Size Separation Techniques for the Characterisation of Cross-Linked Casein: A Review of Methods and Their Applications

Abstract
:1. Introduction
2. Enzymatic and Non-Enzymatic Cross-Linking Reactions
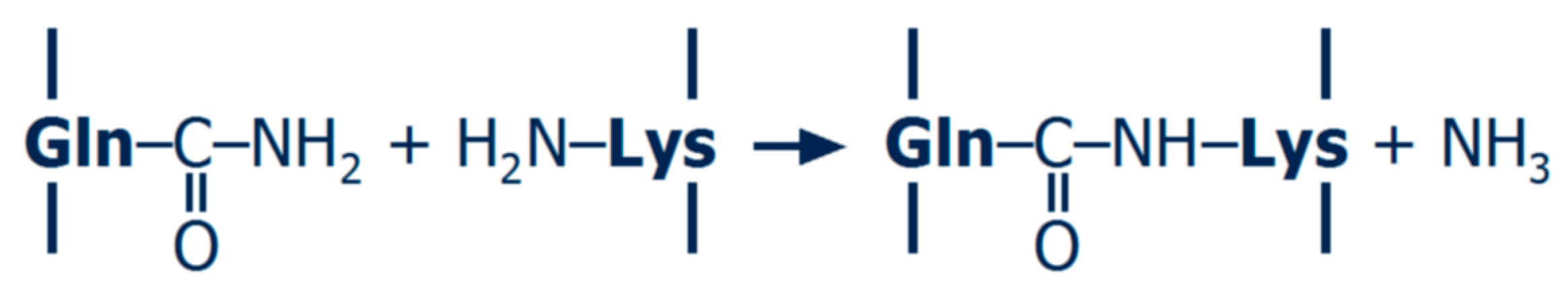
2.1. Cross-Linking Enzymes
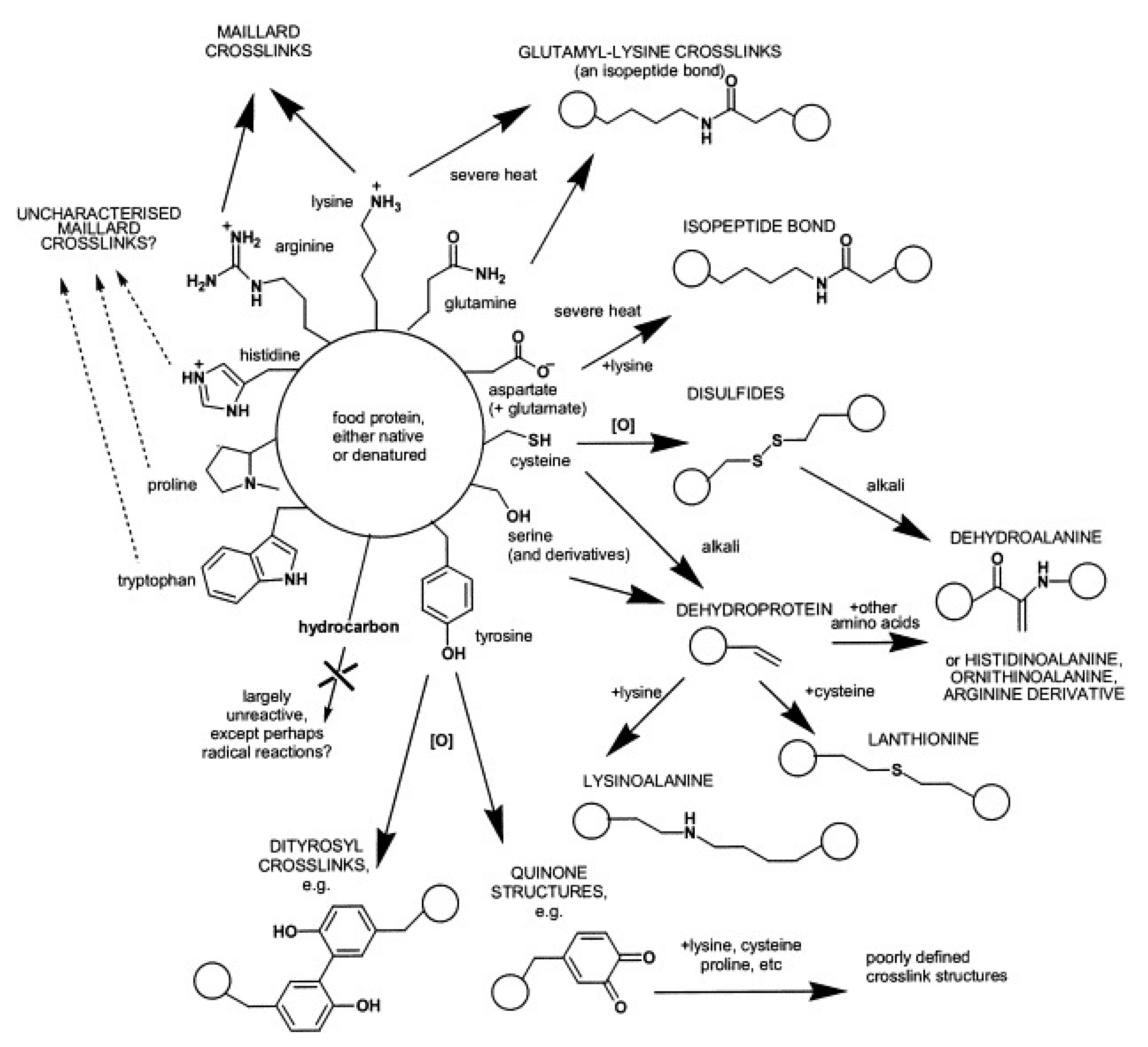
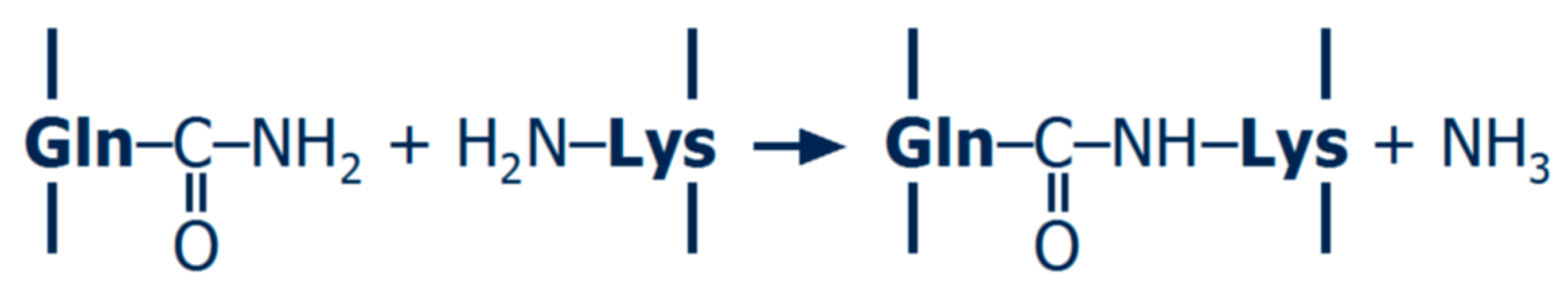
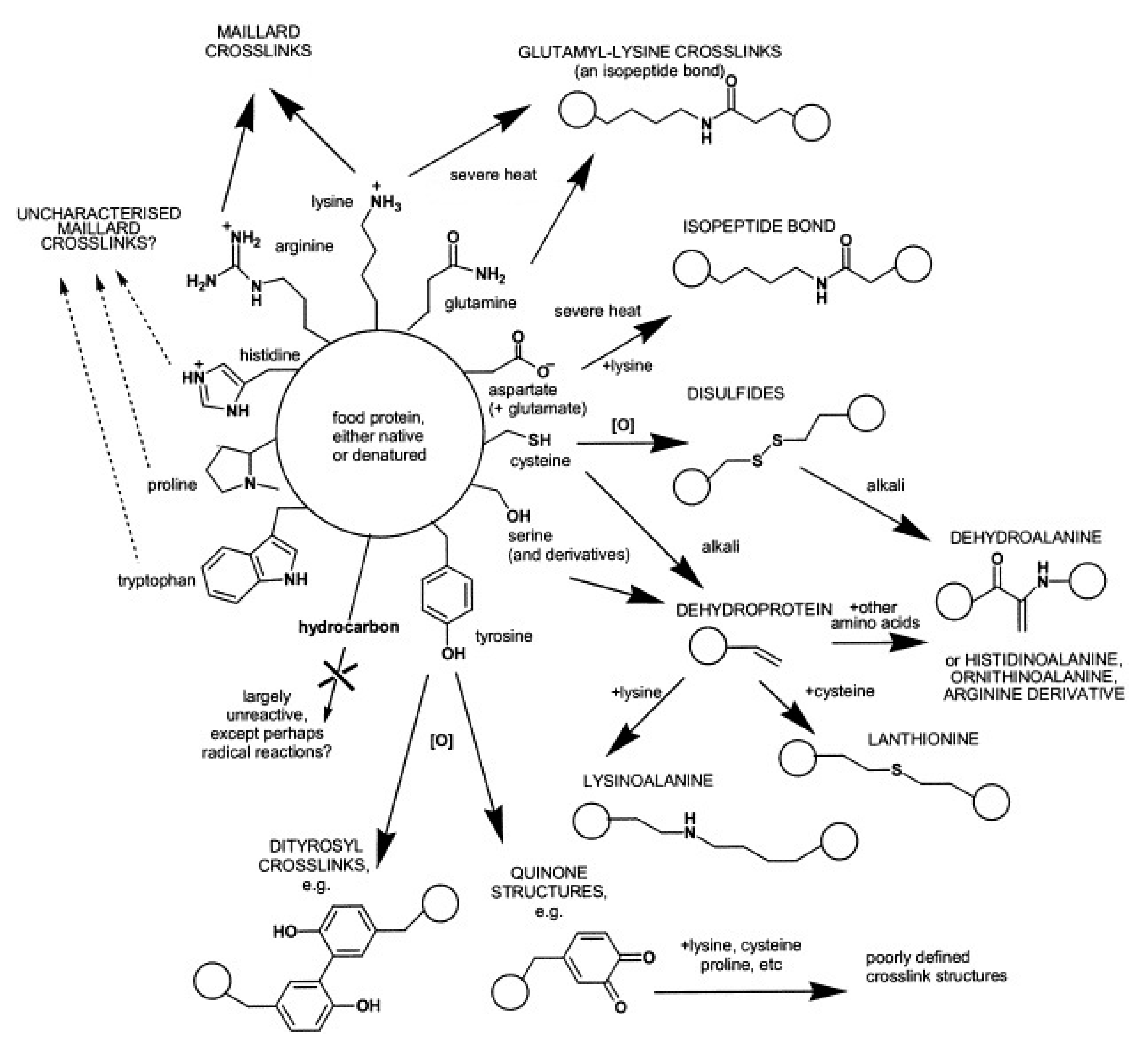
2.2. Non-Enzymatic Cross-Linking
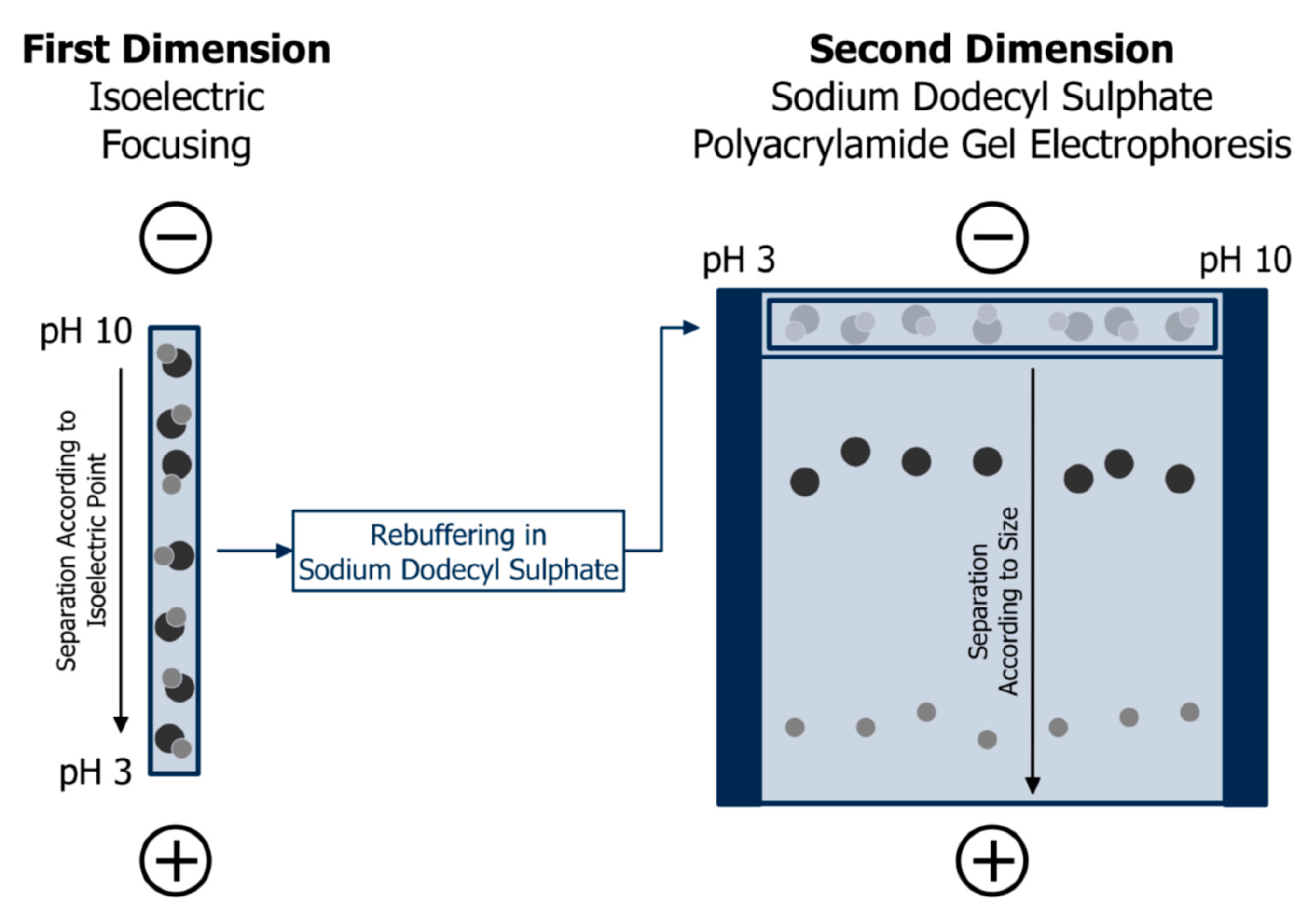
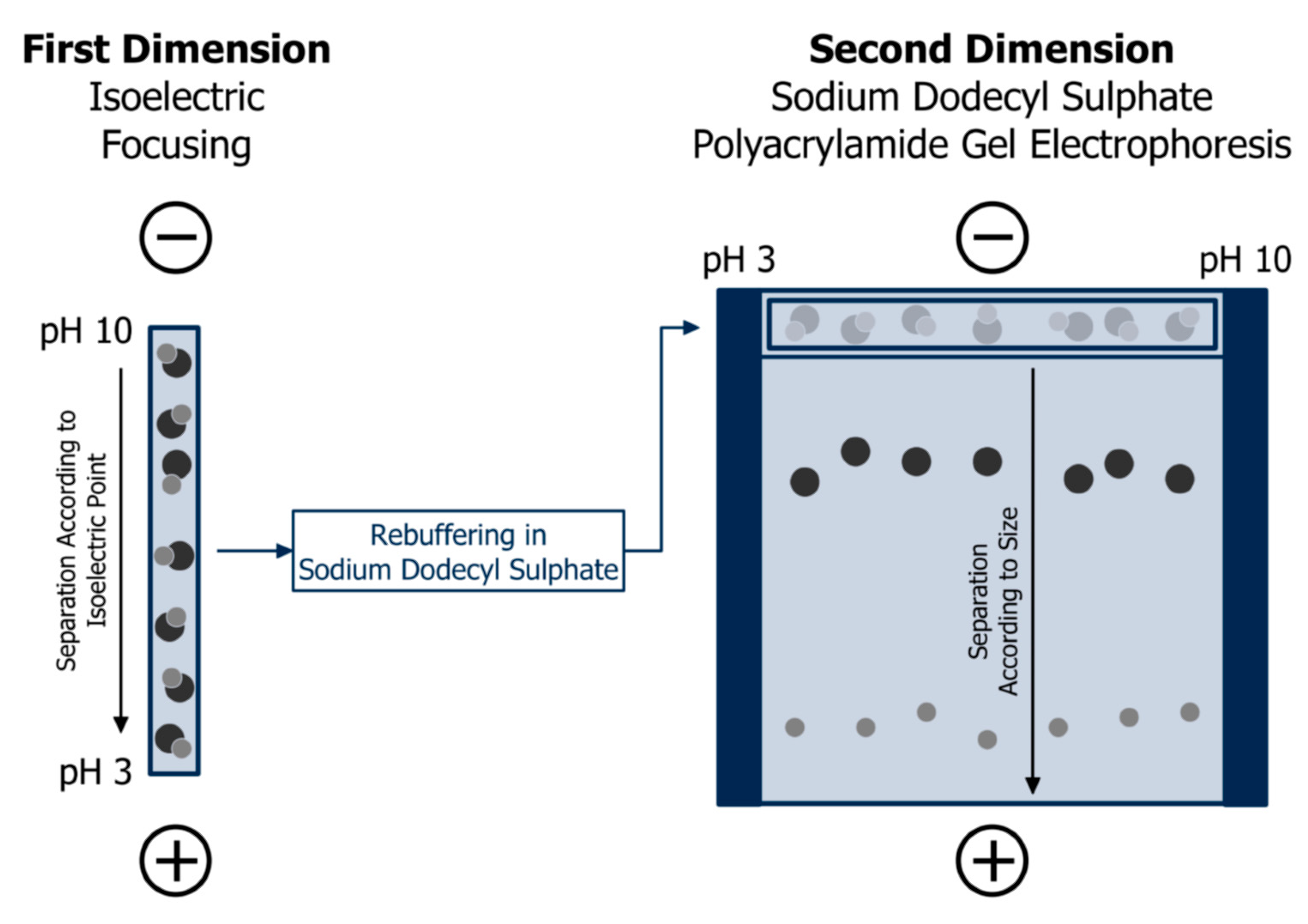
3. Gel Electrophoresis and Capillary Electrophoresis
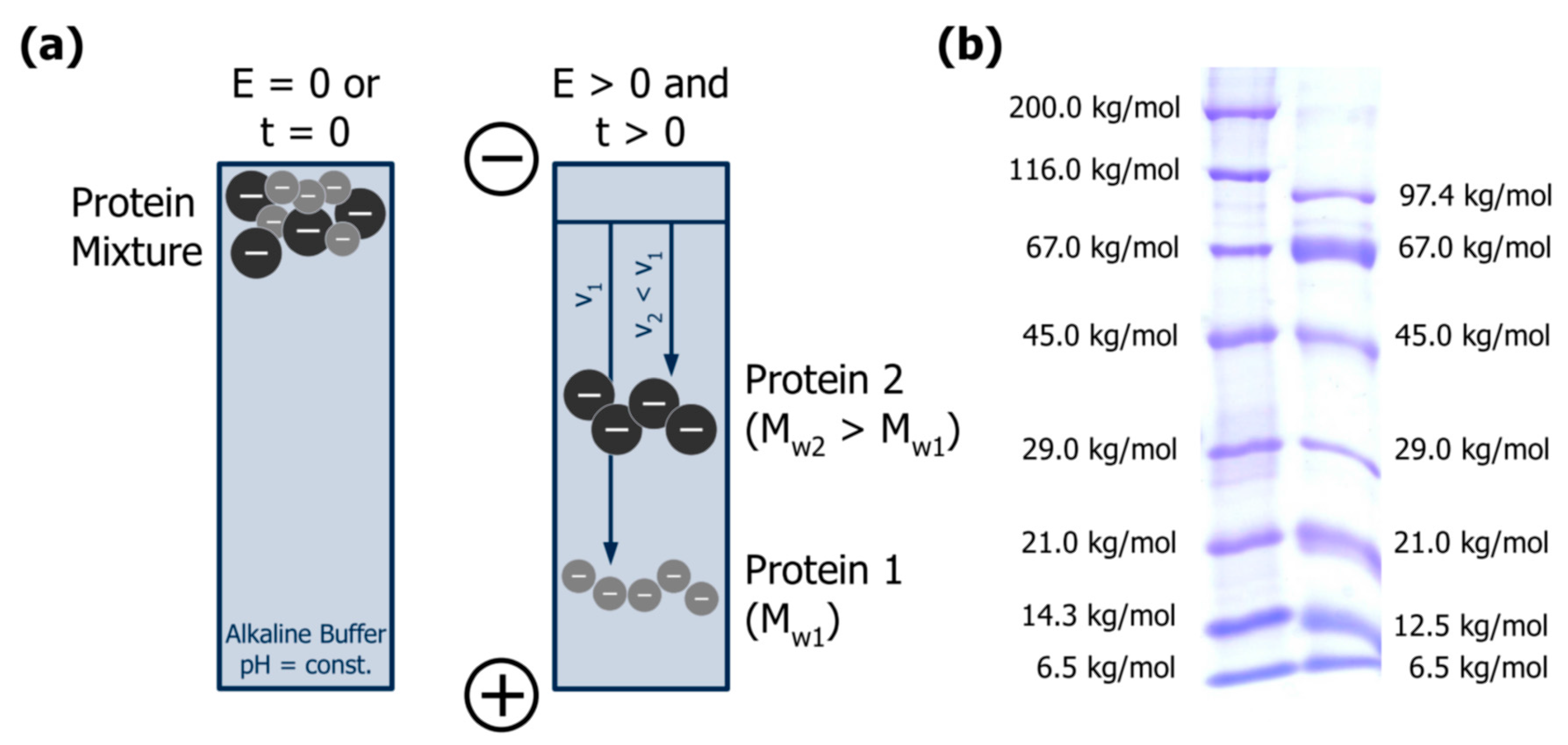
3.1. Separation Principle
3.2. Advantages and Limitations in the Context of Casein Investigation
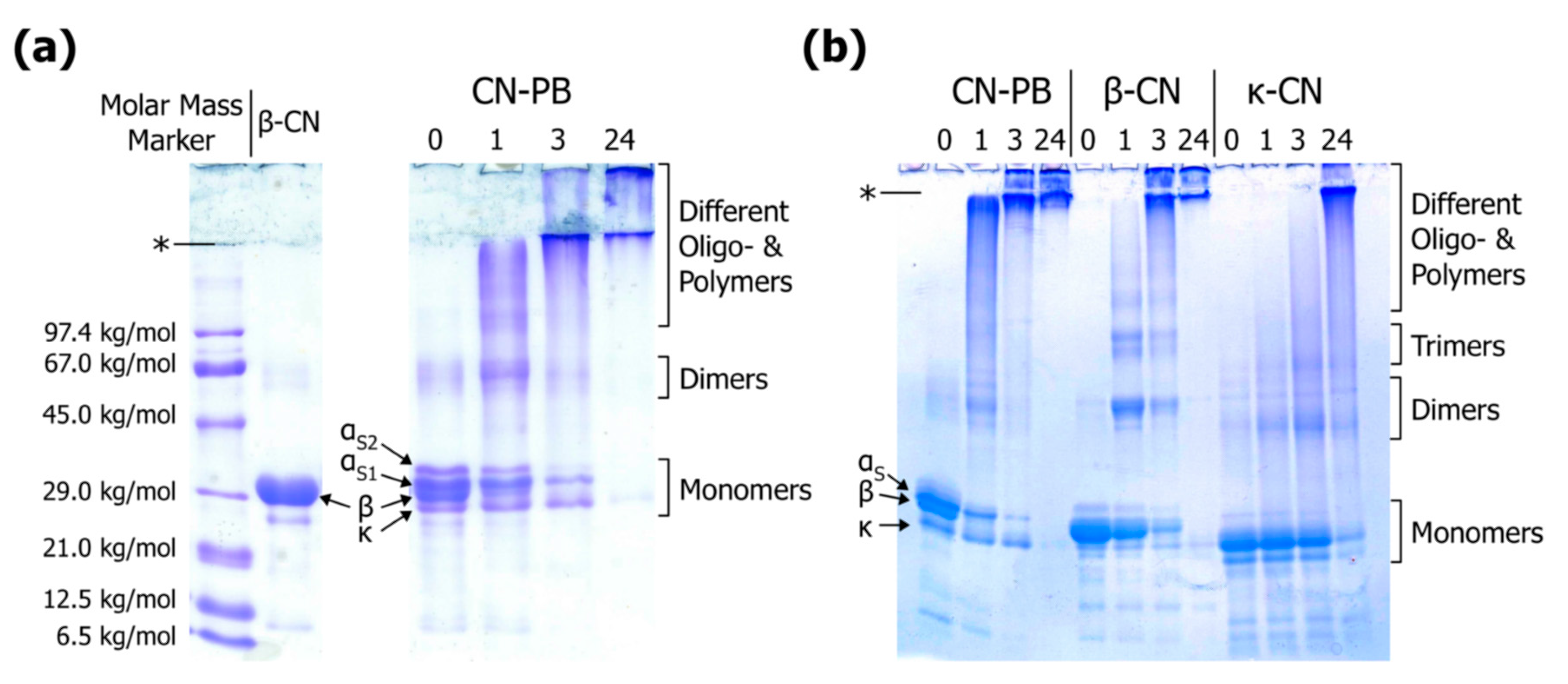
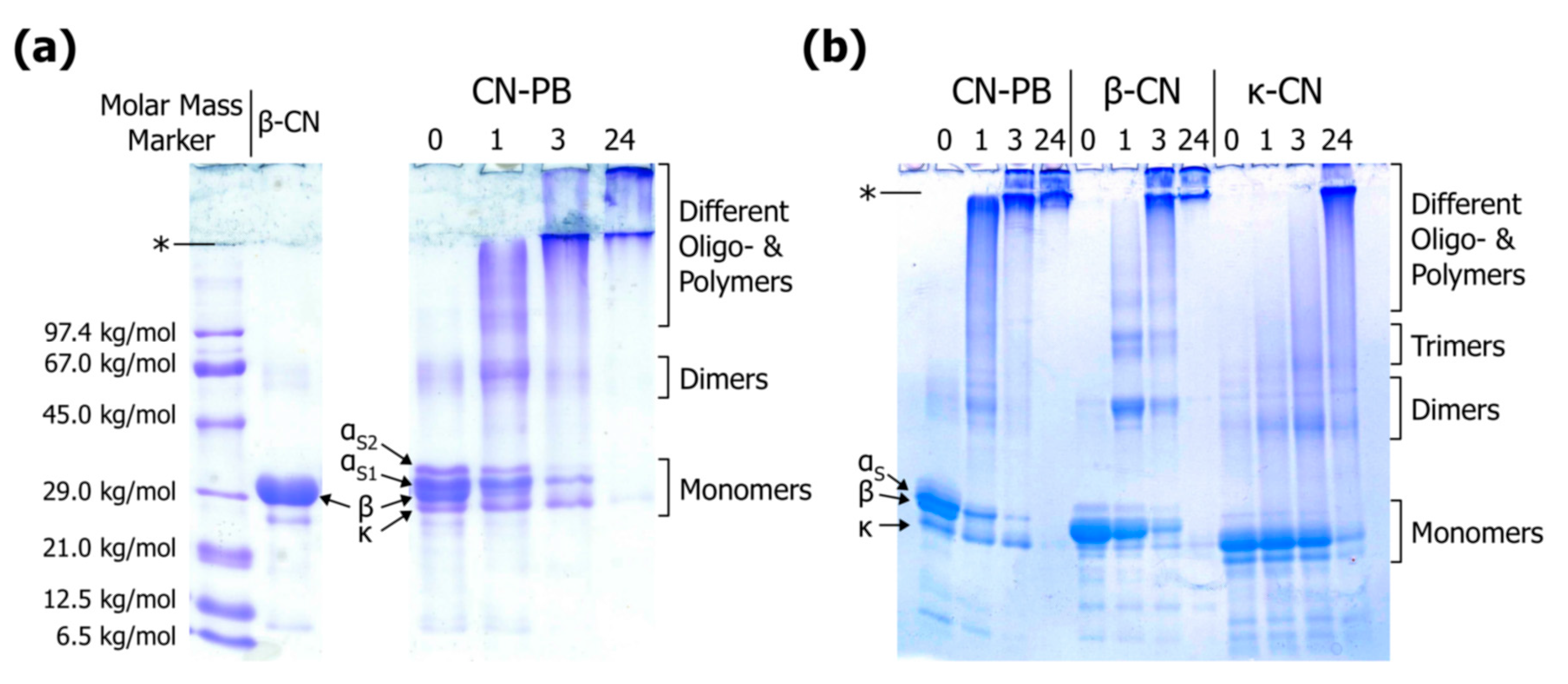
3.3. Literature Review of Studies on Cross-Linked Casein
4. Size Exclusion Chromatography
4.1. Separation Principle
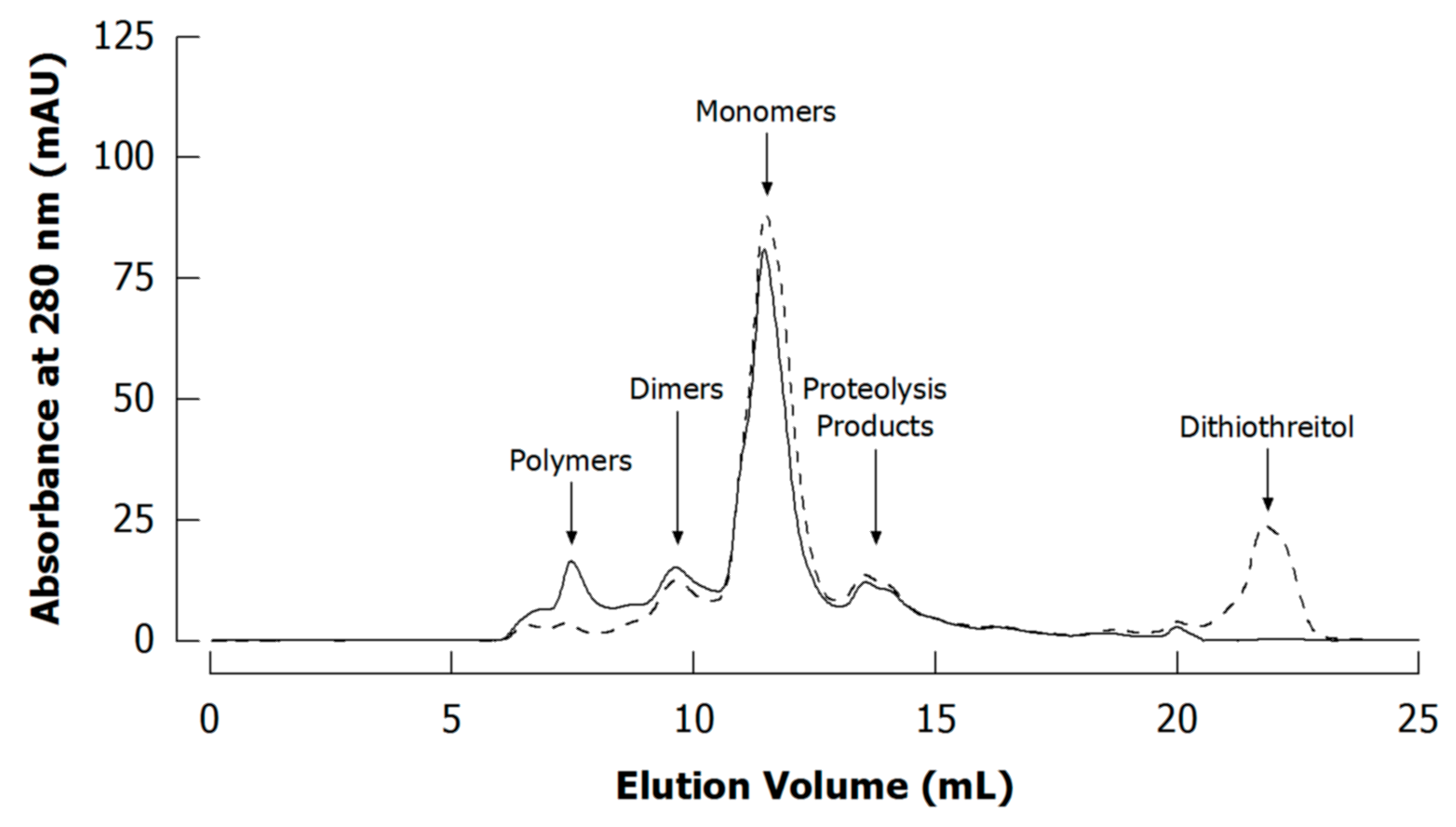
4.2. Advantages and Limitations in the Context of Casein Investigation
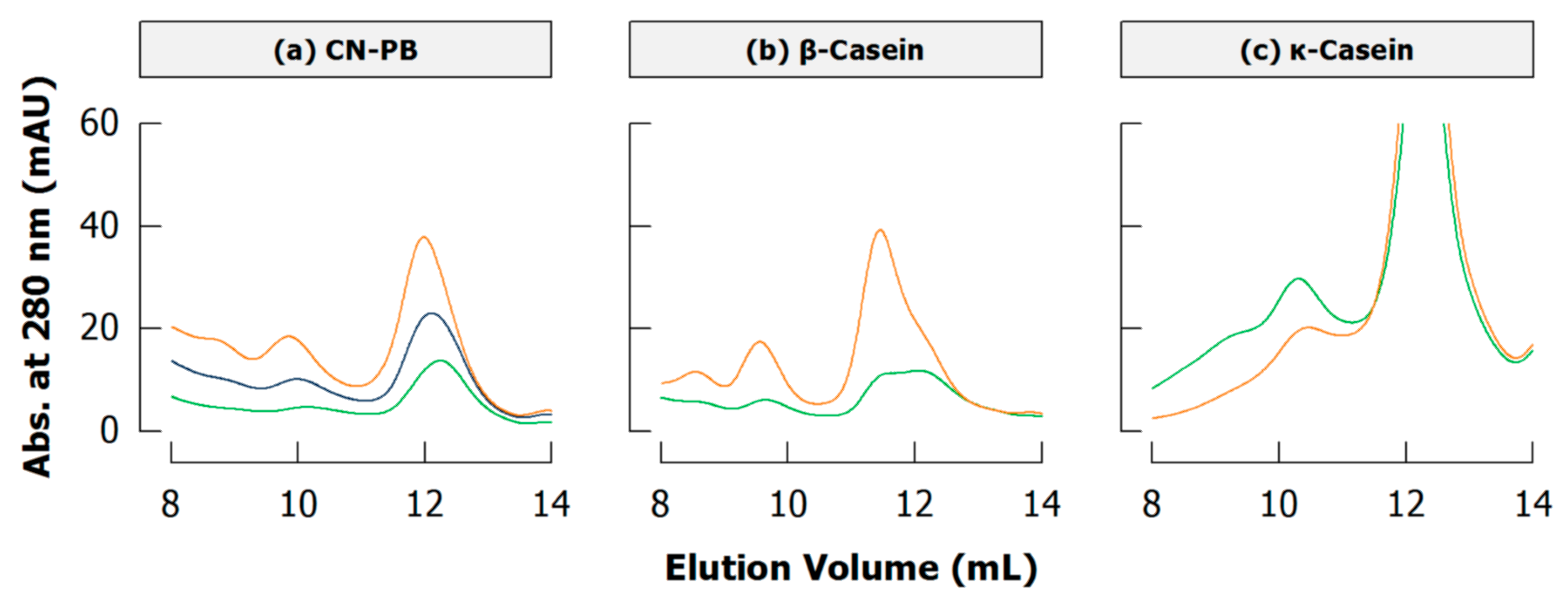
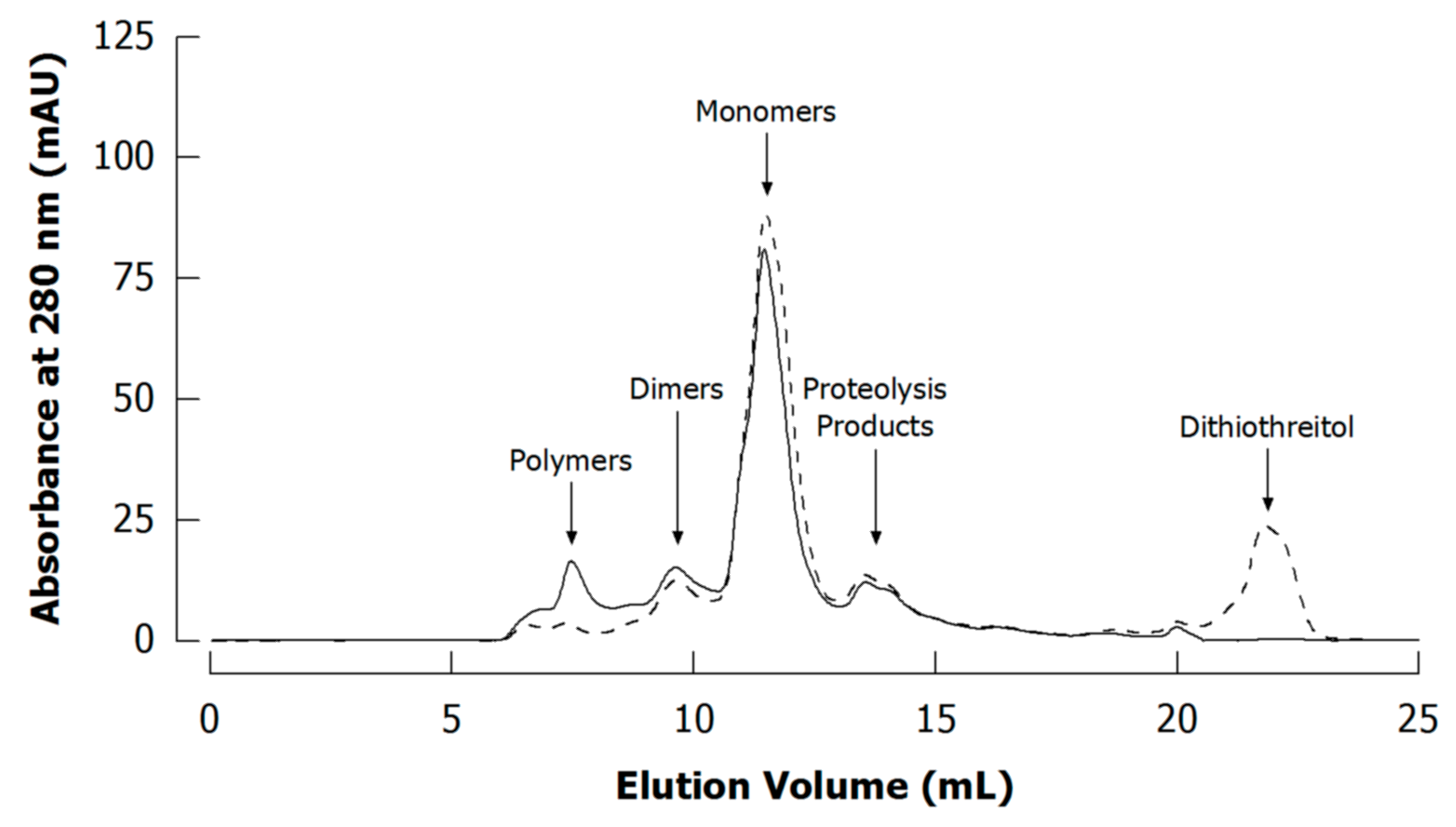
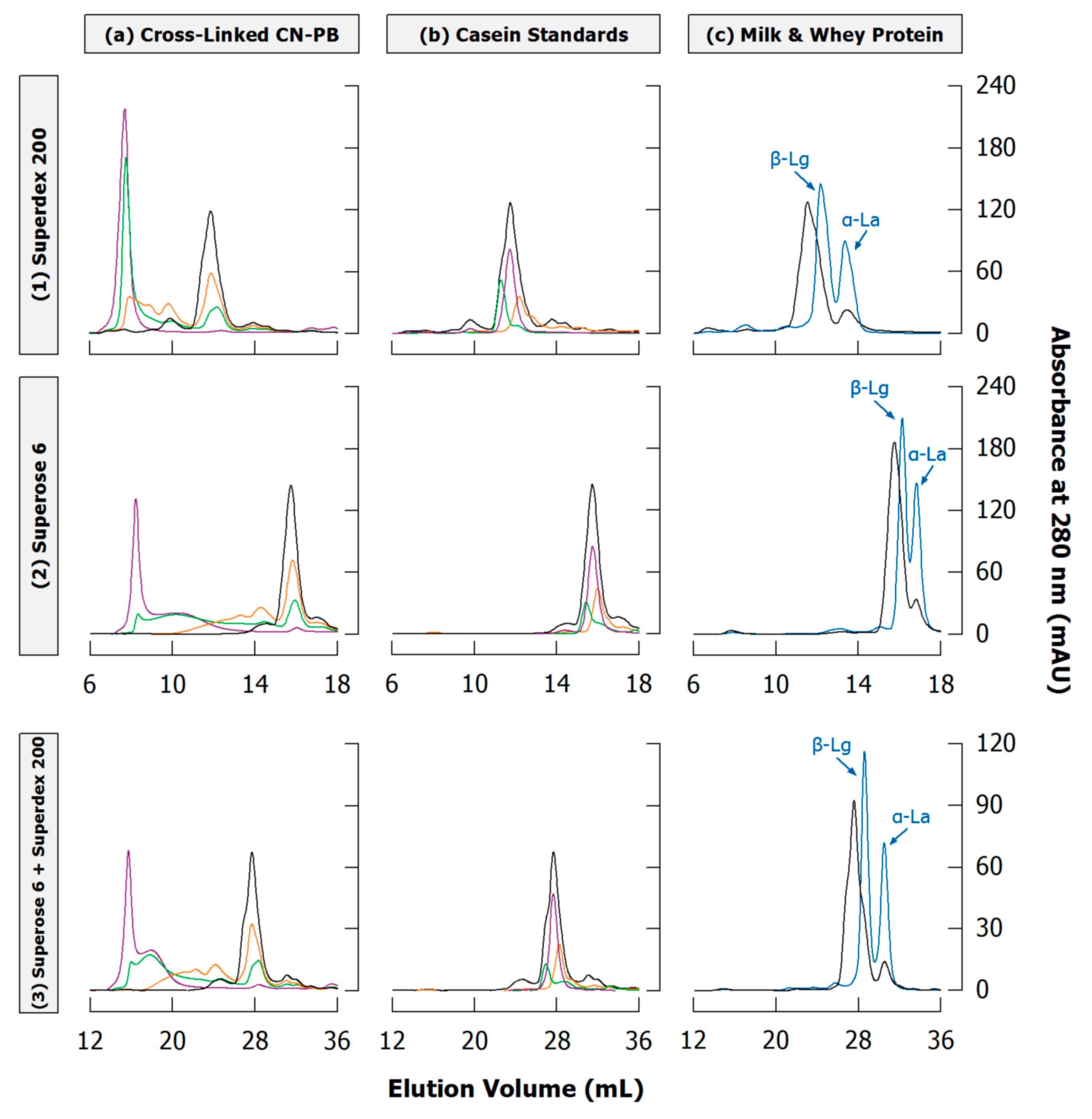
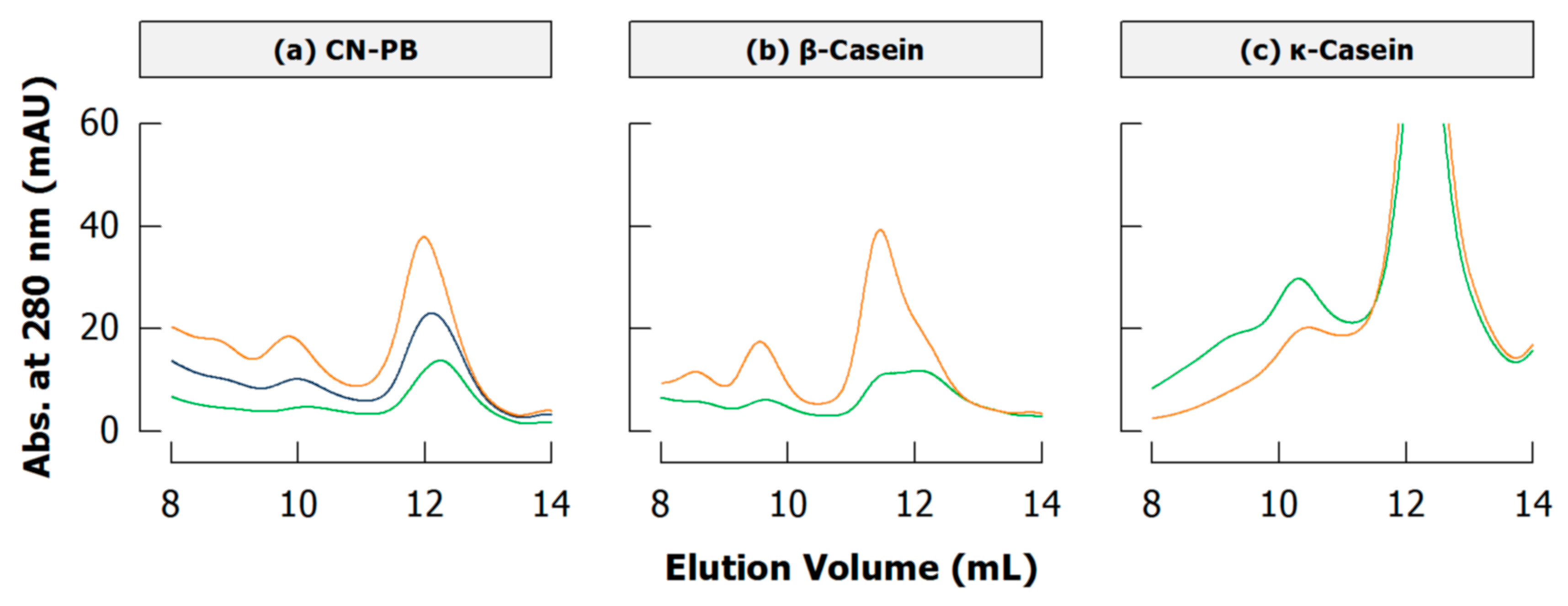
4.3. Literature Review of Studies on Cross-Linked Casein
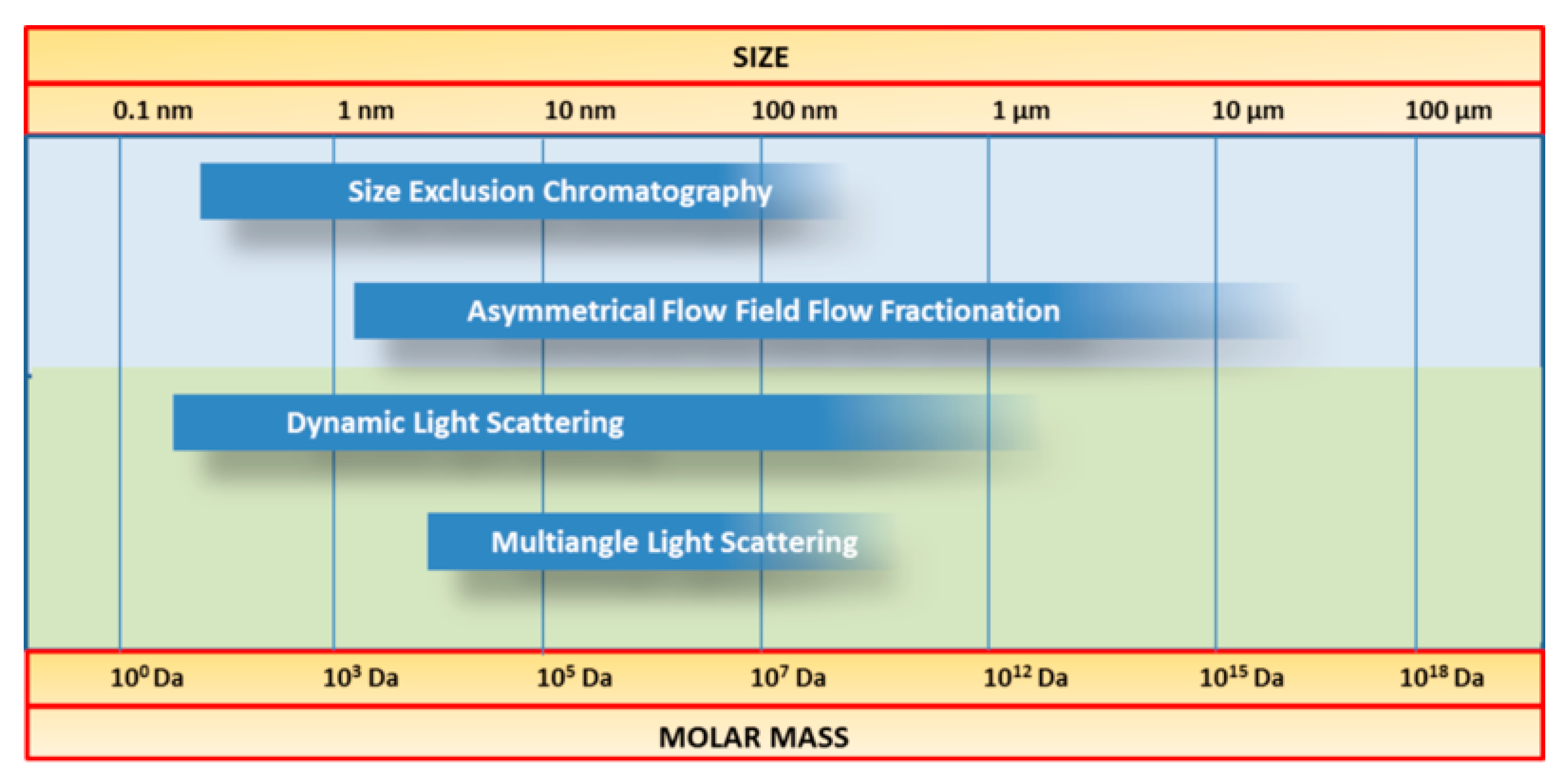
5. Field Flow Fractionation
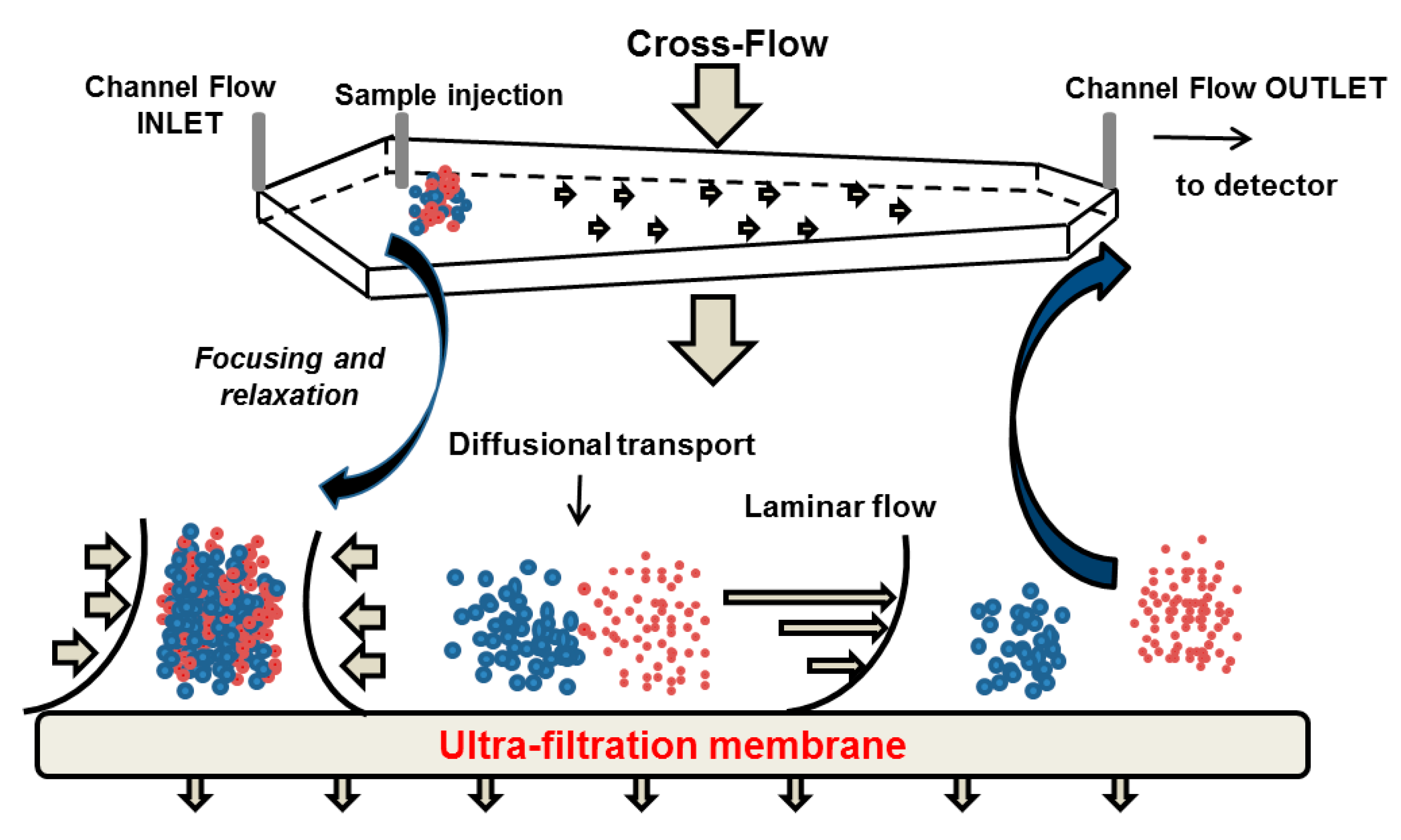
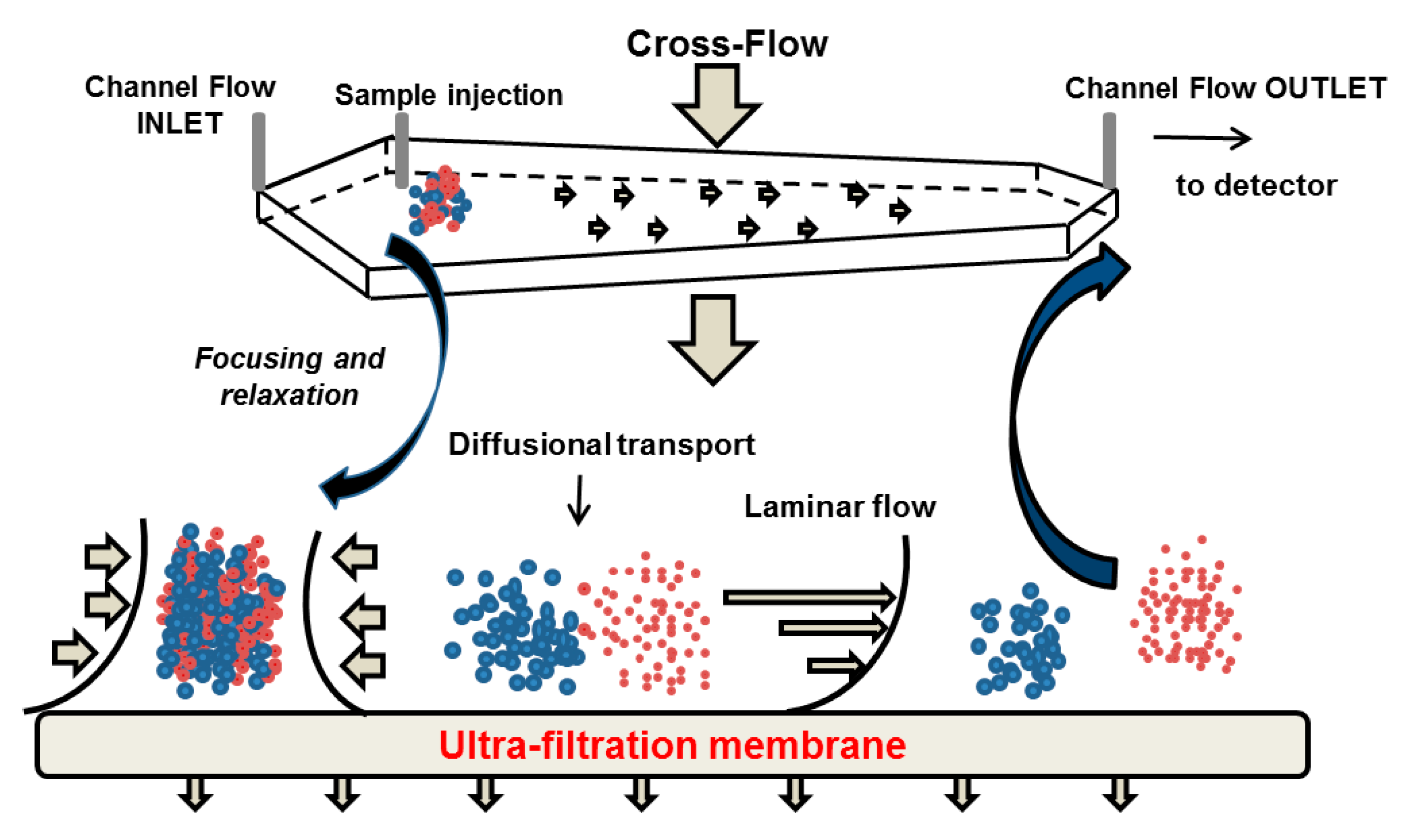
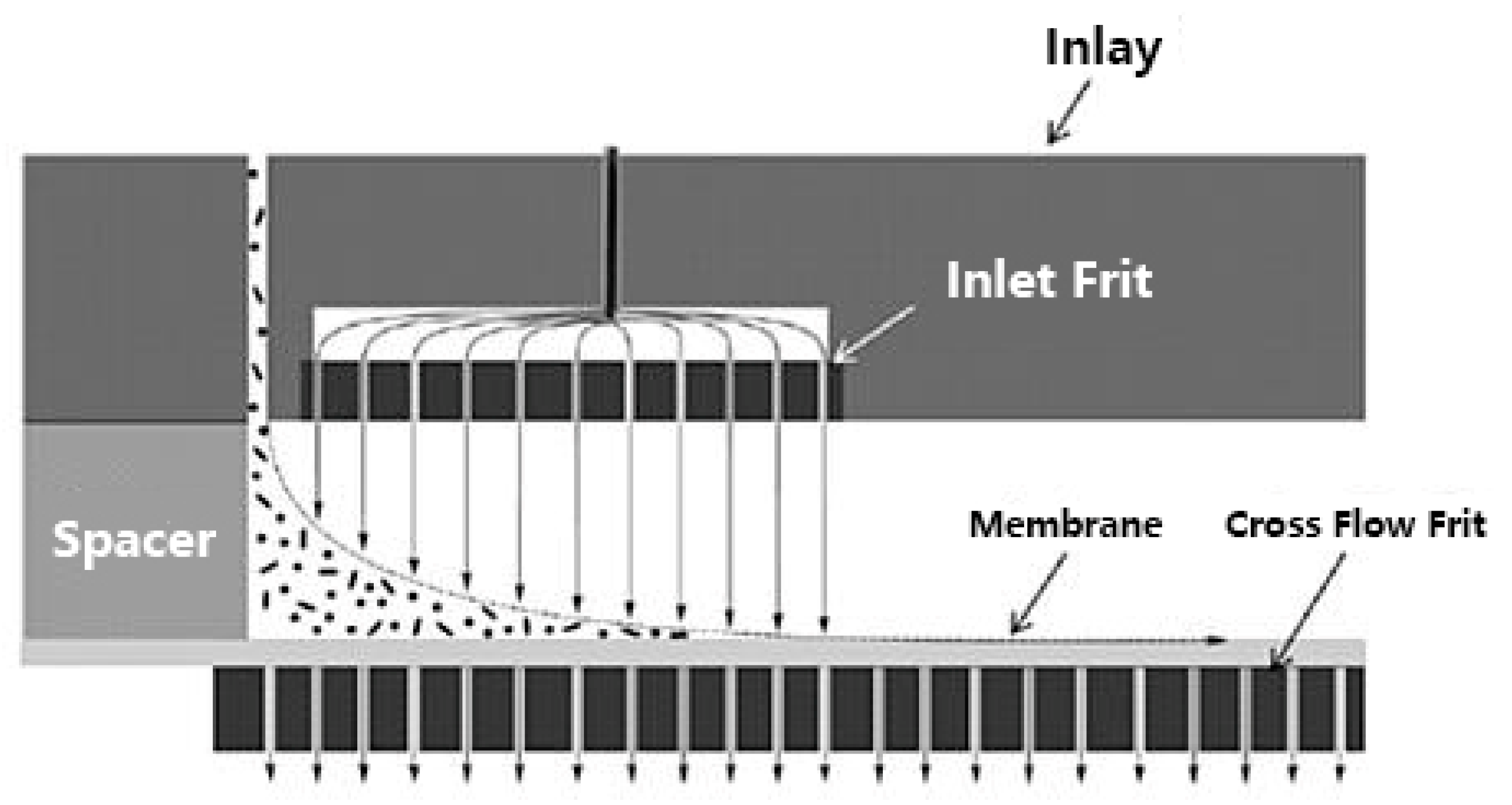
5.1. Separation Principle
5.2. Potential Advantages and Limitations in the Context of Casein Investigation
5.3. Literature Review of Studies on Casein
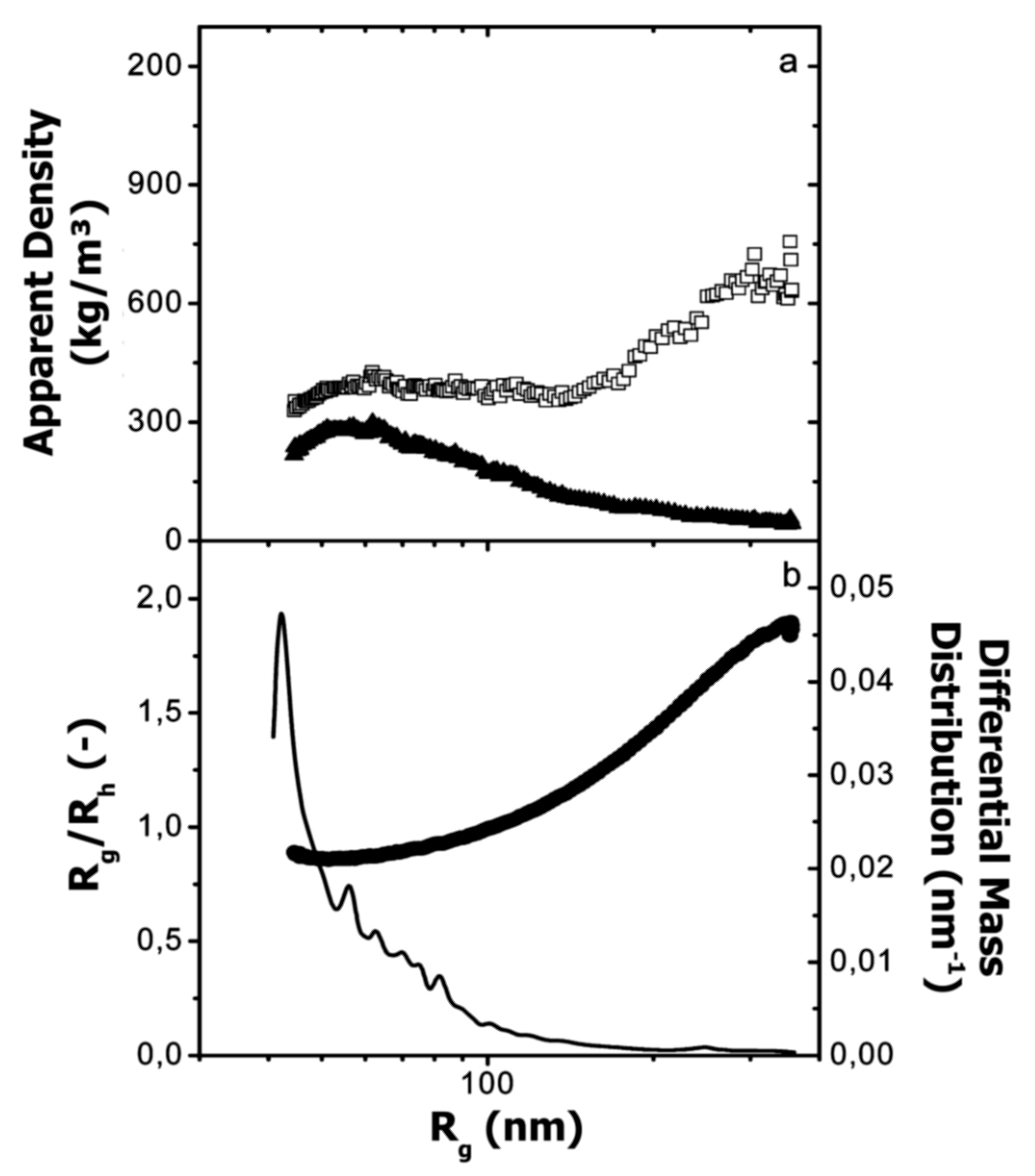
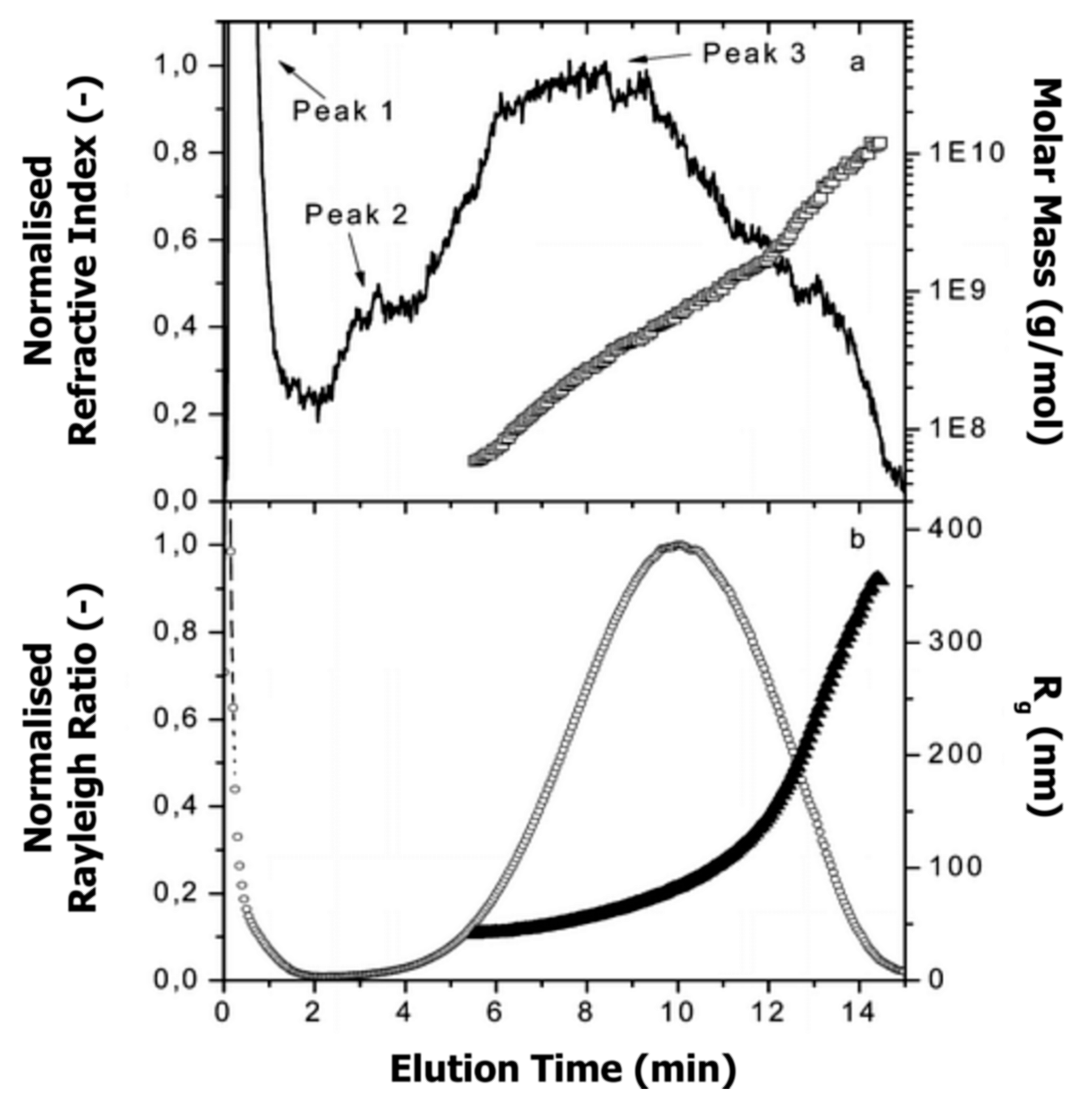
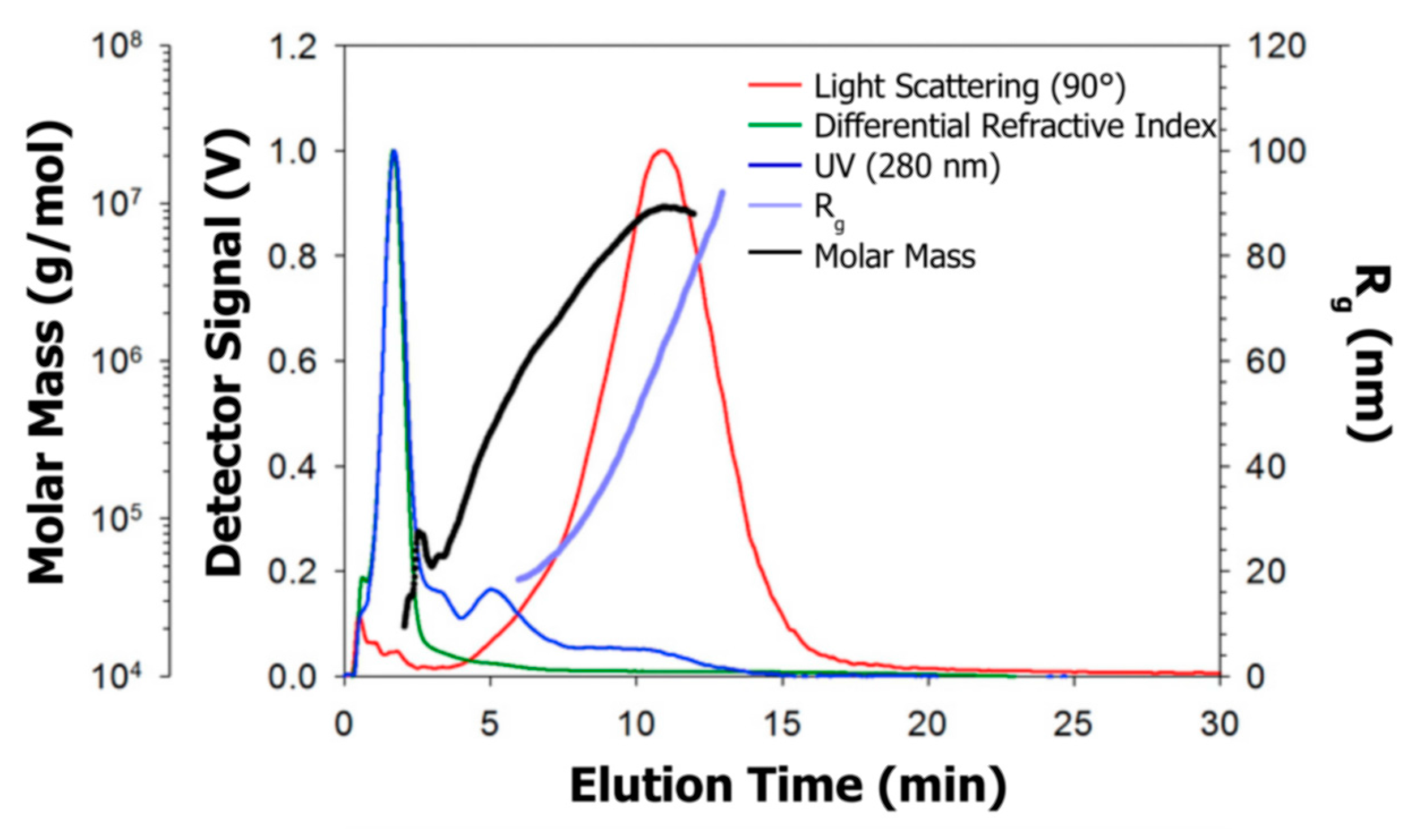
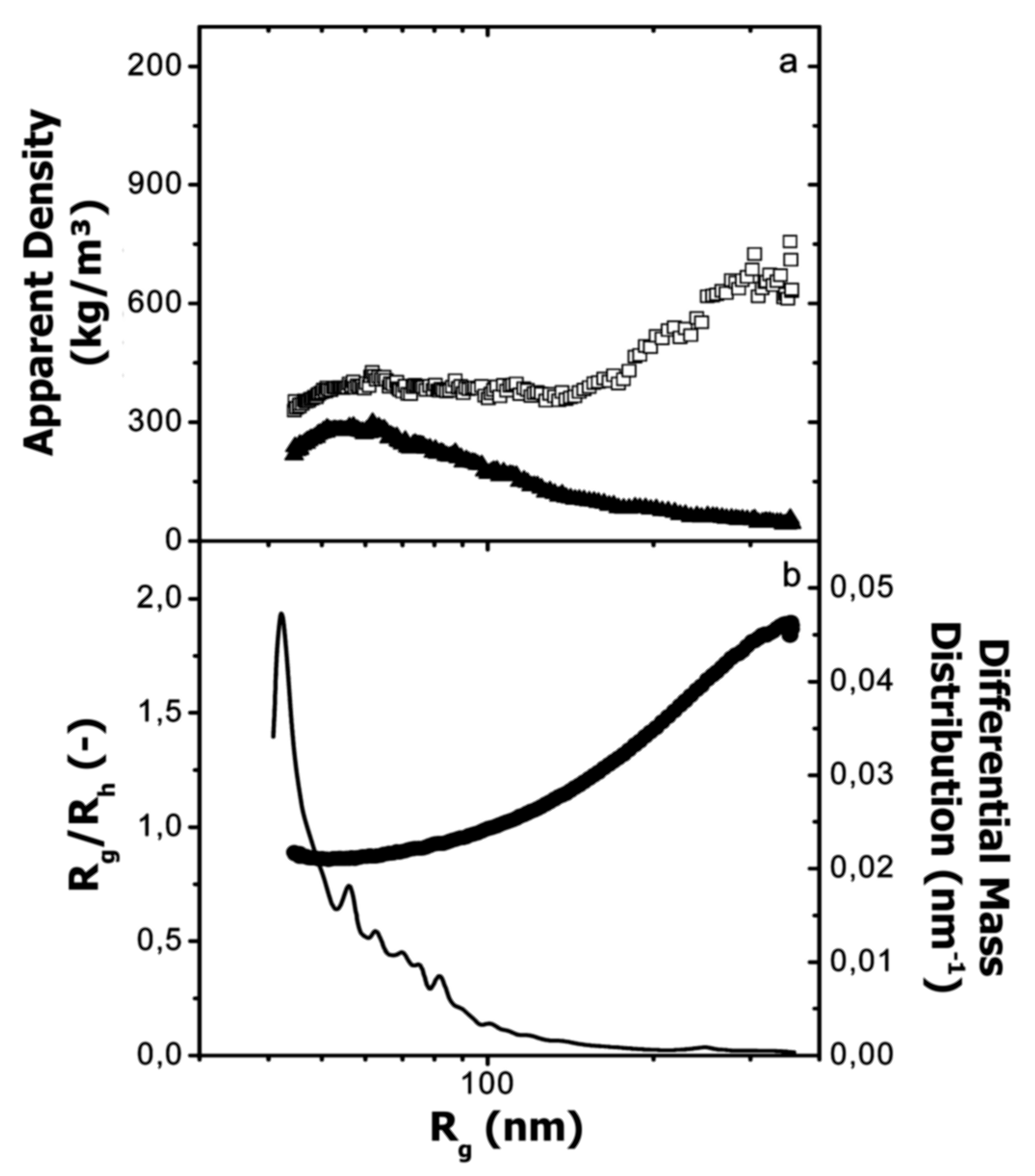
6. Absolute Molar Mass and Size Determination
6.1. Determination Principle and Requirements
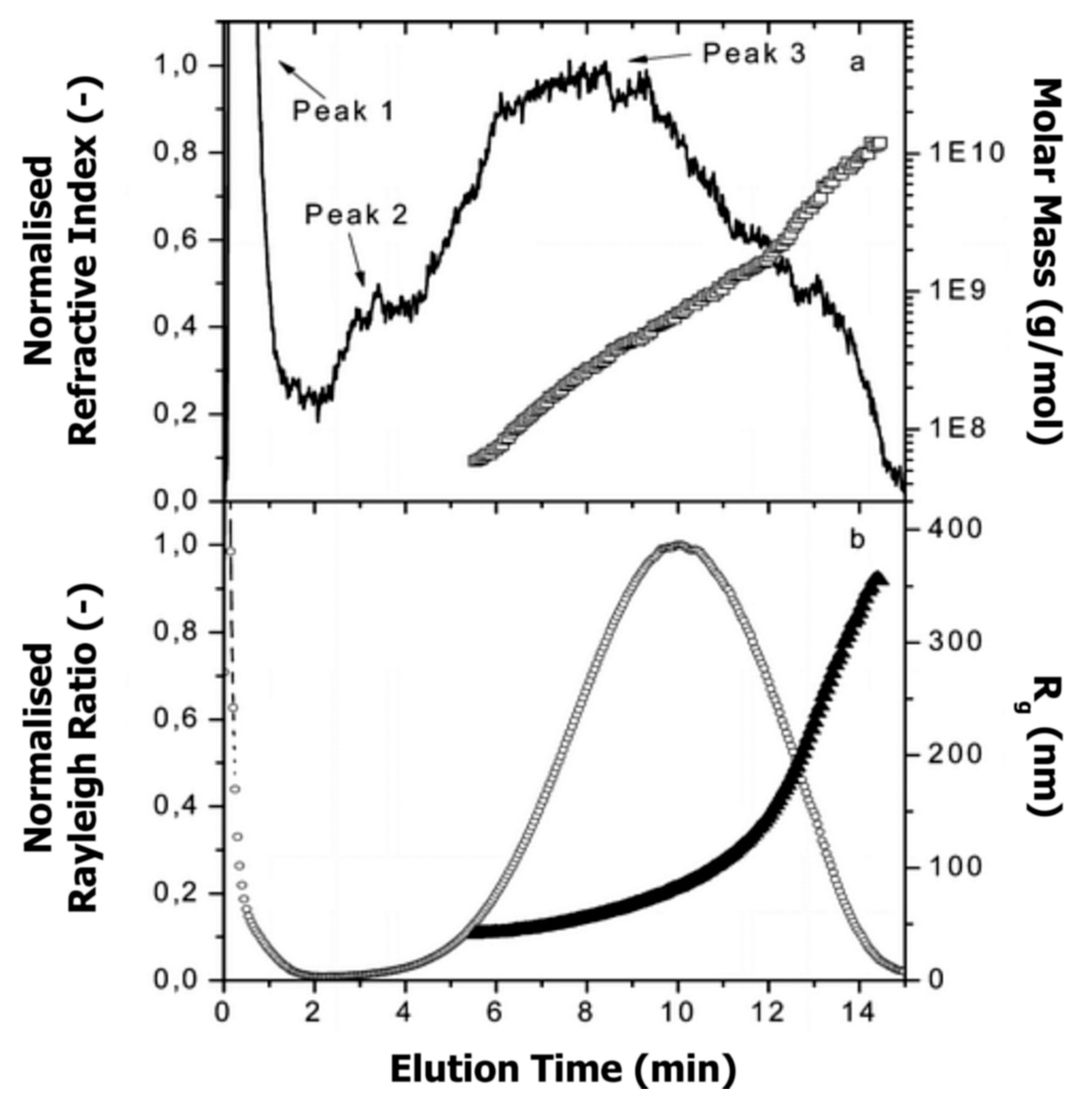
6.2. Literature Review of Studies on Casein
7. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Experimental Details
Appendix A.1. Casein Sample Preparation
Appendix A.2. Linear Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis
Appendix A.3. Gradient Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis
Appendix A.4. Size Exclusion Chromatography
References
- Farrell, H.M., Jr.; Jimenez-Flores, R.; Bleck, G.T.; Brown, E.M.; Butler, J.E.; Creamer, L.K.; Hicks, C.L.; Hollar, C.M.; Ng-Kwai-Hang, K.F.; Swaisgood, H.E. Nomenclature of the Proteins of Cows’ Milk—Sixth Revision. J. Dairy Sci. 2004, 87, 1641–1674. [Google Scholar] [CrossRef]
- O’Mahony, J.A.; Fox, P.F. Milk Proteins: Introduction and Historical Aspects. In Advanced Dairy Chemistry, 4th ed.; McSweeney, P.L.H., Fox, P.F., Eds.; Springer: Boston, MA, USA, 2013; pp. 43–85. [Google Scholar]
- Dalgleish, D.G. On the Structural Models of Bovine Casein Micelles—Review and Possible Improvements. Soft Matter 2011, 7, 2265–2272. [Google Scholar] [CrossRef]
- De Kruif, C.G.; Huppertz, T.; Urban, V.S.; Petukhov, A.V. Casein Micelles and Their Internal Structure. Adv. Colloid Interface Sci. 2012, 171–172, 36–52. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.J.; Oommen, B.S. Supramolecular Structure of the Casein Micelle. J. Dairy Sci. 2008, 91, 1709–1721. [Google Scholar] [CrossRef] [PubMed]
- De Kruif, C.G. Supra-Aggregates of Casein Micelles as a Prelude to Coagulation. J. Dairy Sci. 1998, 81, 3019–3028. [Google Scholar] [CrossRef]
- Horne, D.S. Casein Structure, Self-Assembly and Gelation. Curr. Opin. Colloid Interface Sci. 2002, 7, 456–461. [Google Scholar] [CrossRef]
- Horne, D.S. Casein Micelle Structure: Models and Muddles. Curr. Opin. Colloid Interface Sci. 2006, 11, 148–153. [Google Scholar] [CrossRef]
- Holt, C.; Carver, J.A.; Ecroyd, H.; Thorn, D.C. Caseins and the Casein Micelle: Their Biological Functions, Structures, and Behavior in Foods. J. Dairy Sci. 2013, 96, 6127–6146. [Google Scholar] [CrossRef] [PubMed]
- Thorn, D.C.; Ecroyd, H.; Carver, J.A.; Holt, C. Casein Structures in the Context of Unfolded Proteins. Int. Dairy J. 2015, 46, 2–11. [Google Scholar] [CrossRef]
- Carver, J.A.; Thorn, D.C.; Ecroyd, H.; Holt, C. Letter to the Editor: A Response to Horne and Lucey (2017). J. Dairy Sci. 2017, 100, 5121–5124. [Google Scholar] [CrossRef] [PubMed]
- Horne, D.S. A Balanced View of Casein Interactions. Curr. Opin. Colloid Interface Sci. 2017, 28, 74–86. [Google Scholar] [CrossRef]
- Horne, D.; Lucey, J.A. Letter to the Editor: Hydrophobic Interactions in the Caseins: Challenging Their Dismissal by Holt et al. (2013). J. Dairy Sci. 2017, 100, 5119–5120. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.J.; Du, H.; McManus, W.R.; Larsen, K.M. Microstructural Changes in Casein Supramolecules during Acidification of Skim Milk. J. Dairy Sci. 2009, 92, 5854–5867. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.J. Letter to the Editor: The Dynamic Casein Supramolecule: A Response to Horne (2010). J. Dairy Sci. 2010, 93, 3404. [Google Scholar] [CrossRef]
- Horne, D.S. Letter to the Editor: Casein Micelles or Casein Supramolecules? J. Dairy Sci. 2010, 93, 3403. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, T. Chemistry of the Caseins. In Advanced Dairy Chemistry, 4th ed.; McSweeney, P.L.H., Fox, P.F., Eds.; Springer: Boston, MA, USA, 2013; pp. 135–160. [Google Scholar]
- Lucey, J.A. Formation and Physical Properties of Milk Protein Gels. J. Dairy Sci. 2002, 85, 281–294. [Google Scholar] [CrossRef]
- Lucey, J.A.; Singh, H. Formation and Physical Properties of Acid Milk Gels: A Review. Food Res. Int. 1997, 30, 529–542. [Google Scholar] [CrossRef]
- O’Regan, J.; Mulvihill, D.M. Caseins and Caseinates, Industrial Production, Compositional Standards, Specifications, and Regulatory Aspects. In Encyclopedia of Dairy Science, 2nd ed.; Fuquay, J.W., Fox, P.F., McSweeney, P.L.H., Eds.; Academic Press: San Diego, CA, USA, 2011; Volume 3, pp. 855–863. [Google Scholar]
- Ando, H.; Adachi, M.; Umeda, K.; Matsuura, A.; Nonaka, M.; Uchio, R.; Tanaka, H.; Motoki, M. Purification and Characteristics of a Novel Transglutaminase Derived from Microorganisms. Agric. Biol. Chem. 1989, 53, 2613–2617. [Google Scholar]
- Martins, I.M.; Matos, M.; Costa, R.; Silva, F.; Pascoal, A.; Estevinho, L.M.; Choupina, A.B. Transglutaminases: Recent Achievements and New Sources. Appl. Microbiol. Biotechnol. 2014, 98, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Buchert, J.; Ercili Cura, D.; Ma, H.; Gasparetti, C.; Monogioudi, E.; Faccio, G.; Mattinen, M.; Boer, H.; Partanen, R.; Selinheimo, E.; et al. Crosslinking Food Proteins for Improved Functionality. Ann. Rev. Food Sci. Technol. 2010, 1, 113–138. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.L.C.; de Góes-Favoni, S.P. Action of Microbial Transglutaminase (MTGase) in the Modification of Food Proteins: A Review. Food Chem. 2015, 171, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Jaros, D.; Partschefeld, C.; Henle, T.; Rohm, H. Transglutaminase in Dairy Products: Chemistry, Physics, Applications. J. Texture Stud. 2006, 37, 113–155. [Google Scholar] [CrossRef]
- Özrenk, E. The Use of Transglutaminase in Dairy Products. Int. J. Dairy Technol. 2006, 59, 1–7. [Google Scholar] [CrossRef]
- Romeih, E.; Walker, G. Recent Advances on Microbial Transglutaminase and Dairy Application. Trends Food Sci. Technol. 2017, 62, 133–140. [Google Scholar] [CrossRef]
- Zeeb, B.; Fischer, L.; Weiss, J. Stabilization of Food Dispersions by Enzymes. Food Funct. 2014, 5, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Nemes, Z.; Petrovski, G.; Fésüs, L. Tools for the Detection and Quantitation of Protein Transglutamination. Anal. Biochem. 2005, 342, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jaros, D.; Schwarzenbolz, U.; Raak, N.; Löbner, J.; Henle, T.; Rohm, H. Cross-Linking with Microbial Transglutaminase: Relationship between Polymerisation Degree and Stiffness of Acid Casein Gels. Int. Dairy J. 2014, 38, 174–178. [Google Scholar] [CrossRef]
- Jaros, D.; Schwarzenbolz, U.; Raak, N.; Löbner, J.; Henle, T.; Rohm, H. Cross-Linking with Microbial Transglutaminase: Relationship between Polymerisation Degree and Stiffness of Acid Casein Gels. Int. Dairy J. 2014, 38, 174–178, reprinted in Int. Dairy J. 2014, 39, 345–347. [Google Scholar] [CrossRef]
- Raak, N.; Rohm, H.; Jaros, D. Cross-Linking with Microbial Transglutaminase: Isopeptide Bonds and Polymer Size as Drivers for Acid Casein Gel Stiffness. Int. Dairy J. 2017, 66, 49–55. [Google Scholar] [CrossRef]
- Monogioudi, E.; Permi, P.; Filpponen, I.; Lienemann, M.; Li, B.; Argyropoulos, D.; Buchert, J.; Mattinen, M.-L. Protein Analysis by 31P NMR Spectroscopy in Ionic Liquid: Quantitative Determination of Enzymatically Created Cross-Links. J. Agric. Food Chem. 2011, 59, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Mokoonlall, A.; Pfannstiel, J.; Struch, M.; Berger, R.G.; Hinrichs, J. Structure Modification of Stirred Fermented Milk Gel Due to Laccase-Catalysed Protein Crosslinking in a Post-Processing Step. Inn. Food Sci. Emerg. Technol. 2016, 33, 563–570. [Google Scholar] [CrossRef]
- Mokoonlall, A.; Sykora, L.; Pfannstiel, J.; Nöbel, S.; Weiss, J.; Hinrichs, J. A Feasibility Study on the Application of a Laccase-Mediator System in Stirred Yoghurt at the Pilot Scale. Food Hydrocoll. 2016, 60, 119–127. [Google Scholar] [CrossRef]
- Mokoonlall, A.; Hippich, M.; Struch, M.; Berger, R.G.; Weiss, J.; Hinrichs, J. Antioxidant Activity of Milk Suppresses Laccase Induced Radicals and the Subsequent Modification of Acidified Milk Protein Gels. Int. Dairy J. 2016, 60, 24–31. [Google Scholar] [CrossRef]
- Maillard, L.C. Action des Acides Aminés sur les Sucres; Formation des Mélanoïdines par voie Méthodique (in French). C. R. Acad. Sci. 1912, 154, 66–68. [Google Scholar]
- De Oliveira, F.C.; dos Reis Coimbra, J.S.; de Oliveira, E.B.; Zuñiga, A.D.G.; Rojas, E.E.G. Food Protein-Polysaccharide Conjugates Obtained via the Maillard Reaction: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1108–1125. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ru, Q.; Ding, Y. Glycation a Promising Method for Food Protein Modification: Physicochemical Properties and Structure, A Review. Food Res. Int. 2012, 49, 170–183. [Google Scholar] [CrossRef]
- Lund, M.N.; Ray, C.A. Control of Maillard Reactions in Foods: Strategies and Chemical Mechanisms. J. Agric. Food Chem. 2017, 65, 4537–4552. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, J.A.; Drapala, K.P.; Mulcahy, E.M.; Mulvihill, D.M. Controlled Glycation of Milk Proteins and Peptides: Functional Properties. Int. Dairy J. 2017, 67, 16–34. [Google Scholar] [CrossRef]
- Gerrard, J.A. Protein–Protein Crosslinking in Food: Methods, Consequences, Applications. Trends Food Sci. Technol. 2002, 13, 391–399. [Google Scholar] [CrossRef]
- Pellegrino, L.; van Boekel, M.A.J.S.; Gruppen, H.; Resmini, P.; Pagani, M.A. Heat-Induced Aggregation and Covalent Linkages in β-Casein Model Systems. Int. Dairy J. 1999, 9, 255–260. [Google Scholar] [CrossRef]
- Lauber, S.; Klostermeyer, H.; Henle, T. On the Influence of Non-Enzymatic Crosslinking of Caseins on the Gel Strength of Yoghurt. Nahrung/Food 2001, 45, 215–217. [Google Scholar] [CrossRef]
- Schwarzenbolz, U.; Henle, T. Non-Enzymatic Modifications of Proteins under High-Pressure Treatment. High Press. Res. 2010, 30, 458–465. [Google Scholar] [CrossRef]
- Al-Saadi, J.M.S.; Easa, A.M.; Deeth, H.C. Effect of Lactose on Cross-Linking of Milk Proteins during Heat Treatments. Int. J. Dairy Technol. 2013, 66, 1–6. [Google Scholar] [CrossRef]
- Bulca, S.; Dumpler, J.; Kulozik, U. Kinetic Description of Heat-Induced Cross-Linking Reactions of Whey Protein-Free Casein Solutions. Int. J. Dairy Technol. 2016, 69, 489–496. [Google Scholar] [CrossRef]
- Moeckel, U.; Duerasch, A.; Weiz, A.; Ruck, M.; Henle, T. Glycation Reactions of Casein Micelles. J. Agric. Food Chem. 2016, 64, 2953–2961. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Chemistry, Biochemistry, Nutrition, and Microbiology of Lysinoalanine, Lanthionine, and Histidinoalanine in Food and Other Proteins. J. Agric. Food Chem. 1999, 47, 1295–1319. [Google Scholar] [CrossRef] [PubMed]
- Singh, H. Modification of Food Proteins by Covalent Crosslinking. Trends Food Sci. Technol. 1991, 2, 196–200. [Google Scholar] [CrossRef]
- Famelart, M.-H.; Le, N.H.T.; Croguennec, T.; Rousseau, F. Are Disulphide Bonds Formed during Acid Gelation of Preheated Milk? Int. J. Food Sci. Technol. 2013, 48, 1940–1948. [Google Scholar] [CrossRef]
- Koch, L.; Hummel, L.; Schuchmann, H.P.; Emin, M.A. Influence of Defined Shear Rates on Structural Changes and Functional Properties of Highly Concentrated Whey Protein Isolate-Citrus Pectin Blends at Elevated Temperatures. Food Biophys. 2017, 12, 309–322. [Google Scholar] [CrossRef]
- Lauber, S.; Henle, T.; Klostermeyer, H. Relationship between the Crosslinking of Caseins by Transglutaminase and the Gel Strength of Yoghurt. Eur. Food Res. Technol. 2000, 210, 305–309. [Google Scholar] [CrossRef]
- Menéndez, O.; Schwarzenbolz, U.; Rohm, H.; Henle, T. Casein Gelation under Simultaneous Action of Transglutaminase and Glucono-δ-Lactone. Nahrung/Food 2004, 48, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Casanova, F.; Nogueira Silva, N.F.; Gaucheron, F.; Nogueira, M.H.; Teixeira, A.V.N.C.; Perrone, I.T.; Alves, M.P.; Fidelis, P.C.; de Carvalho, A.F. Stability of Casein Micelles Cross-Linked with Genipin: A Physicochemical Study as a Function of pH. Int. Dairy J. 2017, 68, 70–74. [Google Scholar] [CrossRef]
- Chou, W.-L.; Lin, L.T.-W.; Shih, Y.-Y.; Li, C.-T.; Kao, C.-Y.; Tsai, W.-B.; Wang, S.S.-S. Aggregation Behavior of Casein is Correlated With the Type of Glycation-Inducing Agent. J. Taiwan Inst. Chem. Eng. 2014, 45, 393–403. [Google Scholar] [CrossRef]
- Ghosh, A.; Ali, M.A.; Dias, G.J. Effect of Cross-Linking on Microstructure and Physical Performance of Casein Protein. Biomacromolecules 2009, 10, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Holland, J.W.; Bhandari, B.; Alewood, P.F.; Deeth, H.C. Direct Evidence for the Role of Maillard Reaction Products in Protein Cross-Linking in Milk Powder during Storage. Int. Dairy J. 2013, 31, 83–91. [Google Scholar] [CrossRef]
- Nogueira Silva, N.F.; Saint-Jalmes, A.; de Carvalho, A.F.; Gaucheron, F. Development of Casein Microgels from Cross-Linking of Casein Micelles by Genipin. Langmuir 2014, 30, 10167–10175. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.J.S.M.; Sousa, F.; Gübitz, G.; Cavaco-Paulo, A. Chemical Modifications on Proteins Using Glutaraldehyde. Food Technol. Biotechnol. 2004, 42, 51–56. [Google Scholar]
- Westermeier, R. Electrophoresis in Gels. In Protein Purification: Principles, High Resolution Methods, and Applications, 3rd ed.; Janson, J.-C., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 365–377. [Google Scholar]
- O’Mahony, J.A.; Ardö, Y.; McSweeney, P.L.H. Analysis, Electrophoresis. In Encyclopedia of Dairy Sciences; Roginski, H., Fuquay, J.W., Fox, P.F., Eds.; Academic Press: New York, 2002; pp. 67–74. [Google Scholar]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Basak, S. Effect of Surfactants on Casein Structure: A Spectroscopic Study. Colloids Surf. B Biointerfaces 2008, 63, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Bjellqvist, B.; Ek, K.; Giorgio Righetti, P.; Gianazza, E.; Görg, A.; Westermeier, R.; Postel, W. Isoelectric Focusing in Immobilized pH Gradients: Principle, Methodology and Some Applications. J. Biochem. Biophys. Meth. 1982, 6, 317–339. [Google Scholar] [CrossRef]
- Righetti, P.G.; Fasoli, E.; Righetti, S.C. Conventional Isoelectric Focusing in Gel Slabs and Capillaries and Immobilized pH Gradients. In Protein Purification: Principles, High Resolution Methods, and Applications; Janson, J.-C., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 365–377. [Google Scholar]
- Westermeier, R.; Görg, A. Two-Dimensional Electrophoresis in Proteomics. In Protein Purification: Principles, High Resolution Methods, and Applications, 3rd ed.; Janson, J.-C., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 411–439. [Google Scholar]
- O’Farrell, P.H. High Resolution Two-Dimensional Electrophoresis of Proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- O’Farrell, P.Z.; Goodman, H.M.; O’Farrell, P.H. High Resolution Two-Dimensional Electrophoresis of Basic as well as Acidic Proteins. Cell 1977, 12, 1133–1142. [Google Scholar] [CrossRef]
- Blomberg, A.; Blomberg, L.; Norbeck, J.; Fey, S.J.; Larsen, P.M.; Larsen, M.; Roepstorff, P.; Degand, H.; Boutry, M.; Posch, A.; et al. Interlaboratory Reproducibility of Yeast Protein Patterns Analyzed by Immobilized pH Gradient Two-Dimensional Gel Electrophoresis. Electrophoresis 1995, 16, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The Current State of Two-Dimensional Electrophoresis with Immobilized pH Gradients. Electrophoresis 2000, 21, 1037–1053. [Google Scholar] [CrossRef]
- O’Donnell, R.; Holland, J.W.; Deeth, H.C.; Alewood, P. Milk Proteomics. Int. Dairy J. 2004, 14, 1013–1023. [Google Scholar] [CrossRef]
- Abd El-Salam, M.H. Application of Proteomics to the Areas of Milk Production, Processing and Quality Control—A Review. Int. J. Dairy Technol. 2014, 67, 153–166. [Google Scholar] [CrossRef]
- Le, T.T.; Deeth, H.C.; Larsen, L.B. Proteomics of Major Bovine Milk Proteins: Novel Insights. Int. Dairy J. 2017, 67, 2–15. [Google Scholar] [CrossRef]
- O’Sullivan, M.M.; Kelly, A.L.; Fox, P.F. Influence of Transglutaminase Treatment on Some Physico-Chemical Properties of Milk. J. Dairy Res. 2002, 69, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.; Gunning, Y.; FitzGerald, R.J. Effect of Cross-Linking with Transglutaminase on the Heat Stability and Some Functional Characteristics of Sodium Caseinate. Food Res. Int. 2003, 36, 267–274. [Google Scholar] [CrossRef]
- Ercili Cura, D.; Lille, M.; Partanen, R.; Kruus, K.; Buchert, J.; Lantto, R. Effect of Trichoderma reesei Tyrosinase on Rheology and Microstructure of Acidified Milk Gels. Int. Dairy J. 2010, 20, 830–837. [Google Scholar] [CrossRef]
- Jaros, D.; Jacob, M.; Otto, C.; Rohm, H. Excessive Cross-Linking of Caseins by Microbial Transglutaminase and Its Impact on Physical Properties of Acidified Milk Gels. Int. Dairy J. 2010, 20, 321–327. [Google Scholar] [CrossRef]
- Creamer, L.K.; Richardson, T. Anomalous Behavior of Bovine αs1- and β-Caseins on Gel Electrophoresis in Sodium Dodecyl Sulfate Buffers. Arch. Biochem. Biophys. 1984, 234, 476–486. [Google Scholar] [CrossRef]
- Macierzanka, A.; Bordron, F.; Rigby, N.M.; Mills, E.N.C.; Lille, M.; Poutanen, K.; Mackie, A.R. Transglutaminase Cross-Linking Kinetics of Sodium Caseinate is Changed After Emulsification. Food Hydrocoll. 2011, 25, 843–850. [Google Scholar] [CrossRef]
- Raak, N.; Rohm, H.; Jaros, D. Enzymatic Cross-Linking of Casein Facilitates Gel Structure Weakening Induced by Overacidification. Food Biophys. 2017, 12, 261–268. [Google Scholar] [CrossRef]
- Liu, M.; Damodaran, S. Effect of Transglutaminase-Catalyzed Polymerization of β-Casein on Its Emulsifying Properties. J. Agric. Food Chem. 1999, 47, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Lauber, S.; Noack, I.; Klostermeyer, H.; Henle, T. Oligomerization of β-Lactoglobulin by Microbial Transglutaminase during High Pressure Treatment. Eur. Food Res. Technol. 2001, 213, 246–247. [Google Scholar] [CrossRef]
- Truong, V.-D.; Clare, D.A.; Catignani, G.L.; Swaisgood, H.E. Cross-Linking and Rheological Changes of Whey Proteins Treated with Microbial Transglutaminase. J. Agric. Food Chem. 2004, 52, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, C.L.; Andersen, M.L.; Degn, P.E.; Nielsen, J.H. Cross-Linking Proteins by Laccase-Catalyzed Oxidation: Importance Relative to Other Modifications. J. Agric. Food Chem. 2008, 56, 12002–12010. [Google Scholar] [CrossRef] [PubMed]
- Heijnis, W.H.; Wierenga, P.A.; van Berkel, W.J.H.; Gruppen, H. Directing the Oligomer Size Distribution of Peroxidase-Mediated Cross-Linked Bovine α-Lactalbumin. J. Agric. Food Chem. 2010, 58, 5692–5697. [Google Scholar] [CrossRef] [PubMed]
- Ercili-Cura, D.; Partanen, R.; Husband, F.; Ridout, M.; Macierzanka, A.; Lille, M.; Boer, H.; Lantto, R.; Buchert, J.; Mackie, A.R. Enzymatic Cross-Linking of β-Lactoglobulin in Solution and at Air–Water Interface: Structural Constraints. Food Hydrocoll. 2012, 28, 1–9. [Google Scholar] [CrossRef]
- Lauber, S.; Krause, I.; Klostermeyer, H.; Henle, T. Microbial Transglutaminase Crosslinks β-Casein and β-Lactoglobulin to Heterologous Oligomers under High Pressure. Eur. Food Res. Technol. 2003, 216, 15–17. [Google Scholar] [CrossRef]
- Tang, C.; Yang, X.-Q.; Chen, Z.; Wu, H.; Peng, Z.-Y. Physicochemical and Structural Characteristics of Sodium Caseinate Biopolymers Induced by Microbial Transglutaminase. J. Food Biochem. 2005, 29, 402–421. [Google Scholar] [CrossRef]
- Ozer, B.; Avni Kirmaci, H.; Oztekin, S.; Hayaloglu, A.; Atamer, M. Incorporation of Microbial Transglutaminase into Non-Fat Yogurt Production. Int. Dairy J. 2007, 17, 199–207. [Google Scholar] [CrossRef]
- Monogioudi, E.; Creusot, N.; Kruus, K.; Gruppen, H.; Buchert, J.; Mattinen, M.-L. Cross-Linking of β-Casein by Trichoderma reesei Tyrosinase and Streptoverticillium mobaraense Transglutaminase Followed by SEC–MALLS. Food Hydrocoll. 2009, 23, 2008–2015. [Google Scholar] [CrossRef]
- Smiddy, M.A.; Martin, J.-E.G.H.; Kelly, A.L.; de Kruif, C.G.; Huppertz, T. Stability of Casein Micelles Cross-Linked by Transglutaminase. J. Dairy Sci. 2006, 89, 1906–1914. [Google Scholar] [CrossRef]
- Moon, J.-H.; Hong, Y.-H.; Huppertz, T.; Fox, P.F.; Kelly, A.L. Properties of Casein Micelles Cross-Linked by Transglutaminase. Int. J. Dairy Technol. 2009, 62, 27–32. [Google Scholar] [CrossRef]
- Raak, N. Aggregatgröße und Polymerisierungsgrad Enzymatisch Verknüpfter Caseine in Säureinduzierten Gelen (in German). Diploma Thesis, Technische Universität Dresden, Dresden, Germany, 30 November 2013. [Google Scholar]
- Ikura, K.; Kometani, T.; Yoshikawa, M.; Sasaki, R.; Chiba, H. Crosslinking of Casein Components by Transglutaminase. Agric. Biol. Chem. 1980, 44, 1567–1573. [Google Scholar]
- De Jong, G.A.H.; Wijngaards, G.; Koppelman, S.J. Transglutaminase Inhibitor from Milk. J. Food Sci. 2003, 68, 820–825. [Google Scholar] [CrossRef]
- Aaltonen, T.; Huumonen, I.; Myllärinen, P. Controlled Transglutaminase Treatment in Edam Cheese-Making. Int. Dairy J. 2014, 38, 179–182. [Google Scholar] [CrossRef]
- Struch, M.; Krahe, N.-K.; Linke, D.; Mokoonlall, A.; Hinrichs, J.; Berger, R.G. Dose Dependent Effects of a Milk Ion Tolerant Laccase on Yoghurt Gel Structure. LWT-Food Sci. Technol. 2016, 65, 1144–1152. [Google Scholar] [CrossRef]
- Struch, M.; Linke, D.; Mokoonlall, A.; Hinrichs, J.; Berger, R.G. Laccase-Catalysed Cross-Linking of a Yoghurt-Like Model System Made from Skimmed Milk With Added Food-Grade Mediators. Int. Dairy J. 2015, 49, 89–94. [Google Scholar] [CrossRef]
- Ercili Cura, D.; Lantto, R.; Lille, M.; Andberg, M.; Kruus, K.; Buchert, J. Laccase-Aided Protein Modification: Effects on the Structural Properties of Acidified Sodium Caseinate Gels. Int. Dairy J. 2009, 19, 737–745. [Google Scholar] [CrossRef]
- Dinnella, C.; Gargaro, M.T.; Rossano, R.; Monteleone, E. Spectrophotometric Assay Using o-Phtaldialdehyde for the Determination of Transglutaminase Activity on Casein. Food Chem. 2002, 78, 363–368. [Google Scholar] [CrossRef]
- Broyard, C.; Gaucheron, F. Modifications of Structures and Functions of Caseins: A Scientific and Technological Challenge. Dairy Sci. Technol. 2015, 95, 831–862. [Google Scholar] [CrossRef]
- Sharma, R.; Lorenzen, P.C.; Qvist, K.B. Influence of Transglutaminase Treatment of Skim Milk on the Formation of ε-(γ-Glutamyl)Lysine and the Susceptibility of Individual Proteins Towards Crosslinking. Int. Dairy J. 2001, 11, 785–793. [Google Scholar] [CrossRef]
- Hinz, K.; Huppertz, T.; Kulozik, U.; Kelly, A.L. Influence of Enzymatic Cross-Linking on Milk Fat Globules and Emulsifying Properties of Milk Proteins. Int. Dairy J. 2007, 17, 289–293. [Google Scholar] [CrossRef]
- Huppertz, T.; de Kruif, C.G. Ethanol Stability of Casein Micelles Cross-Linked with Transglutaminase. Int. Dairy J. 2007, 17, 436–441. [Google Scholar] [CrossRef]
- Huppertz, T.; de Kruif, C.G. Rennet-Induced Coagulation of Enzymatically Cross-Linked Casein Micelles. Int. Dairy J. 2007, 17, 442–447. [Google Scholar] [CrossRef]
- Ardelean, A.I.; Otto, C.; Jaros, D.; Rohm, H. Transglutaminase Treatment to Improve Physical Properties of Acid Gels from Enriched Goat Milk. Small Ruminant Res. 2012, 106, 47–53. [Google Scholar] [CrossRef]
- Ardelean, A.I.; Jaros, D.; Rohm, H. Influence of Microbial Transglutaminase Cross-Linking on Gelation Kinetics and Texture of Acid Gels Made from Whole Goats and Cows Milk. Dairy Sci. Technol. 2013, 93, 63–71. [Google Scholar] [CrossRef]
- Rodriguez-Nogales, J.M. Enzymatic Cross-Linking of Ewe’s Milk Proteins by Transglutaminase. Eur. Food Res. Technol. 2005, 221, 692–699. [Google Scholar] [CrossRef]
- Rodriguez-Nogales, J.M. Effect of Preheat Treatment on the Transglutaminase-Catalyzed Cross-Linking of Goat Milk Proteins. Process Biochem. 2006, 41, 430–437. [Google Scholar] [CrossRef]
- Rodriguez-Nogales, J.M. Enhancement of Transglutaminase-Induced Protein Cross-Linking by Preheat Treatment of Cows’ Milk: A Statistical Approach. Int. Dairy J. 2006, 16, 26–32. [Google Scholar] [CrossRef]
- Feng, P.; Fuerer, C.; McMahon, A. Quantification of Whey Protein Content in Infant Formulas by Sodium Dodecyl Sulfate-Capillary Gel Electrophoresis (SDS-CGE): Single-Laboratory Validation, First Action 2016.15. J. AOAC Int. 2017, 100, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Lilla, S.; Mamone, G.; Nicolai, M.A.; Chianese, L.; Picariello, G.; Caira, S.; Addeo, F. Structural Characterization of Transglutaminase-Catalyzed Casein Cross-Linking. J. Chromatogr. Sep. Tech. 2012, 3, 1000122. [Google Scholar] [CrossRef]
- Zacharius, R.M.; Zell, T.E.; Morrison, J.H.; Woodlock, J.J. Glycoprotein Staining Following Electrophoresis on Acrylamide Gels. Anal. Biochem. 1969, 30, 148–152. [Google Scholar] [CrossRef]
- Selinheimo, E.; Lampila, P.; Mattinen, M.-L.; Buchert, J. Formation of Protein−Oligosaccharide Conjugates by Laccase and Tyrosinase. J. Agric. Food Chem. 2008, 56, 3118–3128. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, J.; Mulvihill, D.M. Preparation, Characterisation and Selected Functional Properties of Sodium Caseinate–Maltodextrin Conjugates. Food Chem. 2009, 115, 1257–1267. [Google Scholar] [CrossRef]
- Song, C.-L.; Zhao, X.-H. Rheological, Gelling and Emulsifying Properties of a Glycosylated and Cross-Linked Caseinate Generated by Transglutaminase. Int. J. Food Sci. Technol. 2013, 48, 2595–2602. [Google Scholar] [CrossRef]
- Zhang, S.; Gong, Y.; Khanal, S.; Lu, Y.; Lucey, J.A. Properties of Acid Gels Made From Sodium Caseinate-Maltodextrin Conjugates Prepared by a Wet Heating Method. J. Dairy Sci. 2017, 100, 8744–8753. [Google Scholar] [CrossRef] [PubMed]
- Partanen, R.; Paananen, A.; Forssell, P.; Linder, M.B.; Lille, M.; Buchert, J.; Lantto, R. Effect of Transglutaminase-Induced Cross-Linking of Sodium Caseinate on the Properties of Equilibrated Interfaces and Foams. Colloids Surf. A Physicochem. Eng. Asp. 2009, 344, 79–85. [Google Scholar] [CrossRef]
- Partanen, R.; Forssell, P.; Mackie, A.; Blomberg, E. Interfacial Cross-Linking of β-Casein Changes the Structure of the Adsorbed Layer. Food Hydrocoll. 2013, 32, 271–277. [Google Scholar] [CrossRef]
- Myllärinen, P.; Buchert, J.; Autio, K. Effect of Transglutaminase on Rheological Properties and Microstructure of Chemically Acidified Sodium Caseinate Gels. Int. Dairy J. 2007, 17, 800–807. [Google Scholar] [CrossRef]
- Partanen, R.; Autio, K.; Myllärinen, P.; Lille, M.; Buchert, J.; Forssell, P. Effect of Transglutaminase on Structure and Syneresis of Neutral and Acidic Sodium Caseinate Gels. Int. Dairy J. 2008, 18, 414–421. [Google Scholar] [CrossRef]
- Chevalier, F.; Hirtz, C.; Sommerer, N.; Kelly, A.L. Use of Reducing/Nonreducing Two-Dimensional Electrophoresis for the Study of Disulfide-Mediated Interactions between Proteins in Raw and Heated Bovine Milk. J. Agric. Food Chem. 2009, 57, 5948–5955. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.W.; Gupta, R.; Deeth, H.C.; Alewood, P.F. Proteomic Analysis of Temperature-Dependent Changes in Stored UHT Milk. J. Agric. Food Chem. 2011, 59, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Bhandari, B.; Holland, J.W.; Deeth, H.C. Maillard Reaction and Protein Cross-Linking in Relation to the Solubility of Milk Powders. J. Agric. Food Chem. 2011, 59, 12473–12479. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.F.; Pan, P.H. Proteomic Profiling of Microbial Transglutaminase-Induced Polymerization of Milk Proteins. J. Dairy Sci. 2012, 95, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Hsieh, J.-F. Microwave-Assisted Cross-Linking of Milk Proteins Induced by Microbial Transglutaminase. Sci. Rep. 2016, 6, 39040. [Google Scholar] [CrossRef] [PubMed]
- Hagel, L. Gel Filtration: Size Exclusion Chromatography. In Protein Purification: Principles, High Resolution Methods, and Applications, 3rd ed.; Janson, J.-C., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 51–91. [Google Scholar]
- Striegel, A.M.; Yau, W.W.; Kirkland, J.J.; Bly, D.D. Modern Size-Exclusion Liquid Chromatography: Practice of Gel Permeation and Gel Filtration Chromatography, 2nd ed.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Pasch, H.; Trathnigg, B. Multidimensional HPLC of Polymers; Springer: Berlin, Germany, 2013. [Google Scholar]
- Hiller, B.; Lorenzen, P.-C. Effect of Buffer Systems on the Extent of Enzymatic Oligomerisation of Milk Proteins. LWT-Food Sci. Technol. 2008, 41, 1140–1144. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Oudgenoeg, G.; Spekking, W.T.J.; Berendsen, L.B.J.M.; Vancon, L.; Boumans, H.; Gruppen, H.; van Berkel, W.J.H.; Laane, C.; Voragen, A.G.J. Horseradish Peroxidase-Catalyzed Cross-Linking of Feruloylated Arabinoxylans with β-Casein. J. Agric. Food Chem. 2004, 52, 6633–6639. [Google Scholar] [CrossRef] [PubMed]
- Corzo-Martínez, M.; Moreno, F.J.; Villamiel, M.; Harte, F.M. Characterization and Improvement of Rheological Properties of Sodium Caseinate Glycated with Galactose, Lactose and Dextran. Food Hydrocoll. 2010, 24, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Lauber, S.; Noack, I.; Klostermeyer, H.; Henle, T. Stability of Microbial Transglutaminase to High Pressure Treatment. Eur. Food Res. Technol. 2001, 213, 273–276. [Google Scholar] [CrossRef]
- Bönisch, M.P.; Lauber, S.; Kulozik, U. Effect of Ultra-High Temperature Treatment on the Enzymatic Cross-linking of Micellar Casein and Sodium Caseinate by Transglutaminase. J. Food Sci. 2004, 69, E398–E404. [Google Scholar] [CrossRef]
- Bönisch, M.P.; Tolkach, A.; Kulozik, U. Inactivation of an Indigenous Transglutaminase Inhibitor in Milk Serum by Means of UHT-Treatment and Membrane Separation Techniques. Int. Dairy J. 2006, 16, 669–678. [Google Scholar] [CrossRef]
- Bönisch, M.P.; Lauber, S.; Kulozik, U. Improvement of Enzymatic Cross-Linking of Casein Micelles with Transglutaminase by Glutathione Addition. Int. Dairy J. 2007, 17, 3–11. [Google Scholar] [CrossRef]
- Bönisch, M.P.; Huss, M.; Lauber, S.; Kulozik, U. Yoghurt Gel Formation by Means of Enzymatic Protein Cross-Linking during Microbial Fermentation. Food Hydrocoll. 2007, 21, 585–595. [Google Scholar] [CrossRef]
- Bönisch, M.P.; Huss, M.; Weitl, K.; Kulozik, U. Transglutaminase Cross-Linking of Milk Proteins and Impact on Yoghurt Gel Properties. Int. Dairy J. 2007, 17, 1360–1371. [Google Scholar] [CrossRef]
- Menéndez, O.; Schwarzenbolz, U.; Partschefeld, C.; Henle, T. Affinity of Microbial Transglutaminase to αs1-, β-, and Acid Casein under Atmospheric and High Pressure Conditions. J. Agric. Food Chem. 2009, 57, 4177–4184. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.; Lorenzen, P.C. Effect of Phosphatase/Transglutaminase Treatment on Molar Mass Distribution and Techno-Functional Properties of Sodium Caseinate. LWT-Food Sci. Technol. 2009, 42, 87–92. [Google Scholar] [CrossRef]
- Hiller, B.; Lorenzen, P.C. Functional Properties of Milk Proteins as Affected by Enzymatic Oligomerisation. Food Res. Int. 2009, 42, 899–908. [Google Scholar] [CrossRef]
- Partschefeld, C.; Schreiner, J.; Schwarzenbolz, U.; Henle, T. Studies on Enzymatic Crosslinking of Casein Micelles. Czech J. Food Sci. 2009, 27, 99–101. [Google Scholar] [CrossRef]
- Guyot, C.; Kulozik, U. Effect of Transglutaminase-Treated Milk Powders on the Properties of Skim Milk Yoghurt. Int. Dairy J. 2011, 21, 628–635. [Google Scholar] [CrossRef]
- Özer, B.; Guyot, C.; Kulozik, U. Simultaneous Use of Transglutaminase and Rennet in Milk Coagulation: Effect of Initial Milk pH and Renneting Temperature. Int. Dairy J. 2012, 24, 1–7. [Google Scholar] [CrossRef]
- Rohm, H.; Ullrich, F.; Schmidt, C.; Löbner, J.; Jaros, D. Gelation of Cross-Linked Casein under Small and Large Shear Strain. J. Texture Stud. 2014, 45, 130–137. [Google Scholar] [CrossRef]
- Kizzie-Hayford, N.; Jaros, D.; Rohm, H. Enrichment of Tiger Nut Milk with Microbial Transglutaminase Cross-Linked Protein Improves the Physico-Chemical Properties of the Fermented System. LWT-Food Sci. Technol. 2017, 81, 226–232. [Google Scholar] [CrossRef]
- Raak, N.; Schöne, C.; Rohm, H.; Jaros, D. Acid-Induced Gelation of Enzymatically Cross-Linked Caseinate in Different Ionic Milieus. Food Hydrocoll. 2018. [Google Scholar] [CrossRef]
- Lucey, J.A.; Srinivasan, M.; Singh, H.; Munro, P.A. Characterization of Commercial and Experimental Sodium Caseinates by Multiangle Laser Light Scattering and Size-Exclusion Chromatography. J. Agric. Food Chem. 2000, 48, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Siegl, T. Studien zur Quervernetzung von Milchproteinen und Zur Bildung Individueller Crosslink-Aminosäuren. Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 16 March 2003. (In German). [Google Scholar]
- Johnson, K.A.A. Century of Enzyme Kinetic Analysis, 1913 to 2013. FEBS Lett. 2013, 587, 2753–2766. [Google Scholar] [CrossRef] [PubMed]
- Holt, C. Casein Micelle Substructure and Calcium Phosphate Interactions Studied by Sephacryl Column Chromatography. J. Dairy Sci. 1998, 81, 2994–3003. [Google Scholar] [CrossRef]
- Monogioudi, E.; Faccio, G.; Lille, M.; Poutanen, K.; Buchert, J.; Mattinen, M.-L. Effect of Enzymatic Cross-Linking of β-Casein on Proteolysis by Pepsin. Food Hydrocoll. 2011, 25, 71–81. [Google Scholar] [CrossRef]
- Hellwig, M.; Löbner, J.; Schneider, A.; Schwarzenbolz, U.; Henle, T. Release of Protein-Bound N-ε-(γ-glutamyl)-Lysine during Simulated Gastrointestinal Digestion. Czech J. Food Sci. 2009, 27, 153–155. [Google Scholar] [CrossRef]
- Hellwig, M.; Matthes, R.; Peto, A.; Löbner, J.; Henle, T. N-ε-Fructosyllysine and N-ε-Carboxymethyllysine, but not Lysinoalanine, are Available for Absorption after Simulated Gastrointestinal Digestion. Amino Acids 2014, 46, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Giddings, J.; Yang, F.; Myers, M. Flow-Field-Flow Fractionation: A Versatile New Separation Method. Science 1976, 193, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Messaud, F.A.; Sanderson, R.D.; Runyon, J.R.; Otte, T.; Pasch, H.; Williams, S.K.R. An Overview on Field-Flow Fractionation Techniques and Their Applications in the Separation and Characterization of Polymers. Prog. Polym. Sci. 2009, 34, 351–368. [Google Scholar] [CrossRef]
- Malik, M.I.; Pasch, H. Field-Flow Fractionation: New and Exciting Perspectives in Polymer Analysis. Prog. Polym. Sci. 2016, 63, 42–85. [Google Scholar] [CrossRef]
- Ratanathanawongs Williams, S.K.; Lee, D. Field-Flow Fractionation of Proteins, Polysaccharides, Synthetic Polymers, and Supramolecular Assemblies. J. Sep. Sci. 2006, 29, 1720–1732. [Google Scholar] [CrossRef]
- Qureshi, R.N.; Kok, W.T. Application of Flow Field-Flow Fractionation for the Characterization of Macromolecules of Biological Interest: A Review. Anal. Bioanal. Chem. 2011, 399, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Baalousha, M.; Stolpe, B.; Lead, J.R. Flow Field-Flow Fractionation for the Analysis and Characterization of Natural Colloids and Manufactured Nanoparticles in Environmental Systems: A Critical Review. J. Chromatogr. A 2011, 1218, 4078–4103. [Google Scholar] [CrossRef] [PubMed]
- Runyon, J.R.; Ulmius, M.; Nilsson, L.A. Perspective on the Characterization of Colloids and Macromolecules Using Asymmetrical Flow Field-Flow Fractionation. Colloids Surf. A Physicochem. Eng. Asp. 2014, 442, 25–33. [Google Scholar] [CrossRef]
- Von der Kammer, F.; Legros, S.; Hofmann, T.; Larsen, E.H.; Loeschner, K. Separation and Characterization of Nanoparticles in Complex Food and Environmental Samples by Field-Flow Fractionation. TrAC Trend. Anal. Chem. 2011, 30, 425–436. [Google Scholar] [CrossRef]
- Roda, B.; Zattoni, A.; Reschiglian, P.; Moon, M.H.; Mirasoli, M.; Michelini, E.; Roda, A. Field-Flow Fractionation in Bioanalysis: A Review of Recent Trends. Anal. Chim. Acta 2009, 635, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L. Separation and Characterization of Food Macromolecules Using Field-Flow Fractionation: A Review. Food Hydrocoll. 2013, 30, 1–11. [Google Scholar] [CrossRef]
- Wahlund, K.G.; Giddings, J.C. Properties of an Asymmetrical Flow Field-Flow Fractionation Channel Having One Permeable Wall. Anal. Chem. 1987, 59, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rea, D.; Zielke, C.; Nilsson, L. Co-Elution Effects Can Influence Molar Mass Determination of Large Macromolecules with Asymmetric Flow Field-Flow Fractionation Coupled to Multiangle Light Scattering. J. Chromatogr. A 2017, 1506, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Einstein, A. Über die von der molekularkinatischen Theorie der Wärma geforderte Bewegung von in ruhenden Flussigkeiten suspendierten Teilchen. Ann. Phys. 1905, 17, 549–560. (In German) [Google Scholar] [CrossRef]
- Wittgren, B.; Wahlund, K.-G. Fast Molecular Mass and Size Characterization of Polysaccharides Using Asymmetrical Flow Field-Flow Fractionation-Multiangle Light Scattering. J. Chromatogr. A 1997, 760, 205–218. [Google Scholar] [CrossRef]
- Nilsson, L.; Leeman, M.; Wahlund, K.-G.; Bergenståhl, B. Mechanical Degradation and Changes in Conformation of Hydrophobically Modified Starch. Biomacromolecules 2006, 7, 2671–2679. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, A.; Magnusson, E.; Bergenståhl, B.; Nilsson, L. Hydrodynamic Radius Determination with Asymmetrical Flow Field-Flow Fractionation Using Decaying Cross-Flows. Part I. A Theoretical Approach. J. Chromatogr. A 2012, 1253, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, E.; Håkansson, A.; Janiak, J.; Bergenståhl, B.; Nilsson, L. Hydrodynamic Radius Determination with Asymmetrical Flow Field-Flow Fractionation Using Decaying Cross-Flows. Part II. Experimental Evaluation. J. Chromatogr. A 2012, 1253, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Leeman, M.; Wahlund, K.G.; Wittgren, B. Programmed Cross-Flow Asymmetrical Flow Field-Flow Fractionation for the Size Separation of Pullulans and Hydroxypropyl Cellulose. J. Chromatogr. A 2006, 1134, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.; Benavente, J. Streaming Potential along the Surface of Polysulfone Membranes: A Comparative Study between Two Different Experimental Systems and Determination of Electrokinetic and Adsorption Parameters. J. Membrane Sci. 2001, 190, 119–132. [Google Scholar] [CrossRef]
- Pontié, M. Effect of Aging on UF Membranes by a Streaming Potential (SP) Method. J. Membrane Sci. 1999, 154, 213–220. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Q.; Shire, S.J.; Demeule, B. Assessing and Improving Asymmetric Flow Field-Flow Fractionation of Therapeutic Proteins. In Field-Flow Fractionation in Biopolymer Analysis; Williams, S.K.R., Caldwell, K.D., Eds.; Springer: Vienna, Austria, 2012; pp. 89–101. [Google Scholar]
- Boye, S.; Ennen, F.; Scharfenberg, L.; Appelhans, D.; Nilsson, L.; Lederer, A. From 1D Rods to 3D Networks: A Biohybrid Topological Diversity Investigated by Asymmetrical Flow Field-Flow Fractionation. Macromolecules 2015, 48, 4607–4619. [Google Scholar] [CrossRef]
- Mes, E.P.C.; de Jonge, H.; Klein, T.; Welz, R.R.; Gillespie, D.T. Characterization of High Molecular Weight Polyethylenes Using High Temperature Asymmetrical Flow Field-Flow Fractionation with On-Line Infrared, Light Scattering, and Viscometry Detection. J. Chromatogr. A 2007, 1154, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Hiller, W.; Sinha, P.; Hehn, M.; Pasch, H. Online LC-NMR—From an Expensive Toy to a Powerful Tool in Polymer Analysis. Prog. Polym. Sci. 2014, 39, 979–1016. [Google Scholar] [CrossRef]
- Pasch, H.; Makan, A.C.; Chirowodza, H.; Ngaza, N.; Hiller, W. Analysis of Complex Polymers by Multidetector Field-Flow Fractionation. Anal. Bioanal. Chem. 2014, 406, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hansen, M.; Giddings, J.C. Advances in Frit-Inlet and Frit-Outlet Flow Field-Flow Fractionation. J. Microcolumn Sep. 1998, 10, 7–18. [Google Scholar] [CrossRef]
- Moon, M.H.; Williams, P.S.; Kang, D.; Hwang, I. Field and Flow Programming in Frit-Inlet Asymmetrical Flow Field-Flow Fractionation. J. Chromatogr. A 2002, 955, 263–272. [Google Scholar] [CrossRef]
- Schimpf, M.E.; Caldwell, K.; Giddings, J.C. Field Flow Fractionation Handbook; Wiley-Interscience: New York, NY, USA, 2000. [Google Scholar]
- HadjSadok, A.; Pitkowski, A.; Nicolai, T.; Benyahia, L.; Moulai-Mostefa, N. Characterisation of Sodium Caseinate as a Function of Ionic Strength, pH and Temperature Using Static and Dynamic Light Scattering. Food Hydrocoll. 2008, 22, 1460–1466. [Google Scholar] [CrossRef]
- Huppertz, T.; Gazi, I.; Luyten, H.; Nieuwenhuijse, H.; Alting, A.; Schokker, E. Hydration of Casein Micelles and Caseinates: Implications for Casein Micelle Structure. Int. Dairy J. 2017, 74, 1–11. [Google Scholar] [CrossRef]
- Lorenzen, P.C.; Schlimme, E.; Roos, N. Crosslinking of Sodium Caseinate by a Microbial Transglutaminase. Nahrung/Food 1998, 42, 151–154. [Google Scholar] [CrossRef]
- McKinnon, I.R.; Beckett, R.; Udabage, P.; Augustin, M.A. The Use of Field Flow Fractionation in the Characterization of Casein Micelles. Int. Dairy J. 1999, 9, 391–392. [Google Scholar] [CrossRef]
- Glantz, M.; Håkansson, A.; Lindmark Månsson, H.; Paulsson, M.; Nilsson, L. Revealing the Size, Conformation, and Shape of Casein Micelles and Aggregates with Asymmetrical Flow Field-Flow Fractionation and Multiangle Light Scattering. Langmuir 2010, 26, 12585–12591. [Google Scholar] [CrossRef] [PubMed]
- Nogueira Silva, N.N.; Piot, M.; de Carvalho, A.F.; Violleau, F.; Fameau, A.-L.; Gaucheron, F. pH-Induced Demineralization of Casein Micelles Modifies Their Physico-Chemical and Foaming Properties. Food Hydrocoll. 2013, 32, 322–330. [Google Scholar] [CrossRef]
- Lazzaro, F.; Saint-Jalmes, A.; Violleau, F.; Lopez, C.; Gaucher-Delmas, M.; Madec, M.-N.; Beaucher, E.; Gaucheron, F. Gradual Disaggregation of the Casein Micelle Improves its Emulsifying Capacity and Decreases the Stability of Dairy Emulsions. Food Hydrocoll. 2017, 63, 189–200. [Google Scholar] [CrossRef]
- Guyomarc’h, F.; Violleau, F.; Surel, O.; Famelart, M.-H. Characterization of Heat-Induced Changes in Skim Milk Using Asymmetrical Flow Field-Flow Fractionation Coupled with Multiangle Laser Light Scattering. J. Agric. Food Chem. 2010, 58, 12592–12601. [Google Scholar] [CrossRef] [PubMed]
- Cragnell, C.; Choi, J.; Segad, M.; Lee, S.; Nilsson, L.; Skepö, M. Bovine β-Casein has a Polydisperse Distribution of Equilibrium Micelles. Food Hydrocoll. 2017, 70, 65–68. [Google Scholar] [CrossRef]
- Wyatt, P.J. Light Scattering and the Absolute Characterization of Macromolecules. Anal. Chim. Acta 1993, 272, 1–40. [Google Scholar] [CrossRef]
- Boye, S.; Polikarpov, N.; Appelhans, D.; Lederer, A. An Alternative Route to Dye–Polymer Complexation Study Using Asymmetrical Flow Field-Flow Fractionation. J. Chromatogr. A 2010, 1217, 4841–4849. [Google Scholar] [CrossRef] [PubMed]
- Boye, S.; Appelhans, D.; Boyko, V.; Zschoche, S.; Komber, H.; Friedel, P.; Formanek, P.; Janke, A.; Voit, B.I.; Lederer, A. pH-Triggered Aggregate Shape of Different Generations Lysine-Dendronized Maleimide Copolymers with Maltose Shell. Biomacromolecules 2012, 13, 4222–4235. [Google Scholar] [CrossRef] [PubMed]
- Yassin, M.A.; Appelhans, D.; Wiedemuth, R.; Formanek, P.; Boye, S.; Lederer, A.; Temme, A.; Voit, B. Overcoming Concealment Effects of Targeting Moieties in the PEG Corona: Controlled Permeable Polymersomes Decorated with Folate-Antennae for Selective Targeting of Tumor Cells. Small 2015, 11, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.; Rakhmatullina, E.; Bleek, K.; Boye, S.; Yuan, J.; Völkel, A.; Gräwert, M.; Cheaib, Z.; Eick, S.; Günter, C.; et al. Poly(ethylene oxide)-b-poly(3-sulfopropyl methacrylate) Block Copolymers for Calcium Phosphate Mineralization and Biofilm Inhibition. Biomacromolecules 2014, 15, 3901–3914. [Google Scholar] [CrossRef] [PubMed]
- Zimm, B.H. The Scattering of Light and the Radial Distribution Function of High Polymer Solutions. J. Chem. Phys. 1948, 16, 1093. [Google Scholar] [CrossRef]
- Andersson, M.; Wittgren, B.; Wahlund, K.G. Accuracy in Multiangle Light Scattering Measurements for Molar Mass and Radius Estimations. Model Calculations and Experiments. Anal. Chem. 2003, 75, 4279–4291. [Google Scholar] [CrossRef] [PubMed]
- Brewer, A.K.; Striegel, A.M. Characterizing the Size, Shape, and Compactness of a Polydisperse Prolate Ellipsoidal Particle via Quadruple-Detector Hydrodynamic Chromatography. Analyst 2011, 136, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Ennen, F.; Boye, S.; Lederer, A.; Cernescu, M.; Komber, H.; Brutschy, B.; Voit, B.; Appelhans, D. Biohybrid Structures Consisting of Biotinylated Glycodendrimers and Proteins: Influence of the Biotin Ligand’s Number and Chemical Nature on the Biotin–Avidin Conjugation. Polym. Chem. 2014, 5, 1323–1339. [Google Scholar] [CrossRef]
- Ennen, F.; Fenner, P.; Boye, S.; Lederer, A.; Komber, H.; Voit, B.; Appelhans, D. Sphere-Like Protein–Glycopolymer Nanostructures Tailored by Polyassociation. Biomacromolecules 2016, 17, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Haladjova, E.; Rangelov, S.; Geisler, M.; Boye, S.; Lederer, A.; Mountrichas, G.; Pispas, S. Asymmetric Flow Field-Flow Fractionation Investigation of Magnetopolyplexes. Macromol. Chem. Phys. 2015, 216, 1862–1867. [Google Scholar] [CrossRef]
- Nzé, R.-P.; Nicolai, T.; Chassenieux, C.; Nicol, E.; Boye, S.; Lederer, A. Effect of Connectivity on the Structure and the Liquid–Solid Transition of Dense Suspensions of Soft Colloids. Macromolecules 2015, 48, 7995–8002. [Google Scholar] [CrossRef]
- Zessin, J.; Fischer, F.; Heerwig, A.; Kick, A.; Boye, S.; Stamm, M.; Kiriy, A.; Mertig, M. Tunable Fluorescence of a Semiconducting Polythiophene Positioned on DNA Origami. Nano Lett. 2017, 17, 5163–5170. [Google Scholar] [CrossRef] [PubMed]
- Burchard, W. Solution Properties of Branched Macromolecules. In Branched Polymers II; Roovers, J., Ed.; Springer: Berlin, Germany, 1999; Volume 143, pp. 113–194. [Google Scholar]
- Marchin, S.; Putaux, J.-L.; Pignon, F.; Léonil, J. Effects of the Environmental Factors on the Casein Micelle Structure Studied by Cryo Transmission Electron Microscopy and Small-Angle X-Ray Scattering/Ultrasmall-Angle X-Ray Scattering. J. Chem. Phys. 2007, 126, 045101. [Google Scholar] [CrossRef] [PubMed]
- Moitzi, C.; Portnaya, I.; Glatter, O.; Ramon, O.; Danino, D. Effect of Temperature on Self-Assembly of Bovine β-Casein above and below Isoelectric pH. Structural Analysis by Cryogenic-Transmission Electron Microscopy and Small-Angle X-ray Scattering. Langmuir 2008, 24, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- International Dairy Federation. Caseins and Caseinates—Determination of Protein Content (Reference Method); IDF Standard 92: Brussels, Belgium, 1979. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Casein Type | Approx. Percentage from Total Casein (%) | Number of Amino Acids | Approx. Molar Mass (kg/mol) | Isoelectric Point (-) | Grand Average of Hydropathicity (-) 1 |
|---|---|---|---|---|---|
| αS1-casein | 40 | 199 | 23.6 | 4.42 | −0.704 |
| αS2-casein | 10 | 207 | 25.2 | 4.95 | −0.918 |
| β-casein | 35 | 209 | 24.0 | 4.65 | −0.355 |
| κ-casein | 15 | 169 | 19.1 | 5.60 | −0.557 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raak, N.; Abbate, R.A.; Lederer, A.; Rohm, H.; Jaros, D. Size Separation Techniques for the Characterisation of Cross-Linked Casein: A Review of Methods and Their Applications. Separations 2018, 5, 14. https://doi.org/10.3390/separations5010014
Raak N, Abbate RA, Lederer A, Rohm H, Jaros D. Size Separation Techniques for the Characterisation of Cross-Linked Casein: A Review of Methods and Their Applications. Separations. 2018; 5(1):14. https://doi.org/10.3390/separations5010014
Chicago/Turabian StyleRaak, Norbert, Raffaele Andrea Abbate, Albena Lederer, Harald Rohm, and Doris Jaros. 2018. "Size Separation Techniques for the Characterisation of Cross-Linked Casein: A Review of Methods and Their Applications" Separations 5, no. 1: 14. https://doi.org/10.3390/separations5010014