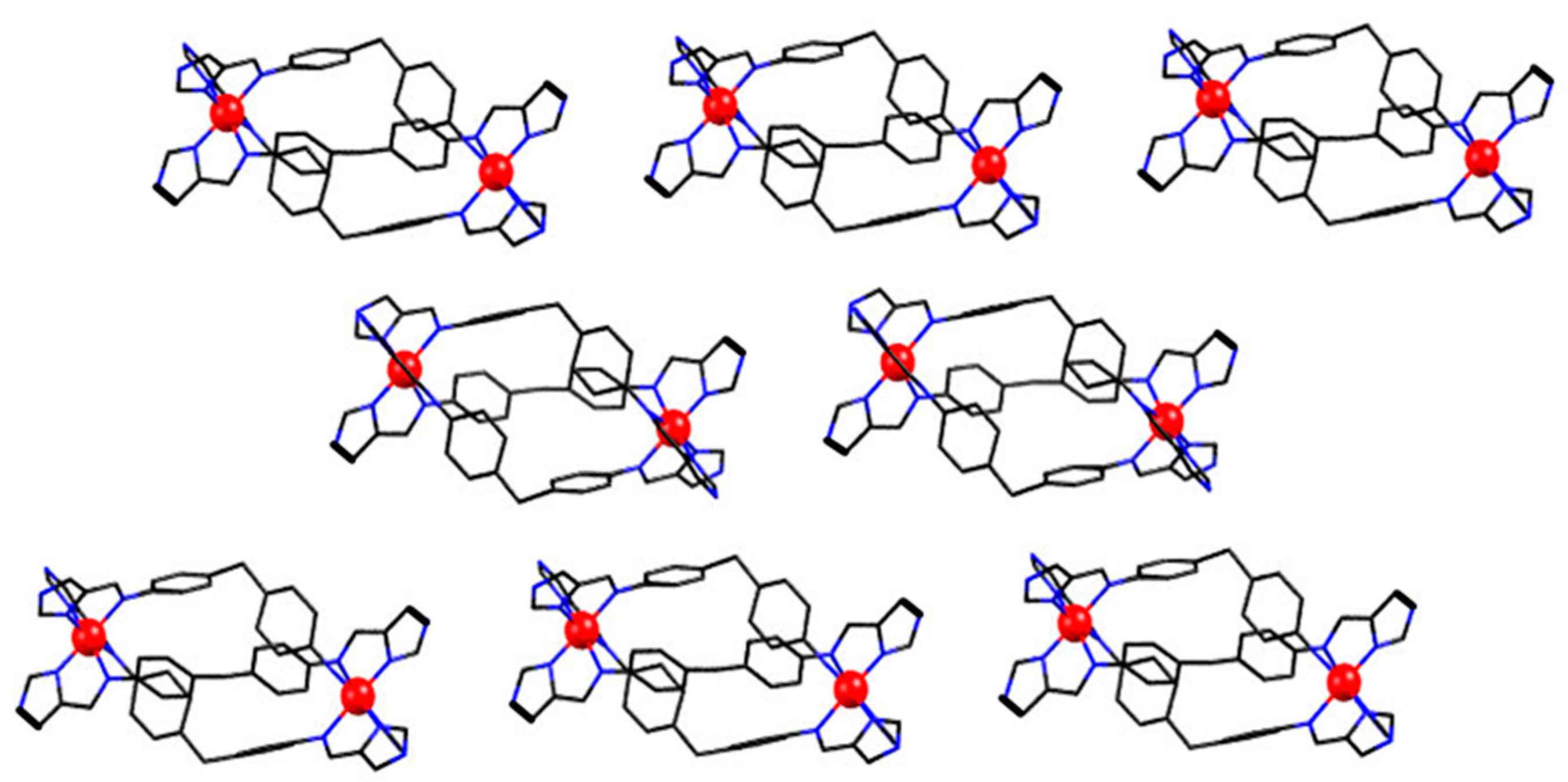
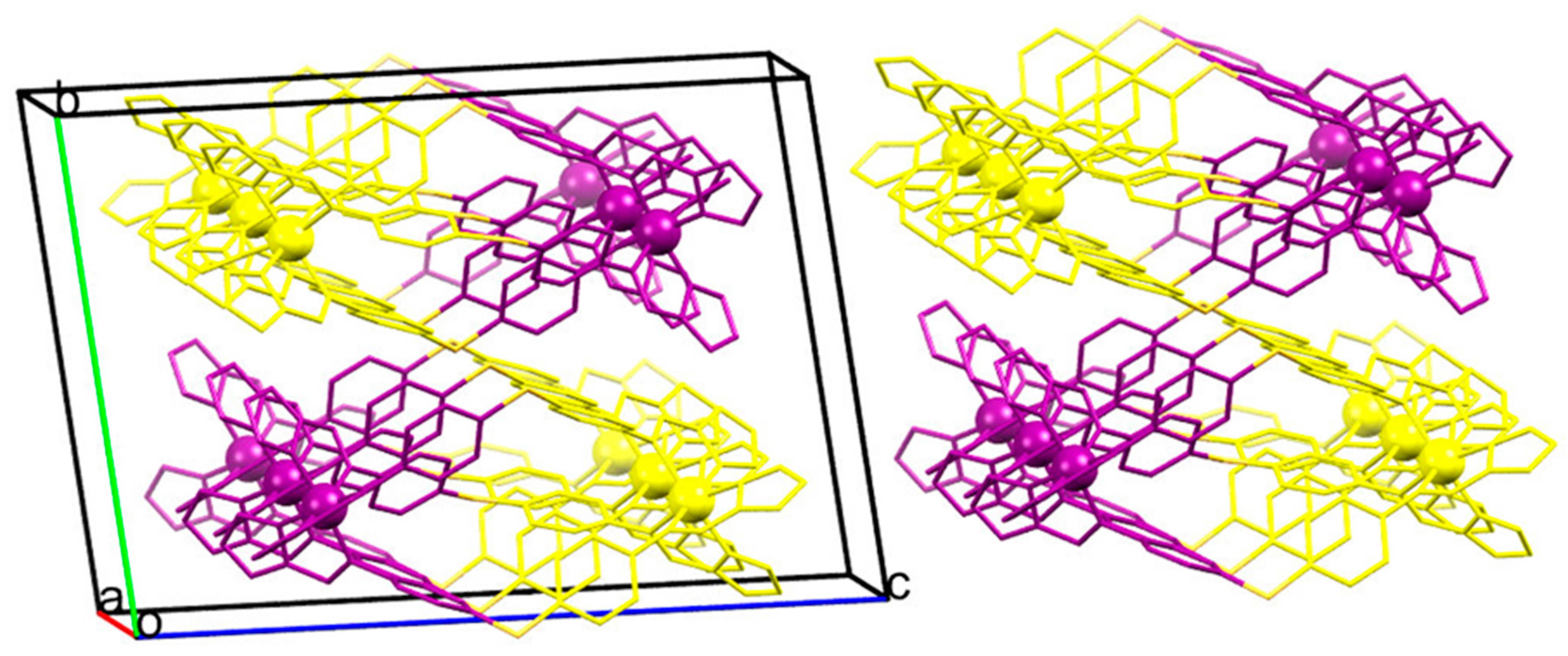
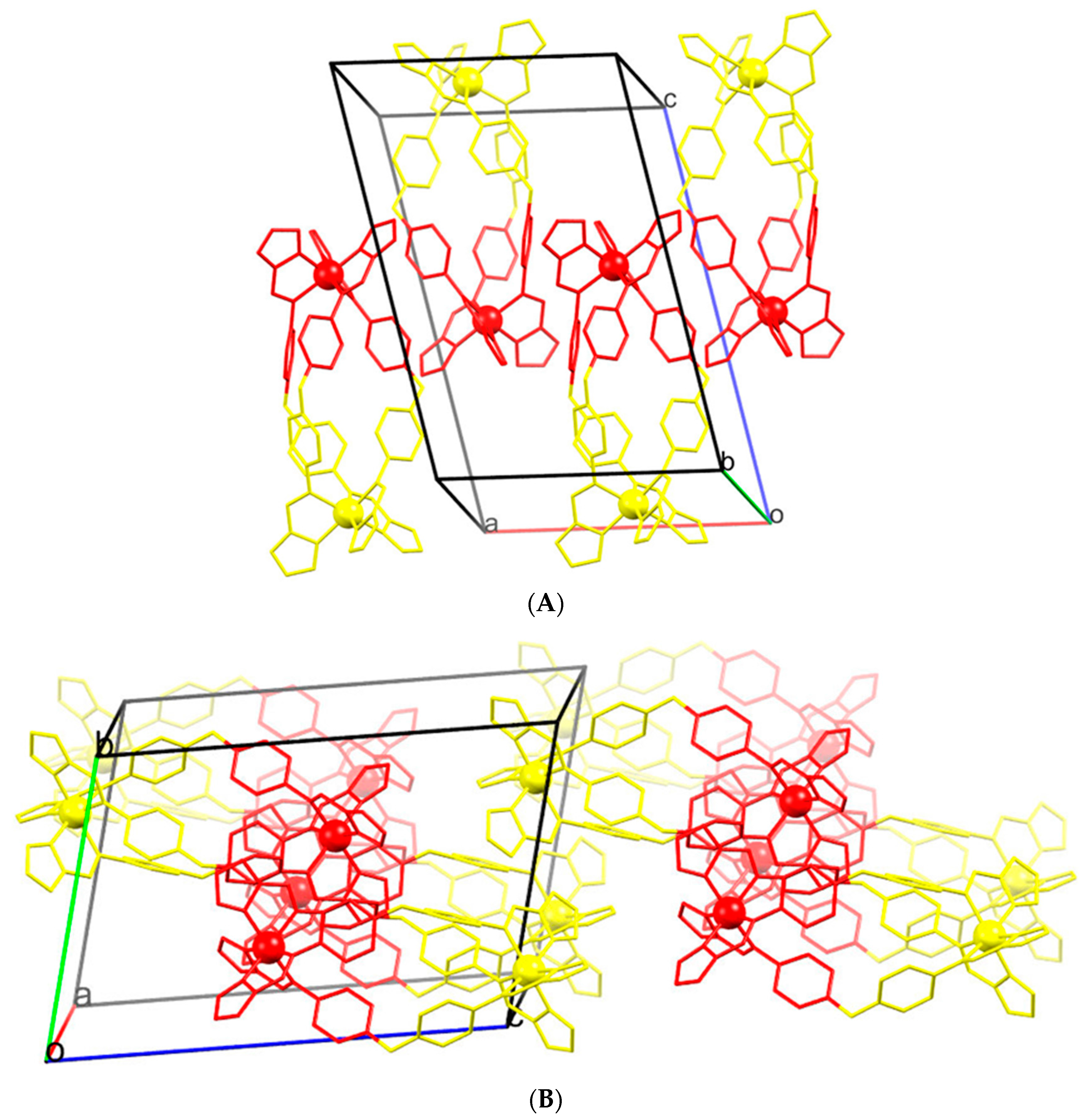
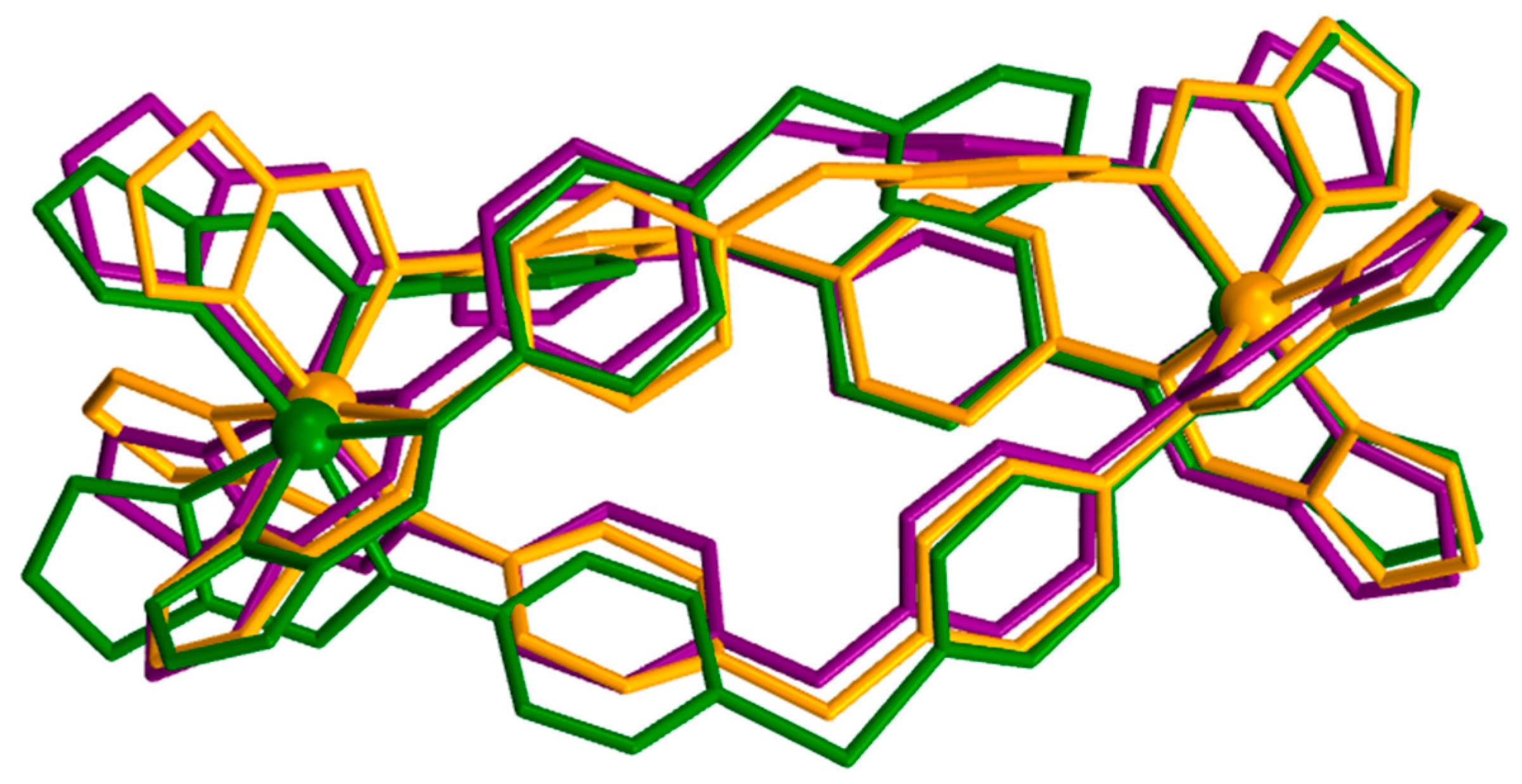
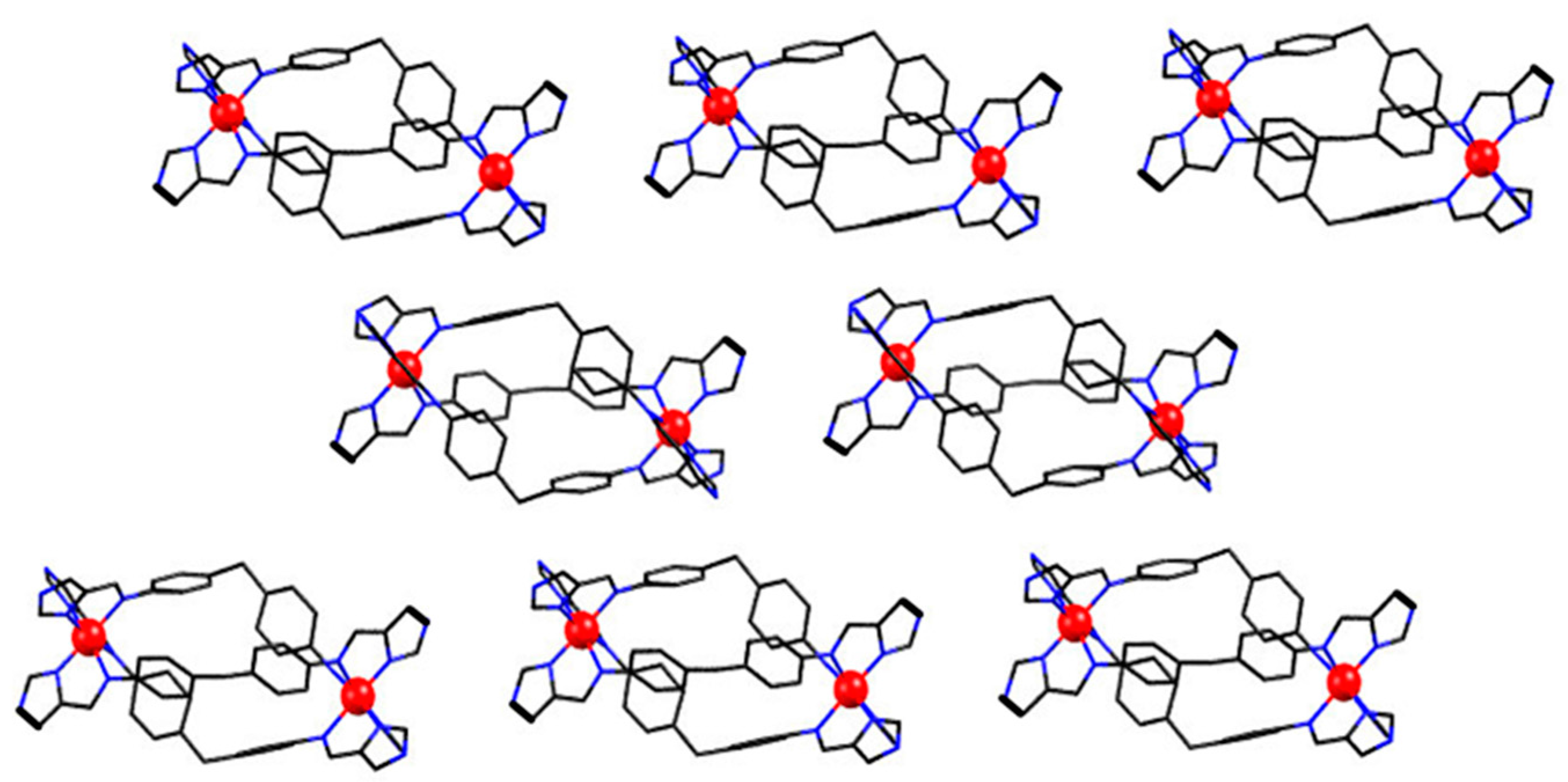
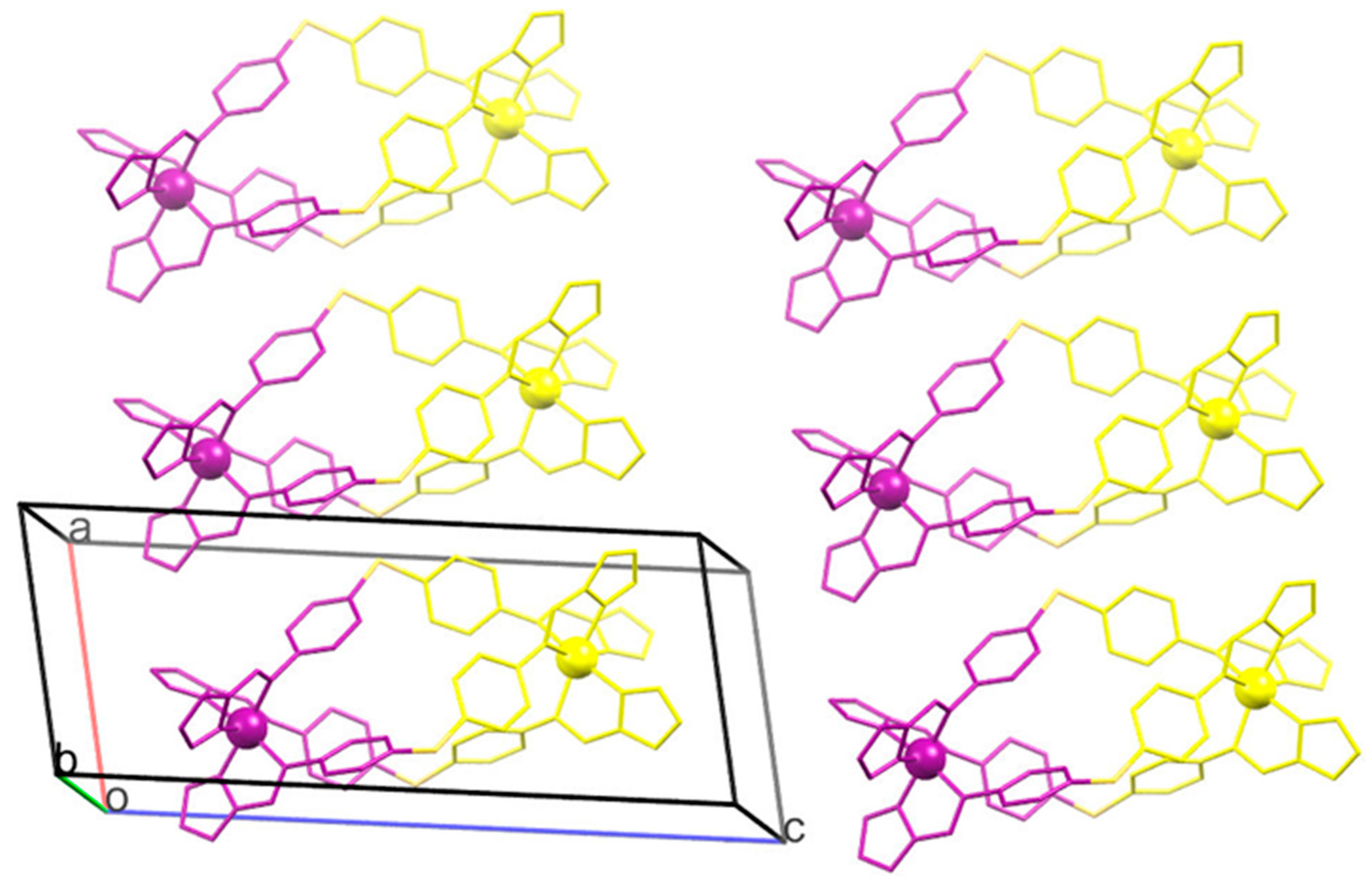
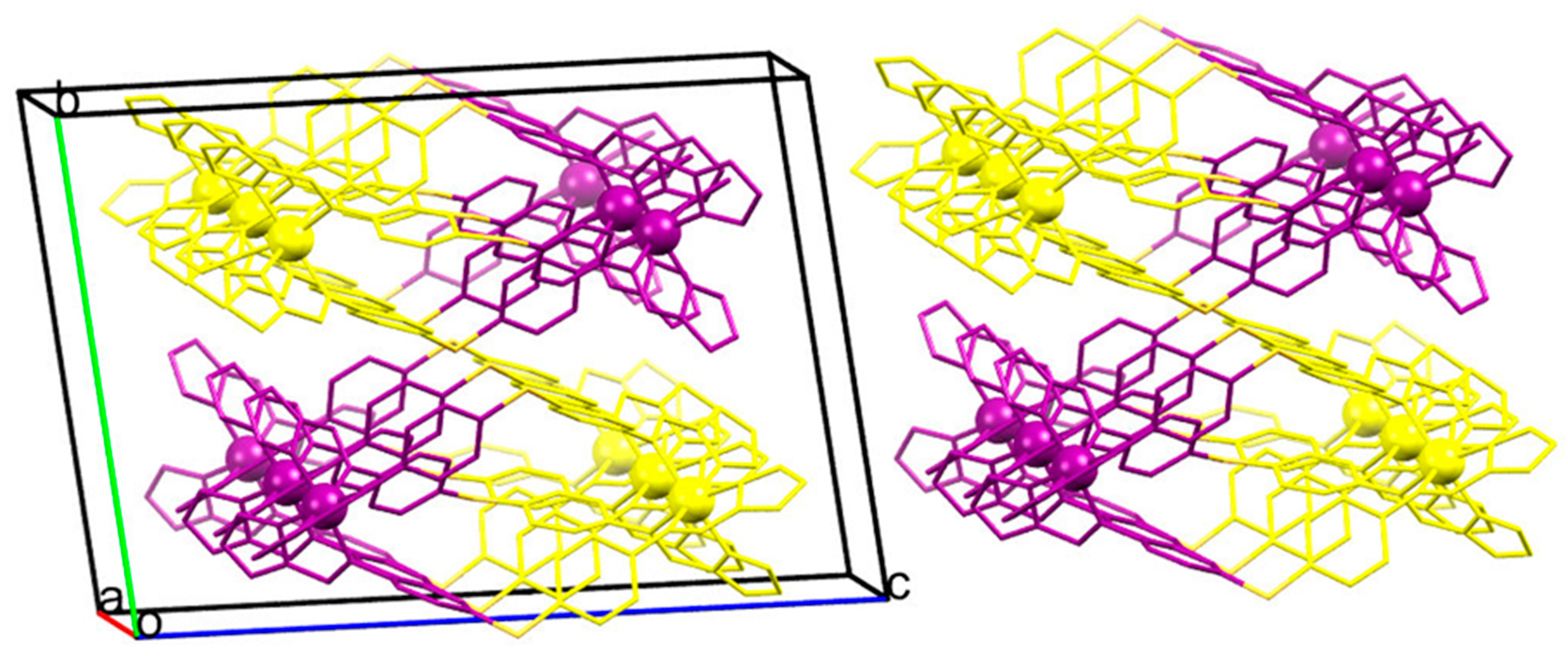
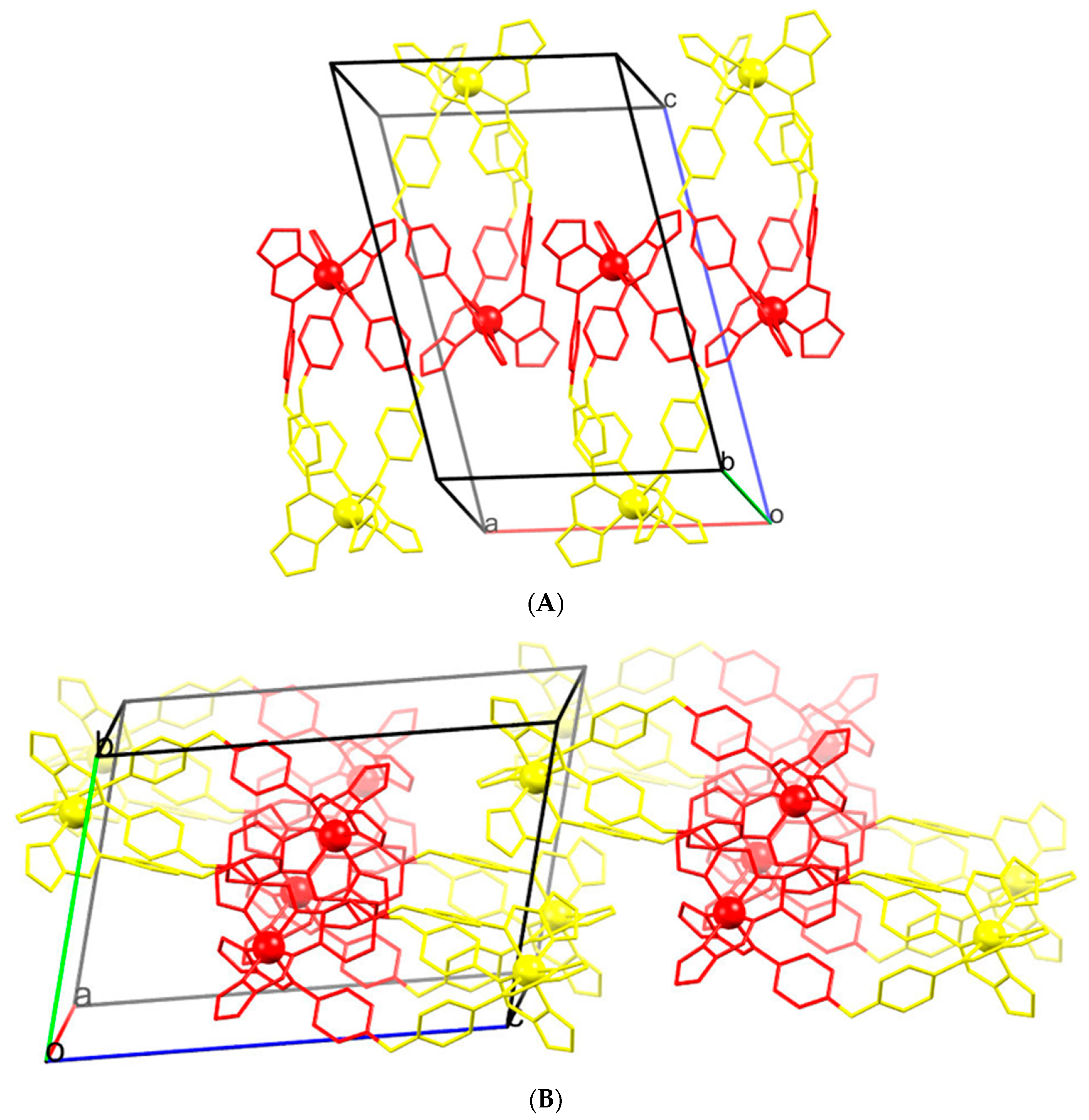
2.3. Magneto-Structural Correlations
3. Materials and Methods
3.1. Preparation of (L1, L2 and L3)
3.2. Preparation of 1, 2 and 3
3.3. X-ray Crystallography
3.4. Magnetic Susceptibility Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [PubMed]
- Brooker, S. Spin crossover with thermal hysteresis: Practicalities and lessons learnt. Chem. Soc. Rev. 2015, 44, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. Structure: Function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119–4142. [Google Scholar] [CrossRef] [PubMed]
- Bousseksou, A.; Molnár, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Leita, B.A.; Moubaraki, B.; Murray, K.S.; Smith, J.P.; Cashion, J.D. Structure and magnetism of a new pyrazolate bridged iron(II) spin crossover complex displaying a single HS–HS to LS–LS transition. Chem. Commun. 2004, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Toftlund, H. Spin equilibrium in solutions. Monatshefte Chem. 2001, 132, 1269–1277. [Google Scholar] [CrossRef]
- Kahn, O.; Martinez, C.J. Spin-Transition Polymers: From Molecular Materials toward Memory Devices. Science 1998, 279, 44–48. [Google Scholar] [CrossRef]
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; John Wiley & Sons Ltd.: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Hayami, S.; Shigeyoshi, Y.; Akita, M.; Inoue, K.; Kato, K.; Osaka, K.; Takata, M.; Kawajiri, R.; Mitani, T.; Maeda, Y. Reverse Spin Transition Triggered by a Structural Phase Transition. Angew. Chem. Int. Ed. Engl. 2005, 44, 4899–4903. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Moriyama, R.; Shigeyoshi, Y.; Kawajiri, R.; Mitani, T.; Akita, M.; Inoue, K.; Maeda, Y. Spin-Crossover Cobalt(II) Compound with Banana-Shaped Structure. Inorg. Chem. 2005, 44, 7295–7297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-H.; Zhang, S.-L.; Shao, D.; Wang, X.-Y. Spin Crossover in [Fe(2-Picolylamine)3](2+) Adjusted by Organosulfonate Anions. Inorg. Chem. 2015, 54, 7857–7867. [Google Scholar] [CrossRef] [PubMed]
- Dupouy, G.; Marchivie, M.; Triki, S.; Sala-Pala, J.; Gómez-García, C.J.; Pillet, S.; Lecomte, C.; Letard, J.-F. Photoinduced HS state in the first spin-crossover chain containing a cyanocarbanion as bridging ligand. Chem. Commun. 2009, 3404–3406. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; Niel, V.; Muñoz, M.C.; Real, J.A. Spin Crossover in 1D, 2D and 3D Polymeric Fe(II) Networks. In Spin Crossover in Transition Metal Compounds I; Gütlich, P., Goodwin, H.A., Eds.; Springer: Heidelberg/Berlin, Germany, 2004; pp. 229–257. [Google Scholar]
- Gütlich, P.; Garcia, Y.; Goodwin, H.A. Spin crossover phenomena in Fe(II) complexes. Chem. Soc. Rev. 2000, 29, 419–427. [Google Scholar] [CrossRef]
- Li, F.; Clegg, J.K.; Lindoy, L.F.; Macquart, R.B.; Meehan, G.V. Metallosupramolecular self-assembly of a universal 3-ravel. Nat. Commun. 2011, 2, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, P.N.W.; Lehn, J.-M.; Sleiman, H.; Rissanen, K. Multicomponent Self-Assembly: Generation and Crystal Structure of a Trimetallic[4]Pseudorotaxane. Angew. Chem. Int. Ed. 1997, 36, 1294–1296. [Google Scholar] [CrossRef]
- Sleiman, H.; Baxter, P.N.W.; Lehn, J.-M.; Airola, K.; Rissanen, K. Multicomponent Self-Assembly: Generation of Rigid-Rack Multimetallic Pseudorotaxanes. Inorg. Chem. 1997, 36, 4734–4742. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.D.; Raithby, P.R. Functional behaviour from controlled self-assembly: Challenges and prospects. Chem. Soc. Rev. 2013, 42, 1619–1636. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Childs, L.J. Helices and Helicates: Beautiful Supramolecular Motifs with Emerging Applications. Supramol. Chem. 2004, 16, 7–22. [Google Scholar] [CrossRef]
- Albrecht, M. “Let’s Twist Again” Double-Stranded, Triple-Stranded, and Circular Helicates. Chem. Rev. 2001, 101, 3457–3498. [Google Scholar] [CrossRef] [PubMed]
- Hora, S.; Hagiwara, H. High-Temperature Wide Thermal Hysteresis of an Iron(II) Dinuclear Double Helicate. Inorganics 2017, 5, 49. [Google Scholar] [CrossRef]
- Breuning, E.; Ruben, M.; Lehn, J.-M.; Renz, F.; Garcia, Y.; Ksenofontov, V.; Gutlich, P.; Wegelius, E.; Rissanen, K. Spin Crossover in a Supramolecular Fe4II [2×2] Grid Triggered by Temperature, Pressure, and Light. Angew. Chem. Int. Ed. 2000, 39, 2504–2507. [Google Scholar] [CrossRef]
- Struch, N.; Bannwarth, C.; Ronson, T.K.; Lorenz, Y.; Mienert, B.; Wagner, N.; Engeser, M.; Bill, E.; Puttready, R.; Rissanen, K.; et al. An Octanuclear Metallosupramolecular Cage Designed To Exhibit Spin-Crossover Behavior. Angew. Chem. Int. Ed. 2017, 56, 4930–4935. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Saigo, N.; Zhang, Y.; Fanna, D.J.; Shepherd, N.D.; Clegg, J.K.; Zheng, R.; Hayami, S.; Lindoy, L.F.; Aldrich-Wright, J.R.; et al. A large spin-crossover [Fe4L4]8+ tetrahedral cage. J. Mater. Chem. C 2015, 3, 7878–7882. [Google Scholar] [CrossRef]
- Tuna, F.; Lees, M.R.; Clarkson, G.J.; Hannon, M.J. Readily prepared metallo-supramolecular triple helicates designed to exhibit spin-crossover behaviour. Chem. Eur. J. 2004, 10, 5737–5750. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.; Hrudka, J.J.; Igimbayeva, D.; Lawson Daku, L.M.; Shatruk, M. A Simple Approach for Predicting the Spin State of Homoleptic Fe(II) Tris-diimine Complexes. J. Am. Chem. Soc. 2017, 139, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, T.; Nishi, K.; Matsumoto, N.; Iijima, S.; Halcrow, M.A.; Sunatsuki, Y.; Kojima, M. 1D and 2D assembly structures by imidazole···chloride hydrogen bonds of iron(II) complexes [FeII(HLn-Pr)3]Cl·Y (HLn-Pr = 2-methylimidazol-4-yl-methylideneamino-n-propyl; Y = AsF6, BF4) and their spin states. Dalton Trans. 2011, 40, 12301–12309. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; Grunert, C.M.; Reiman, S.; van Campenhoudt, O.; Gütlich, P. The Two-Step Spin Conversion in a Supramolecular Triple Helicate Dinuclear Iron(II) Complex Studied by Mössbauer Spectroscopy. Eur. J. Inorg. Chem. 2006, 2006, 3333–3339. [Google Scholar] [CrossRef]
- Pelleteret, D.; Clérac, R.; Mathonière, C.; Harté, E.; Schmitt, W.; Kruger, P.E. Asymmetric spin crossover behaviour and evidence of light-induced excited spin state trapping in a dinuclear iron(II) helicate. Chem. Commun. 2009, 2, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Archer, R.J.; Hawes, C.S.; Jameson, G.N.L.; McKee, V.; Moubaraki, B.; Chilton, N.F.; Murray, K.S.; Kruger, P.E. Partial spin crossover behaviour in a dinuclear iron(II) triple helicate. Dalton Trans. 2011, 40, 12368–12373. [Google Scholar] [CrossRef] [PubMed]
- Telfer, S.G.; Bocquet, B.; Williams, A.F. Thermal Spin Crossover in Binuclear Iron(II) Helicates: Negative Cooperativity and a Mixed Spin State in Solution. Inorg. Chem. 2001, 40, 4818–4820. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Kawamoto, R.; Tsubouchi, R.; Sunatsuki, Y.; Kojima, M.; Iijima, S.; Matsumoto, N. Spin States of Mono- and Dinuclear Iron(II) Complexes with Bis(imidazolylimine) Ligands. Chem. Lett. 2007, 36, 1284–1285. [Google Scholar] [CrossRef]
- Sun, Q.-F.; Iwasa, J.; Ogawa, D.; Ishido, Y.; Sato, S.; Ozeki, T.; Sei, T.; Yamaguchi, K.; Fujita, M. Self-Assembled M24L48 Polyhedra and Their Sharp Structural Switch upon Subtle Ligand Variation. Science 2010, 328, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Marchivie, M.; Guionneau, P.; Létard, J.-F.; Chasseau, D. Photo-induced spin-transition: The role of the iron(II) environment distortion. Acta Crystallogr. Sect. B 2005, 61, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Dupouy, G.; Marchivie, M.; Triki, S.; Sala-Pala, J.; Salaün, J.-Y.; Gómez-García, C.J.; Guionneau, P. The Key Role of the Intermolecular π-π Interactions in the Presence of Spin Crossover in Neutral [Fe(abpt)2A2] Complexes (A = Terminal Monoanion N Ligand). Inorg. Chem. 2008, 47, 8921–8931. [Google Scholar] [CrossRef] [PubMed]
- Milin, E.; Benaicha, B.; El Hajj, F.; Patinec, V.; Triki, S.; Marchivie, M.; Gomez-Garcia, C.J.; Pillet, S. Magnetic Bistability in Macrocycle-Based FeII Spin-Crossover Complexes: Counter Ion and Solvent Effects. Eur. J. Inorg. Chem. 2016, 34, 5305–5314. [Google Scholar] [CrossRef]
- Guionneau, P.; Gac, F.L.; Lakhoufi, S.; Kaiba, A.; Chasseau, D.; Létard, J.-F.; Negrier, P.; Mondieig, D.; Howard, J.A.K.; Leger, J.-M. X-ray diffraction investigation of a spin crossover hysteresis loop. J. Phys. 2007, 19, 326211. [Google Scholar] [CrossRef]
- Harding, D.J.; Phonsri, W.; Harding, P.; Gass, I.A.; Murray, K.S.; Moubaraki, B.; Cashion, J.D.; Liu, L.; Telfer, S.G. Abrupt spin crossover in an iron(III) quinolylsalicylaldimine complex: Structural insights and solvent effects. Chem. Commun. 2013, 49, 6340–6342. [Google Scholar] [CrossRef] [PubMed]
- Dorbes, S.; Valade, L.; Real, J.A.; Faulmann, C. [Fe(sal2-trien)][Ni(dmit)2]: Towards switchable spin crossover molecular conductors. Chem. Commun. 2005, 69–71. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalex, A.; Sauter, N.K.; Phizackerly, R.P.; et al. Blu-Ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Bruker. SADABS, Version 2.11; Bruker AXS Inc.: Fitchburg, WI, USA, 2001.
- Sheldrick, G.M. SHELX-2014: Programs for Crystal Structure Analysis; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Σ (Degrees) | Average Fe(II)–N Bond Lengths (Å) | High Spin or Low Spin Fe(II) in the Asymmetric Unit |
|---|---|---|---|
| 1 100 K | Fe01—76.30 | Fe01—2.13 | 1 mixed HS/LS-state population |
| 2 100 K | Fe01—59.4 | Fe01—2.00 | 1 HS, 1 LS |
| Fe02—90.3 | Fe02—2.18 | ||
| 3 100 K | Fe01—77.2 | Fe01—2.10 | 1 HS, 1 mixed |
| Fe02—85.2 | Fe02—2.18 | HS/LS state population |
| Complex | –X– Angle (Degrees) | Intra-Helical Separation (Å) | Closest Inter-Helical Separation (Å) |
|---|---|---|---|
| 1 | 113.6 | 11.72 | 8.71 |
| 2 | 104.9 | 11.78 | 8.31 |
| 3 | 115.8 | 11.62 | 7.96 |
| 1·(CH3CN) | 2·2 (CH3CN) | 3·2 (CH3CN) | |
|---|---|---|---|
| Empirical formula | C65H57B4F16Fe2N19 | C64H54B4F16Fe2N20S3 | C64H54B4F16Fe2N20O3 |
| Formula weight | 1563.23 | 1658.39 | 1610.21 |
| Temperature/K | 100 | 100 | 100 |
| Crystal system | monoclinic | triclinic | triclinic |
| Space group | C2/c | P | P |
| a/Å | 21.131(4) | 9.5210(19) | 13.837(3) |
| b/Å | 17.391(4) | 16.723(3) | 13.855(3) |
| c/Å | 21.069(4) | 23.621(5) | 20.684(4) |
| α/° | 90 | 95.29(3) | 77.43(3) |
| β/° | 107.00(3) | 100.29(3) | 77.58(3) |
| γ/° | 90 | 92.93(3) | 86.73(3) |
| Volume/Å3 | 7404(3) | 3675.8(13) | 3779.5(15) |
| Z | 4 | 2 | 2 |
| ρcalc.g/cm3 | 1.402 | 1.498 | 1.415 |
| μ/mm 1 | 0.485 | 0.576 | 0.481 |
| F(000) | 3184.0 | 1684.0 | 1636.0 |
| 2Θ range for data collection/° | 0.02 × 0.01 × 0.01 | 0.015 × 0.0075 × 0.0075 | 0.02 × 0.01 × 0.01 |
| Index ranges | Synchrotron (λ = 0.7108) | Synchrotron (λ = 0.7108) | Synchrotron (λ = 0.7108) |
| Reflections collected | 3.09 to 52.998 | 1.762 to 52.742 | 2.062 to 53.998 |
| Independent reflections | −26 ≤ h ≤ 26, −21 ≤ k ≤ 21, −26 ≤ l ≤ 26 | −11 ≤ h ≤ 11, −20 ≤ k ≤ 20, −29 ≤ l ≤ 29 | −17 ≤ h ≤ 17, −17 ≤ k ≤ 17, −26 ≤ l ≤ 26 |
| Data/restraints/parameters | 50,460 | 45,853 | 57,096 |
| Goodness-of-fit on F2 | 6995 [Rint = 0.0697, Rsigma = 0.0341] | 13,521 [Rint = 0.0971, Rsigma = 0.0898] | 14,860 [Rint = 0.0446, Rsigma = 0.0360] |
| Final R indexes [I >= 2σ (I)] | 6995/218/630 | 13,521/202/1047 | 14,860/33/1095 |
| Final R indexes [all data] | 1.119 | 1.157 | 1.072 |
| Largest diff. peak/hole/e Å-3 | R1 = 0.1050, wR2 = 0.2997 | R1 = 0.1103, wR2 = 0.2958 | R1 = 0.0691, wR2 = 0.1967 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craze, A.R.; Sciortino, N.F.; Badbhade, M.M.; Kepert, C.J.; Marjo, C.E.; Li, F. Investigation of the Spin Crossover Properties of Three Dinulear Fe(II) Triple Helicates by Variation of the Steric Nature of the Ligand Type. Inorganics 2017, 5, 62. https://doi.org/10.3390/inorganics5040062
Craze AR, Sciortino NF, Badbhade MM, Kepert CJ, Marjo CE, Li F. Investigation of the Spin Crossover Properties of Three Dinulear Fe(II) Triple Helicates by Variation of the Steric Nature of the Ligand Type. Inorganics. 2017; 5(4):62. https://doi.org/10.3390/inorganics5040062
Chicago/Turabian StyleCraze, Alexander R., Natasha F. Sciortino, Mohan M. Badbhade, Cameron J. Kepert, Christopher E. Marjo, and Feng Li. 2017. "Investigation of the Spin Crossover Properties of Three Dinulear Fe(II) Triple Helicates by Variation of the Steric Nature of the Ligand Type" Inorganics 5, no. 4: 62. https://doi.org/10.3390/inorganics5040062