On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization
by
Hossein Riazi
1,†, 
Ahmad Arabi Shamsabadi
1,†,
Patrick Corcoran
1,
Michael C. Grady
2,
Andrew M. Rappe
3 and
Masoud Soroush
1,* 1
Department of Chemical and Biological Engineering, Drexel University, Philadelphia, PA 19104, USA
2
Axalta Coating Systems, Wilmington, DE 19803, USA
3
Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Processes 2018, 6(1), 3; https://doi.org/10.3390/pr6010003
Submission received: 13 December 2017
/
Revised: 30 December 2017
/
Accepted: 31 December 2017
/
Published: 4 January 2018
(This article belongs to the Special Issue Feature Papers for the Fifth Year Anniversary of the Founding of Processes)
Abstract
:This experimental and theoretical study deals with the thermal spontaneous polymerization of n-butyl acrylate (n-BA). The polymerization was carried out in solution (n-heptane as the solvent) at 200 and 220 °C without adding any conventional initiators. It was studied with the five different n-BA/n-heptane volume ratios: 50/50, 70/30, 80/20, 90/10, and 100/0. Extensive experimental data presented here show significant monomer conversion at all temperatures and concentrations confirming the occurrence of the thermal self-initiation of the monomer. The order, frequency factor, and activation energy of the thermal self-initiation reaction of n-BA were estimated from n-BA conversion, using a macroscopic mechanistic model. The estimated reaction order agrees well with the order obtained via our quantum chemical calculations. Furthermore, the frequency factor and activation energy estimates agree well with the corresponding values that we already reported for bulk polymerization of n-BA.
1. Introduction
Free-radical polymerization has been initiated by a thermal initiator like ammonium persulfate [1] and azobisisobutyronitrile [2], or a method such as plasma [3], ultrasound [4], UV-irradiation [5], ionizing-irradiation [6], and redox initiation [7]. A monomer can also initiate free-radical polymerization by itself. Examples are vinyl monomers that undergo self-initiation in bulk [8,9], gas [10], and (mini)emulsion media [11,12]. However, the latter needs a large amount of surfactants to stabilize monomer droplets and polymer particles [12]. The monomer self-initiation reactions can also be exploited as a radical generation source in other radical polymerization mechanisms like reversible addition-fragmentation chain transfer (RAFT) polymerization where a RAFT agent exists. Among the vinyl monomers, styrene has shown high tendency for thermal self-initiation. The proposed mechanism is the formation of two monoradicals from two styrene monomers via self-Diels-Alder cycloaddition [13,14]. Monomer self-initiation strongly depends on temperature; styrene needs more than one year to self-polymerize to reach 50% conversion at 29 °C, however, the same conversion will be obtained after only 4 h at 127 °C. In addition to styrene, thermal self-initiation of acrylate and methacrylate monomers have been studied [15,16,17]. For methyl methacrylate, the rate of thermal self-initiation is two orders of magnitude lower than styrene [18].
Purity is an important advantage of thermally self-initiated polymers. Pure polymers have many applications in medicine and food industries [19,20,21], because initiator and catalyst leftovers in polymers reduce their applicability in such areas. For example, molecularly imprinted polymers (MIPs) as antibodies are synthesized through thermal self-initiation free-radical polymerization [22]. The absence of any kind of functional groups in the microstructure of self-polymerized polymers prevents them from participating in many kinds of side reactions. Thus, the so-called dormant chains can be used as a characterization tool in some living polymerizations [23]. In general, in any kind of application where fully-pure polymer chains are required, polymerization through thermal monomer self-initiation is a good choice. Another advantage of thermally self-polymerized polymers is their low molecular weight [24] especially where they are used as paints and coatings [25].
Environmental regulations constantly have forced resin manufacturers to reduce the level of organic solvents in their products. However, solvents are needed in coatings and paints to endow them with good flowability and brushability. Low molecular weight acrylate resins synthesized through thermal monomer self-initiation polymerization can obviate the need for high amount of solvent. Thermally self-polymerized acrylate polymers usually have Mw < 10,000 g/mol and thereby do not need a high amount of solvent to gain flowability [26]. In the synthesis of hybrid organic/inorganic nanoparticles, thermal monomer self-initiation may also contribute. Hui et al. [27] synthesized polystyrene/silica hybrid brushes via ATRP technique, however, they reported that some pure untethered polystyrene homopolymers are formed due to the thermal self-initiation of styrene. Their results showed that nearly 23 weight % of the styrene undergoes self-initiation reactions and these untethered homopolymer chains, though are formed unintentionally and are considered as impurity, improved elastic modulus and toughness of the polystyrene/silica brushes. Another advantage of thermally monomer-self-initiated polymerization comes into play when conventional thermal initiators are not able to polymerize some monomers. For example, it has been reported that benzoyl peroxide is not able to polymerize dimethylaminoethyl methacrylate, while this monomer undergoes thermal self-initiation polymerization [28]. Moreover, thermally monomer-self-initiated polymerization is a good method for the synthesis of macromonomers as the occurrence of a β-Scission reaction, which is a prevalent reaction at high temperatures, leads to the formation of an unsaturated carbon double bond at the backbone of the produced shortened chain [29]. Such macromonomers have wide applications in the synthesis of brush polymers and graft copolymers [30]. Similar to controlled/living polymerization, thermally monomer-self-initiated polymerization is capable of producing macromonomers without a need for an initiator or a mediating agent [29,30]. Monomer-self-initiated polymerization has also been utilized to synthesize graphene/polystyrene nanocomposites. Graphene was functionalized to improve its lipophilicity, while neither external initiator nor immobilization of initiating groups on the surface of the graphene were required to initiate the polymerization [31].
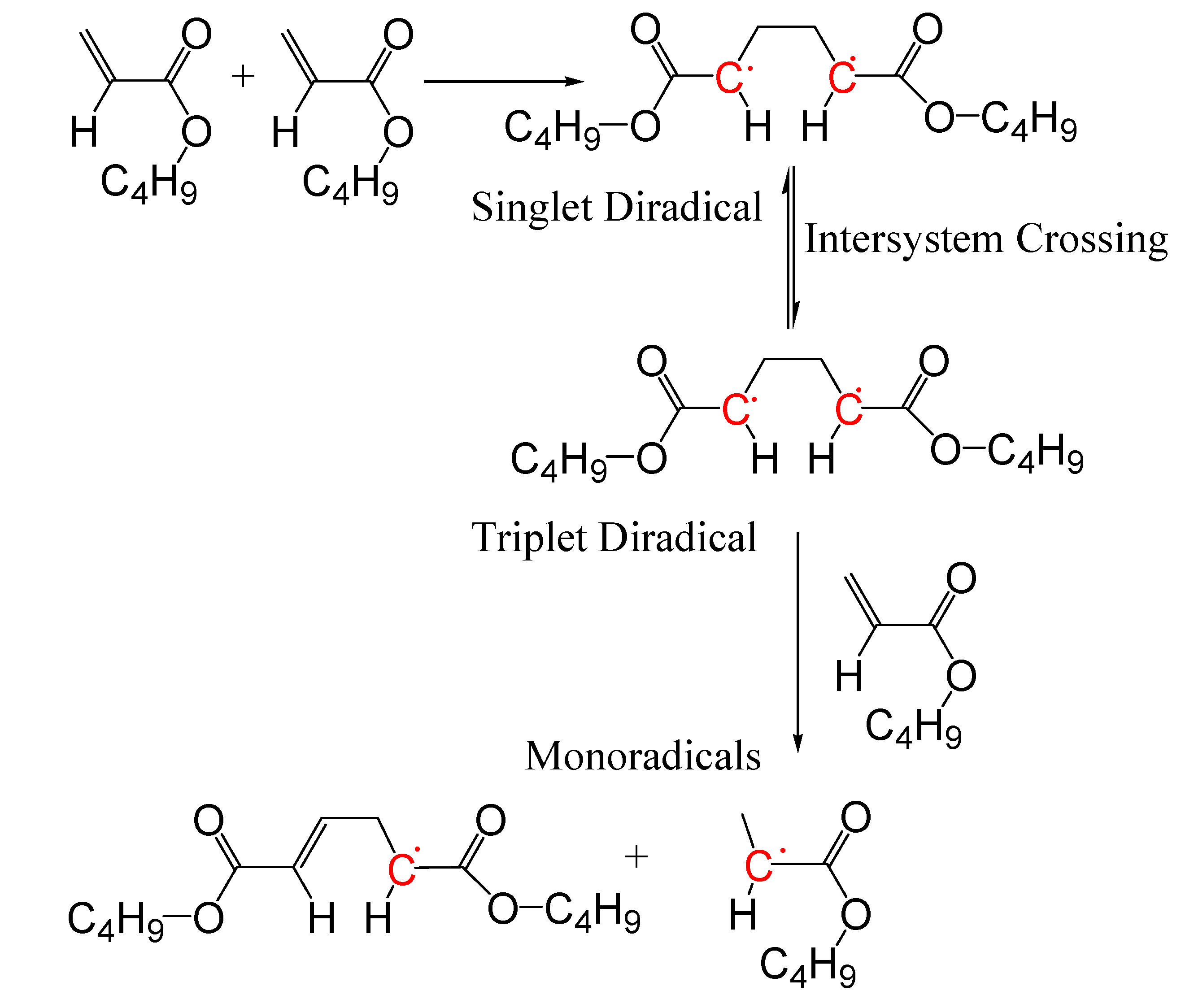
To fully exploit potentials of the monomer self-initiation reaction, a detailed understanding of its kinetics is needed [32]. Monomer self-initiation reaction affects polymer average molecular weights, molecular weight distribution, and consequently many properties of the polymers [10,15]. At high temperatures, styrene undergoes self-initiation reaction through a third order reaction [33]. For acrylate and methacrylate monomers, a kinetic study of the polymerization at high temperatures is more complicated as many side reactions take place at high temperatures. These side reactions are transfer to monomer, transfer to polymer, midchain radical propagation, midchain radical termination, backbiting, and β-Scission. In general, for non-styrenic monomers, the monomer self-initiation mechanism occurs through Flory’s diradical mechanism [16,34,35]. For alkyl acrylate monomers, computational quantum chemistry studies [35,36,37] showed that two monomer molecules (M) react and form a singlet diradical (*MM*S) (Figure 1):
M + M → *MM*S
The singlet diradical includes two monomer units. A singlet diradical has a pair of electrons, one spin-up and one spin-down (+1/2 and −1/2), in one orbital with the second, equal energy orbital, empty [38]. The singlet diradical then goes through a spin crossover reaction and changes into a triplet diradical (*MM*T):
*MM*S → *MM*T
The triplet diradical also includes two monomer units. A triplet diradical has two “spin-up” electrons in adjacent, degenerate (equal energy) orbitals (two electrons of the same spin, +1/2 and +1/2) [38]. Finally, a third monomer molecule abstracts a hydrogen atom from the triplet diradical and forms two monoradicals, one with two monomer units (MM*) and the other with one monomer unit (M*):
*MM*T + M → MM* + M*
Thus, the overall (apparent) monomer self-initiation reaction is:
which according to quantum chemical calculations [35,36,37], is second order. In this paper, experimentally we investigate this theoretical finding (reaction order) via estimating the order of reaction from monomer conversion measurements made in solution and bulk polymerizations. In addition, we estimate the frequency factor and activation energy of the n-BA self-initiation reaction and compare them with those estimated in bulk polymerization of n-BA.
2. Materials and Methods
n-BA with purity of 98% containing 50 ppm of inhibitor (4-methoxyphenol) was purchased from Alfa-Aesar. Anhydrous n-heptane (99%) was supplied by Sigma-Aldrich, MO, USA. Stainless steel Swagelok tubes (Swagelok Inc., Huntingdon Valley, PA, USA) with length of 4-inch were used as batch reactors. Both ends of the reactors were capped with Swagelok stainless steel caps. These tubes provide 4.8 mL reaction volume and bear pressures as high as 3300 psig. Before polymerization, n-BA was passed through an inhibitor removal column to remove the manufacturer-added inhibitor from the monomer. The columns were DHR-4, supplied by Scientific Polymer Products of Ontario, New York, NY, USA. n-BA/n-heptane mixtures with different volume ratios of 50/50, 70/30, 80/20, 90/10, and 100/0 were then prepared by adding the inhibitor-free n-BA to n-heptane followed by magnet stirring. The mixtures were then transferred to a sealed round flask, where its inlet was blocked by a rubber cap, to be purged by ultra-high pure nitrogen. After 1 h purging, the nitrogen flow was stopped and the rubber cap was covered with an aluminum adhesive tape very quickly to make sure no oxygen diffused into the flask contents. Next, the flask, tubes and caps were transferred to a nitrogen-atmosphere glove box (LC Technology Solutions, Salisbury, MD, USA). Before transferring the items to the main chamber of the glove box, several vacuum-nitrogen purging cycles were repeated. When the concentration of oxygen in the glove box was lower than 0.1 ppm, the sealed flask was opened and around 3 mL of the flask contents was added to each tube. Afterwards, the caps were tightened and the tubes were removed from the glovebox and weighed, and their weights were recorded.
To carry out the polymerization, a fluidized sand bath was heated to 200 or 220 °C, and left in that temperature for a while to reach a steady state condition. Each polymerization reaction was started by fastening two tubes to a metal basket and putting it in the stabilized sand bath. The reaction times were 55, 110, 165, and 220 min. After staying in the sand bath for the desired time, the basket was removed from the sand bath and immersed in a cold-water bath to cool down the tubes and stop polymerization. The tubes were then detached from the basket, dried, and weighed again to compare their weights with the values recorded after removing them from the glove box. In case of weight difference, the tube was not considered for monomer conversion measurements. Finally, each tube’s contents were discharged to an aluminum petri dish and left in a vacuum oven at 50 °C to allow the contents to dry for monomer conversion measurements. It is worth noting here that, our previous studies showed that nearly 1 min is required for the monomer inside the tubes to reach the sand bath temperature. Thus, the tubes were allowed to stay for one minute extra in the sand bath beyond each nominal reaction time. The monomer conversion was measured using the gravimetric method [39].
3. Results and Discussion
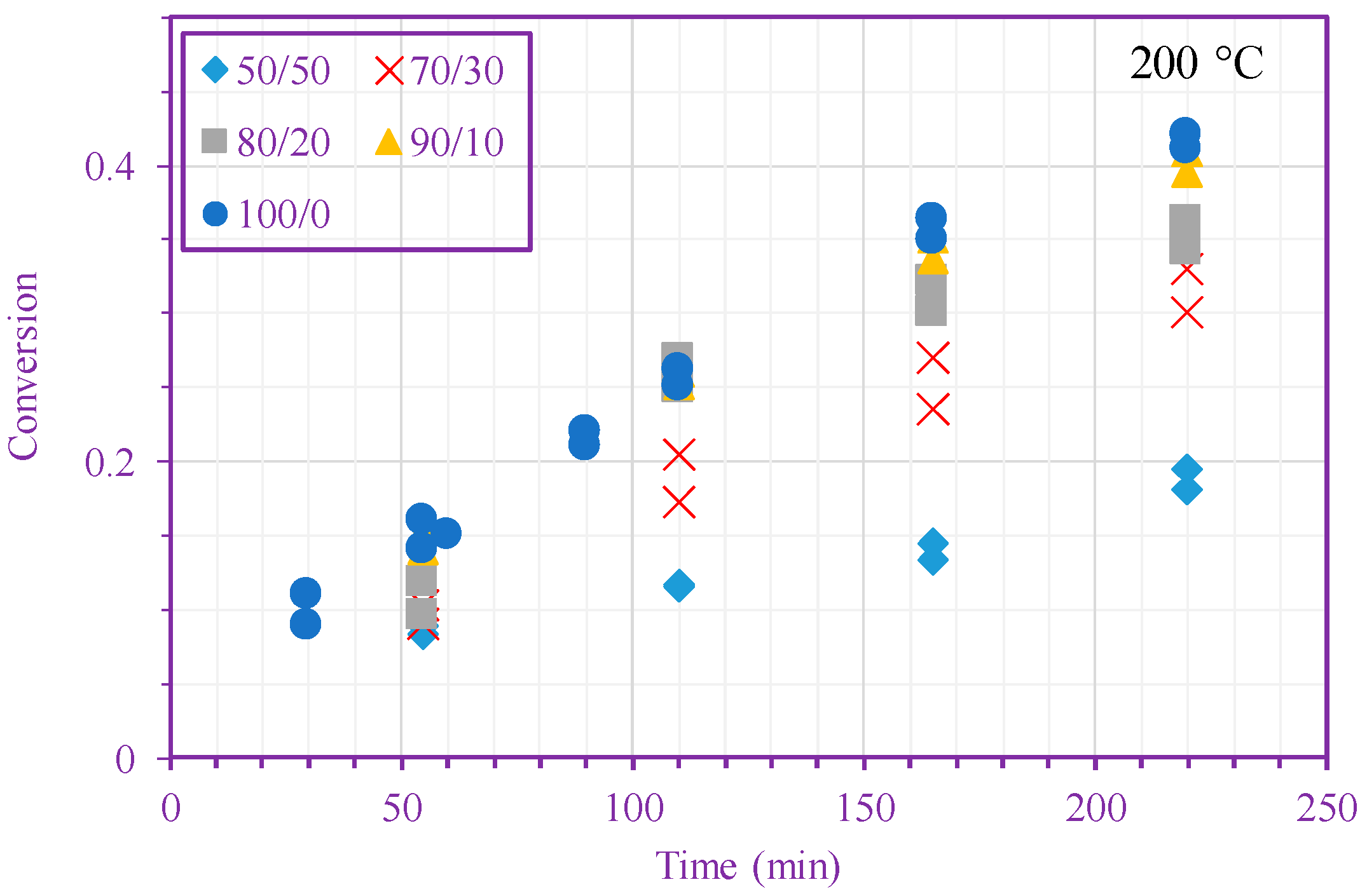
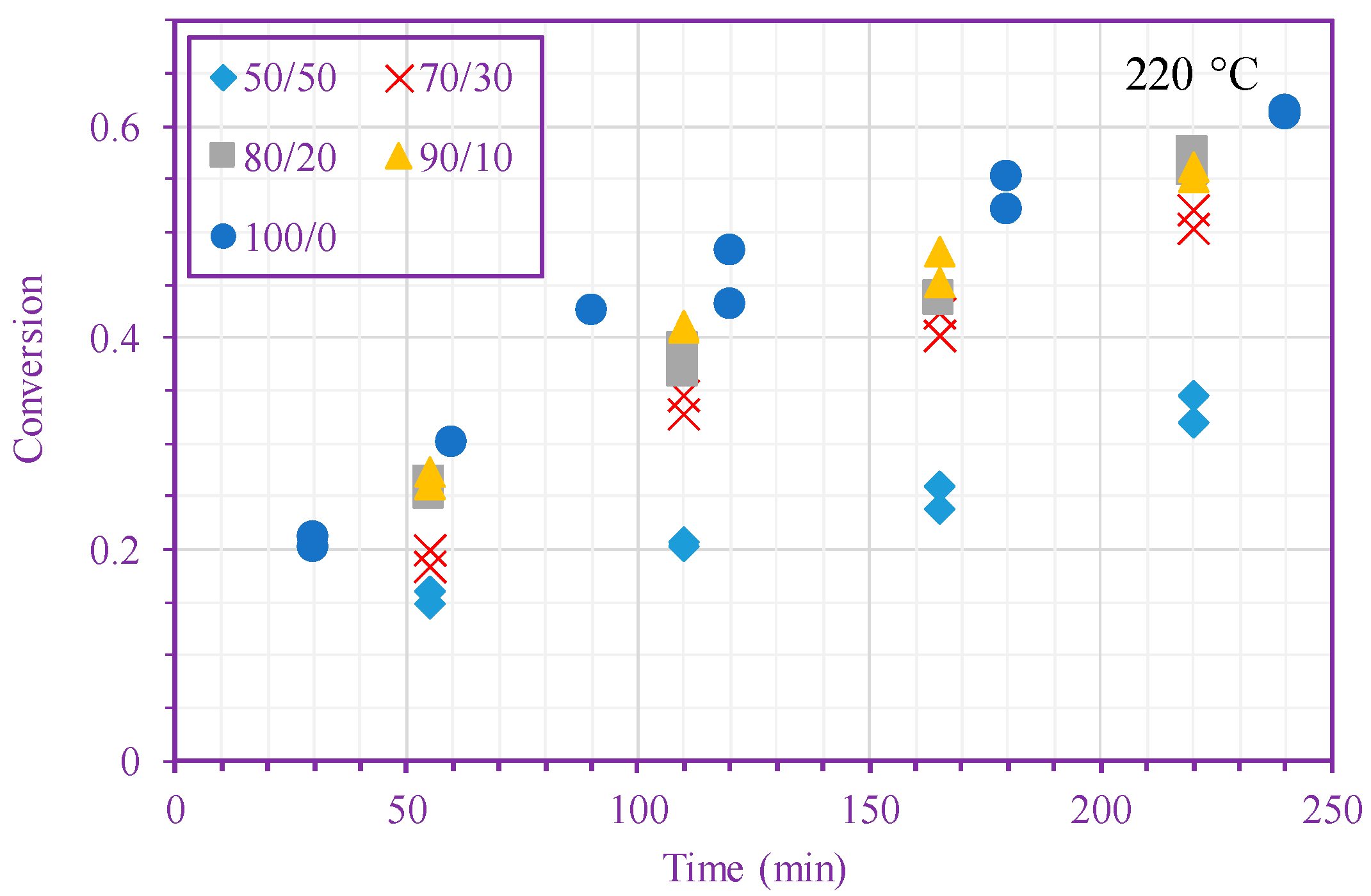
Figure 2 shows measured monomer conversion at 200 and 220 °C. These results agree with what one expects from free-radical polymerization; that is, the conversion increases with time, monomer concentration, and temperature. In addition, this figure shows that the n-BA self-initiation reaction in both solution and bulk media [39] is so strong that the monomer conversion can exceed 60% after only 220 min in the absence of any external initiators. Monomer self-initiation reactions of both acrylate and methacrylate families were investigated in our previous quantum chemical calculation studies [35,36,37]; it was found that the second reaction step, transition from a singlet diradical to a triplet one (Equation (2)), is the rate limiting step in the monomer self-initiation. We also found theoretically that the overall monomer self-initiation reaction is second order [35,36,37]. To validate the overall-reaction order obtained via the quantum chemical calculations, we estimate the overall-reaction order from monomer conversion measurements using a macroscopic mechanistic model.
In thermally monomer-self-initiated polymerization, various reactions happen simultaneously. A list of the most likely reactions is presented in Table A1 in the Appendix A; more details can be found in Ref. [39]. The reactions include, propagation by secondary and tertiary radicals, termination by secondary and tertiary radicals, monomer self-initiation, back-biting, β-Scission, inter-chain transfer to polymer, chain transfer to monomer, chain transfer to solvent, termination by combination, and termination by disproportion. Except for monomer self-initiation reaction, all other reactions are considered as elementary reactions and their reaction rate equations are derived by the method of moments. Writing component mass balances leads to 21 differential equations [39], describing monomer and solvent concentrations; concentrations of secondary radicals containing zero, one, two and three monomer unit(s); the zeroth, first and second moments of the secondary radical chain length distribution; the zeroth, first, second and third moments of the chain length distribution of tertiary radicals formed through intermolecular chain transfer to polymer reactions; the zeroth, first, second and third moments of the chain length distribution of tertiary radicals created by backbiting reactions; and the zeroth, first, second and third moments of the chain length distribution of live and dead polymer chains. Given the initial monomer concentration and the monomer concentration, and the monomer conversion,, is calculated using:
To simulate the model, we use the rate coefficients of all reactions except for the overall self-initiation reaction, reported in Ref. [39]. Although frequency factor and activation energy of n-BA self-initiation reaction in bulk had been estimated in our previous article [39], we re-estimate them here in solution to compare these results with our previous findings. In our previous bulk study [39], to simulate the macroscopic mechanistic model, the order of n-BA overall self-initiation reaction was set to 2, obtained from quantum chemical calculations [35,36,37]. Here, in addition to re-estimating the frequency factor and activation energy, we estimate the order of n-BA overall self-initiation reaction from the conversion measurements. We use ode15s command of MATLAB to integrate the differential equations of the macroscopic mechanistic model and use ga command of MATLAB to minimize the sum of squared residuals:
subject to:
where and (J/mol) are the frequency factor and activation energy values for n-BA bulk polymerization reported in [39], and is the theoretical value of reported in [35,36,37]. Here, is the measured monomer conversion value at the reaction time , the solvent concentration , and the temperature , while is the model-predicted monomer conversion at the same time, solvent concentration and temperature. and are the order, frequency factor, and activation energy of the n-BA overall self-initiation reaction, respectively. The monomer conversion measurements are available at two different temperatures (200 and 220 °C), five different n-BA/n-heptane volume ratios (50/50, 70/30, 80/20, 90/10, and 100/0), and four different times (55, 110, 165, and 220 min) twice.
In high temperature free-radical polymerization, secondary reactions like transfer to solvent are possible. In this work, n-heptane was used as the solvent, because transfer reactions to this solvent are negligible. For other vinyl monomers such as styrene and methyl methacrylate, the transfer to n-heptane constant is also very low [40]. On the other hand, solvents like n-butanol, methyl ethyl ketone, and p-xylene participate in chain transfer reactions [41]. The macroscopic mechanistic model showed that the concentration of n-heptane does not change under the experimental conditions used in this study. For example, the highest transfer to n-heptane constant found in [40] is while values as low as are also reported in the same reference. In the worst scenario when the transfer-to-solvent constant is , n-BA/n-heptane ratio is 50/50 volume%, and the temperature is 220 °C, our model showed that the solvent concentration decreased by 0.39% from 3.4169 to 3.4033 (mol/L) after 250 min of the polymerization, indicating the insignificance of chain-transfer-to n-heptane reactions. Thus, it is logical to assume that transfer-to-n-heptane reactions are negligible.
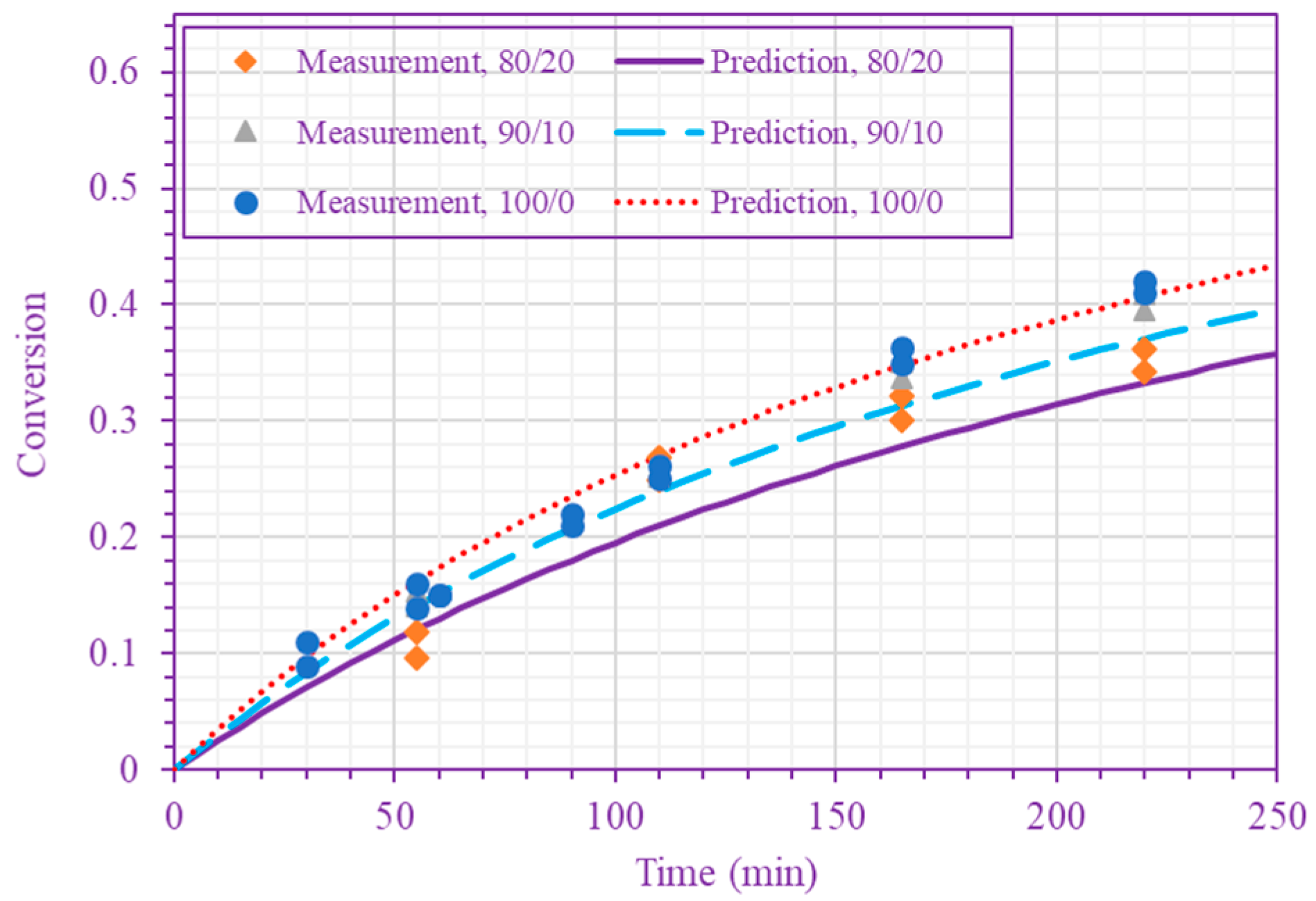
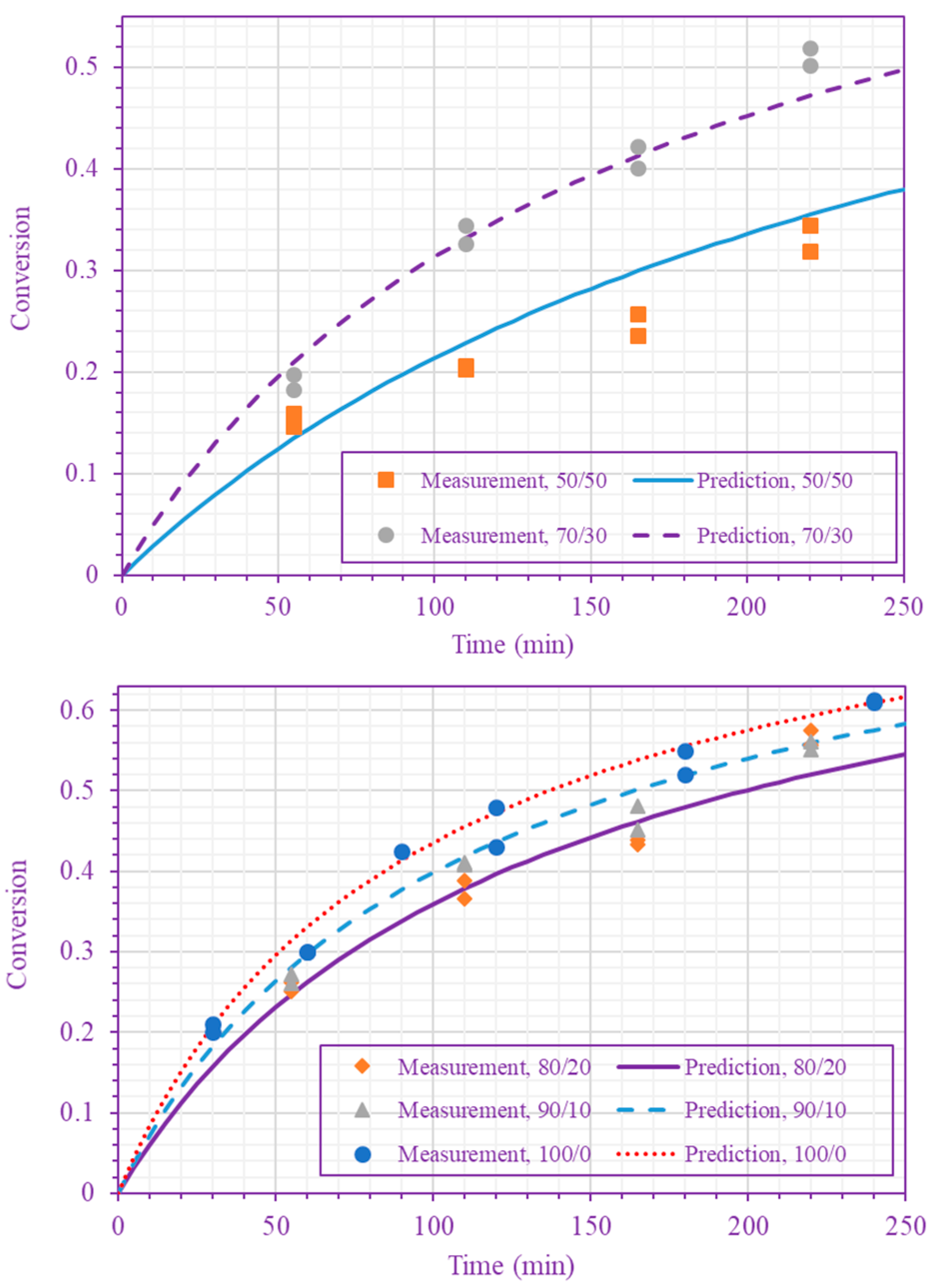
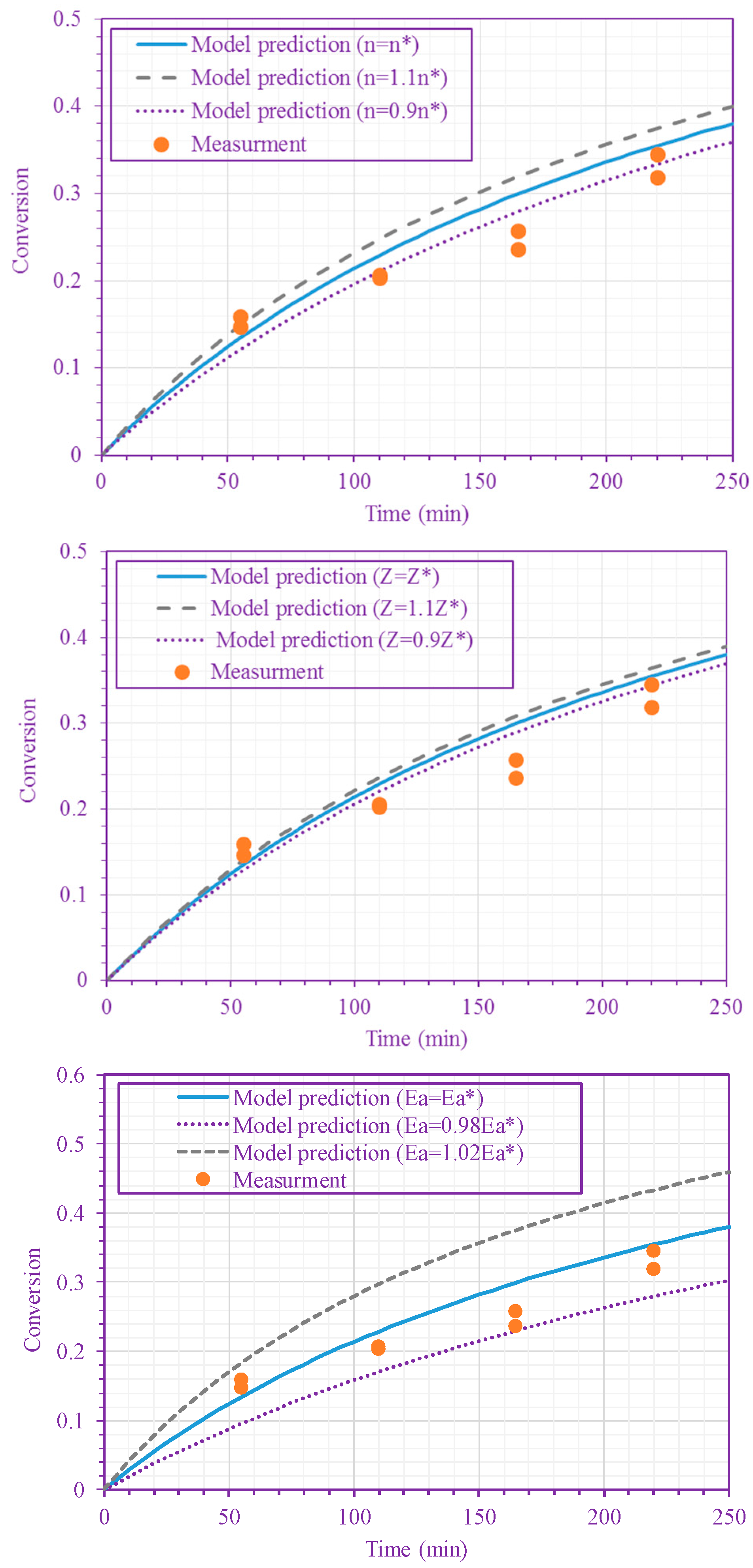
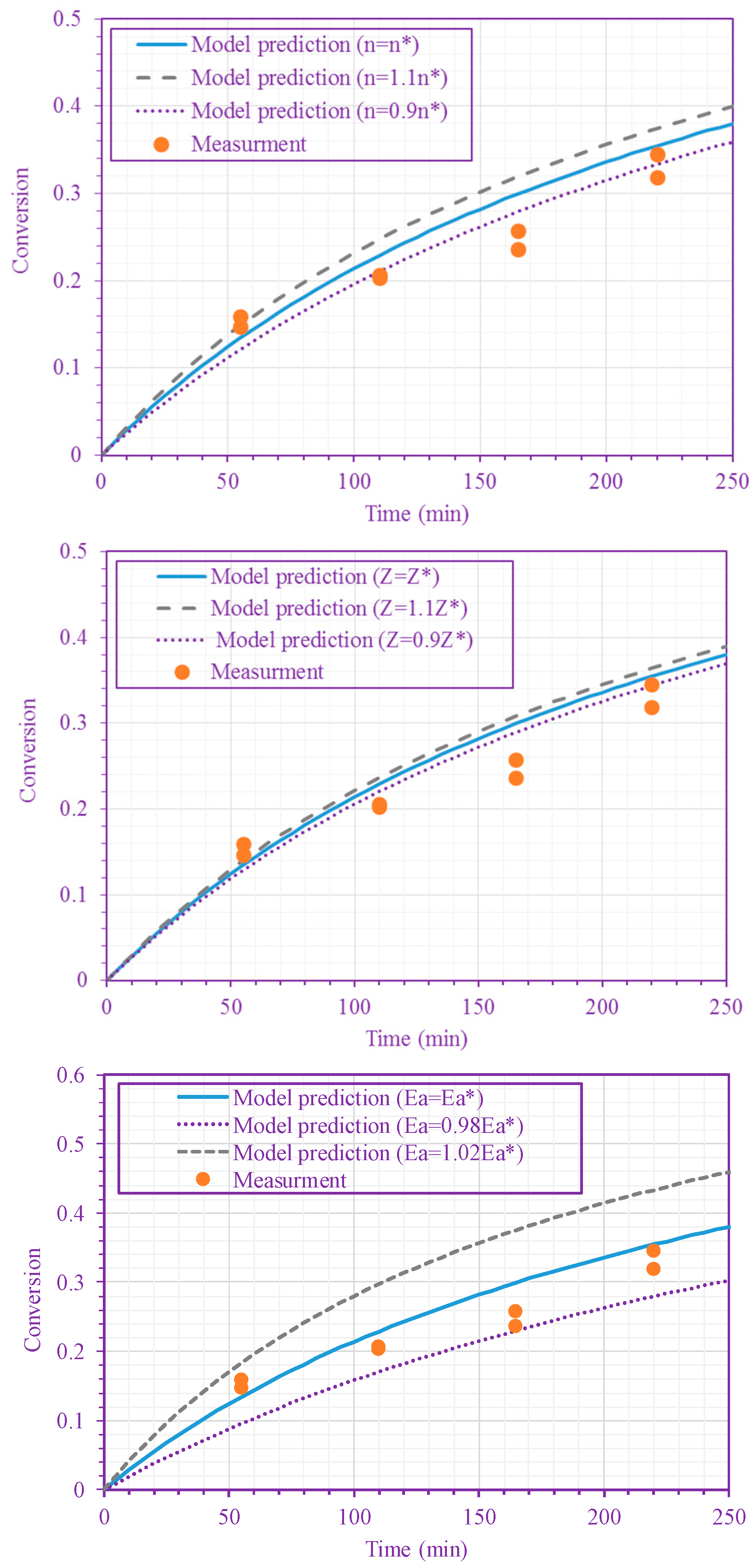
The frequency factor, activation energy, and the order of n-BA overall self-initiation reaction estimated from the conversion measurements are: , and . A possible reason for this frequency factor value (estimated here for solution polymerization) being less than the value obtained for bulk polymerization [39] is the cage effect of the solvent. Figure 3 and Figure 4 show the model prediction of n-BA conversion with these estimated values. They indicate that the model can predict the conversion well at all temperatures and solvent concentrations. The frequency factor and activation energy values reported here for the solution polymerization agree well with the values already reported for the bulk polymerization: and (J/mol) [39]. The order of the monomer self-initiation reaction found here also agrees well with the one obtained theoretically through quantum chemical calculations [35,36,37]. Figure 5 shows the sensitivity of our macroscopic mechanistic model to changes in the order, activation energy, and frequency factor. It indicates that the predicted conversion has the lowest sensitivity to the frequency factor and the highest sensitivity to the activation energy, .
4. Conclusions
Experimental results showed that n-BA undergoes self-initiation reaction at high temperatures in the presence and absence of the solvent. Macroscopic mechanistic model predictions showed that n-heptane does not participate in the polymerization; that is, chain-transfer-to-n-heptane reactions are negligible under the experimental conditions. The monomer conversions predicted by the model agree well with the experimental data. In addition, the order of the n-BA overall self-initiation reaction estimated from conversion is 2.08 0.28, which is in agreement with the theoretical value of 2 calculated using quantum chemical calculations [35,36,37]. Moreover, the frequency factor and activation energy of n-BA overall self-initiation reaction in solution polymerization are in agreement with those estimated for the same reaction in bulk. The sensitivity analysis showed that the macroscopic mechanistic model is adequately sensitive to the three estimated parameters.
Acknowledgments
The authors would like to thank Aaron Fafarman for his lab facility support.
Author Contributions
H.R. and A.A.S. jointly carried out the experiments and polymer analyses. H.R. prepared the initial draft of this paper. A.A.S., M.S., P.C., M.C.G., and A.M.R. reviewed and revised the manuscript. P.C., M.S., M.C.G. and A.M.R. conceived and designed the experiments. A.A.S. and M.S. analyzed the data.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
n-BA polymerization reactions.
|
References
- Adelnia, H.; Gavgani, J.N.; Riazi, H.; Bidsorkhi, H.C. Transition behavior, su characteristics and film formation of functionalized poly (methyl methacrylate-co-butyl acrylate) particles. Prog. Org. Coat. 2014, 77, 1826–1833. [Google Scholar] [CrossRef]
- Adelnia, H.; Riazi, H.; Saadat, Y.; Hosseinzadeh, S. Synthesis of monodisperse anionic submicron polystyrene particles by stabilizer-free dispersion polymerization in alcoholic media. Colloid Polym. Sci. 2013, 291, 1741–1748. [Google Scholar] [CrossRef]
- Yasuda, H.; Wang, C. Plasma polymerization investigated by the substrate temperature dependence. J. Polym. Sci. A1 1985, 23, 87–106. [Google Scholar] [CrossRef]
- Price, G.J.; Norris, D.J.; West, P.J. Polymerization of methyl methacrylate initiated by ultrasound. Macromolecules 1992, 25, 6447–6454. [Google Scholar] [CrossRef]
- Decker, C. The use of UV irradiation in polymerization. Polym. Int. 1998, 45, 133–141. [Google Scholar] [CrossRef]
- Marans, N.S. Ionizing Irradiation Polymerization of Trioxane with Copolymerizable Stabilizing Comonomers. Google Patents. U.S. Patent 3,366,561, 30 January 1968. [Google Scholar]
- Riazi, H.; Mohammadi, N.; Mohammadi, H. Emulsion copolymerization of methyl methacrylate/butyl acrylate/iodine system to monosize rubbery nanoparticles containing iodine and triiodide mixture. Ind. Eng. Chem. Res. 2013, 52, 2449–2456. [Google Scholar] [CrossRef]
- Riazi, H.; Arabi Shamsabadi, A.; Grady, M.; Rappe, A.; Soroush, M. Experimental and Macroscopic Mechanistic Modeling Studies of the Methyl Acrylate Self Initiation Reaction; AIChE Annual Meeting: Minneapolis, MN, USA, 2017. [Google Scholar]
- Riazi, H.; Arabi Shamsabadi, A.; Grady, M.; Rappe, A.M.; Soroush, M. Experimental and Theoretical Study of the Self-Initiation Reaction of Methyl Acrylate in Free-Radical Polymerization. Ind. Eng. Chem. Res. 2017. [Google Scholar] [CrossRef]
- Alsharaeh, E.H.; Ibrahim, Y.M.; El-Shall, M.S. Direct evidence for the gas phase thermal polymerization of styrene. Determination of the initiation mechanism and structures of the early oligomers by ion mobility. J. Am. Chem. Soc. 2005, 127, 6164–6165. [Google Scholar] [PubMed]
- Pan, G.; Sudol, E.D.; Dimonie, V.L.; El-Aasser, M.S. Thermal self-initiation of styrene in the presence of TEMPO radicals: Bulk and miniemulsion. J. Polym. Sci. A1 2004, 42, 4921–4932. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. Handbook of Radical Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2003; p. 878. [Google Scholar]
- Vana, P.; Barner-Kowollik, C.; Davis, T.P.; Matyjaszewski, K. Radical Polymerization; Encyclopedia of Polymer Science and Technology; John Wiely & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Moad, G.; Solomon, D.H. The Chemistry of Radical Polymerization, 2rd ed.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 108–109. [Google Scholar]
- Wang, W.; Hutchinson, R.A. Free-Radical Acrylic Polymerization Kinetics at Elevated Temperatures. Chem. Eng. Technol. 2010, 33, 1745–1753. [Google Scholar] [CrossRef]
- Srinivasan, S.; Kalfas, G.; Petkovska, V.I.; Bruni, C.; Grady, M.C.; Soroush, M. Experimental study of the spontaneous thermal homopolymerization of methyl and n-butyl acrylate. J. Appl. Polym. Sci. 2010, 118, 1898–1909. [Google Scholar] [CrossRef]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of cyclohexanone-monomer co-initiation mechanism in thermal homo-polymerization of methyl acrylate and methyl methacrylate. J. Phys. Chem. A 2012, 116, 5337–5348. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Yagci, Y. Handbook of Radical Vinyl Polymerization; CRC Press: Boca Raton, FL, USA, 1998; p. 48. [Google Scholar]
- Mattamal, G.J. US FDA perspective on the regulations of medical-grade polymers: Cyanoacrylate polymer medical device tissue adhesives. Exp. Rev. Med. Devices 2008, 5, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Frommelt, H. Polymers for medical applications. Macromol. Symp. 1987, 12, 281–301. [Google Scholar] [CrossRef]
- Jin, T.; Zhang, H. Biodegradable polylactic acid polymer with nisin for use in antimicrobial food packaging. J. Food Sci. 2008, 73, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, M.; Beyazit, S.; Nestora, S.; Haupt, K.; Bui, B.T.S. Initiator-free synthesis of molecularly imprinted polymers by polymerization of self-initiated monomers. Polymer 2015, 66, 43–51. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, Y.; Chen, L.; Fu, Z.; Shi, Y. Thermal Self-Initiation in Stable Free-Radical Polymerization of Styrene. Polym. J. 2009, 41, 954–960. [Google Scholar] [CrossRef]
- Peck, A.N.; Hutchinson, R.A. Secondary reactions in the high-temperature free radical polymerization of butyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Yu, X.; Pfaendtner, J.; Broadbelt, L.J. Ab initio study of acrylate polymerization reactions: Methyl methacrylate and methyl acrylate propagation. J. Phys. Chem. A 2008, 112, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, N.; Liu, S.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical study of intermolecular chain transfer to polymer reactions of alkyl acrylates. Ind. Eng. Chem. Res. 2015, 54, 4148–4165. [Google Scholar] [CrossRef]
- Hui, C.M.; Dang, A.; Chen, B.; Yan, J.; Konkolewicz, D.; He, H.; Ferebee, R.; Bockstaller, M.R.; Matyjaszewski, K. Effect of thermal self-initiation on the synthesis, composition, and properties of particle brush materials. Macromolecules 2014, 47, 5501–5508. [Google Scholar] [CrossRef]
- Shalati, M.D.; Scott, R.M. Thermal polymerization of dimethylaminoethyl methacrylate. Macromolecules 1975, 8, 127–130. [Google Scholar] [CrossRef]
- Zorn, A.M.; Junkers, T.; Barner-Kowollik, C. Synthesis of a Macromonomer Library from High-Temperature Acrylate Polymerization. Macromol. Rapid Commun. 2009, 30, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Junkers, T.; Bennet, F.; Koo, S.P.; Barner-Kowollik, C. Self-directed formation of uniform unsaturated macromolecules from acrylate monomers at high temperatures. J. Polym. Sci. Pol. Chem. 2008, 46, 3433–3437. [Google Scholar] [CrossRef]
- Beckert, F.; Rostas, A.M.; Thomann, R.; Weber, S.; Schleicher, E.; Friedrich, C.; Mulhaupt, R. Self-initiated free radical grafting of styrene homo-and copolymers onto functionalized graphene. Macromolecules 2013, 46, 5488–5496. [Google Scholar] [CrossRef]
- Soroush, M.; Grady, M.C.; Kalfas, G.A. Free-radical polymerization at higher temperatures: Systems impacts of secondary reactions. Comput. Chem. Eng. 2008, 32, 2155–2167. [Google Scholar] [CrossRef]
- Meyer, T.; Keurentjes, J. Handbook of Polymer Reaction Engineering; Wiley-VCH: Weinheim, Germany, 2005; p. 169. [Google Scholar]
- Zhang, C.; Wang, X.; Liu, L.; Wang, Y.; Peng, X. Modeling the spontaneous initiation of the polymerization of methyl methacrylate. J. Mol. Model. 2008, 14, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Self-initiation mechanism in spontaneous thermal polymerization of ethyl and n-butyl acrylate: A theoretical study. J. Phys. Chem. A 2010, 114, 7975–7983. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Tao, J.; Grady, M.C.; Soroush, M.; Rappe, A.M. Modeling spin-forbidden monomer self-initiation reactions in spontaneous free-radical polymerization of acrylates and methacrylates. J. Phys. Chem. A 2014, 118, 9310–9318. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of the self-initiation mechanism in thermal polymerization of methyl acrylate. J. Phys. Chem. A 2009, 113, 10787–10794. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Backbiting and β-scission reactions in free-radical polymerization of methyl acrylate. Int. J. Quantum Chem. 2014, 114, 345–360. [Google Scholar] [CrossRef]
- Shamsabadi, A.A.; Moghadam, N.; Srinivasan, S.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling. Processes 2016, 4, 15. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook; Wiley: New York, NY, USA, 1989; p. II/120. [Google Scholar]
- Moghadam, N.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical study of chain transfer to solvent reactions of alkyl acrylates. J. Phys. Chem. A 2014, 118, 5474–5487. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
n-BA self-initiation mechanism [35].
Figure 1.
n-BA self-initiation mechanism [35].
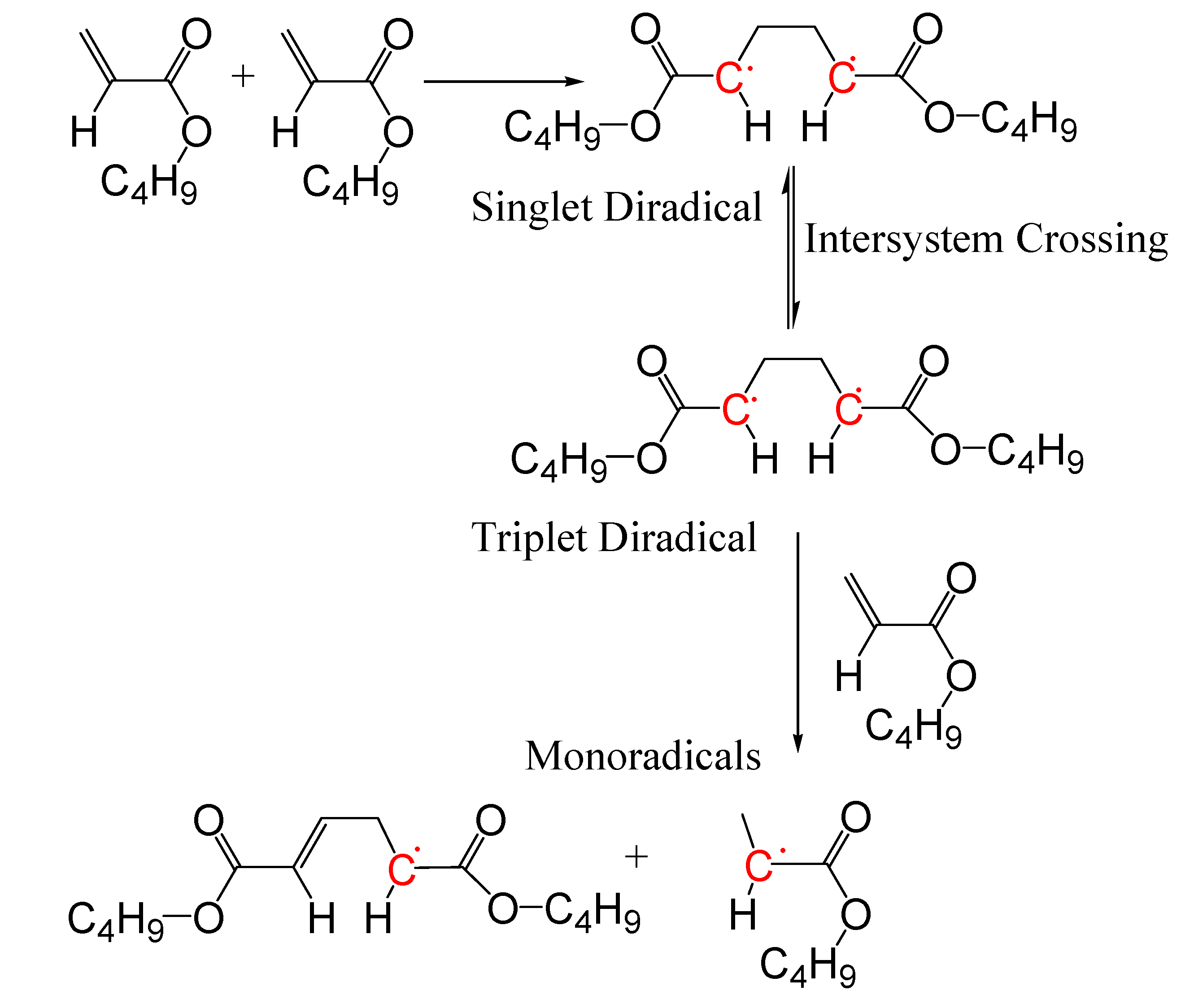
Figure 2.
Measured n-BA conversion vs. time (different n-BA/n-heptane ratios) at 200 °C (top) and 220 °C (bottom).
Figure 2.
Measured n-BA conversion vs. time (different n-BA/n-heptane ratios) at 200 °C (top) and 220 °C (bottom).
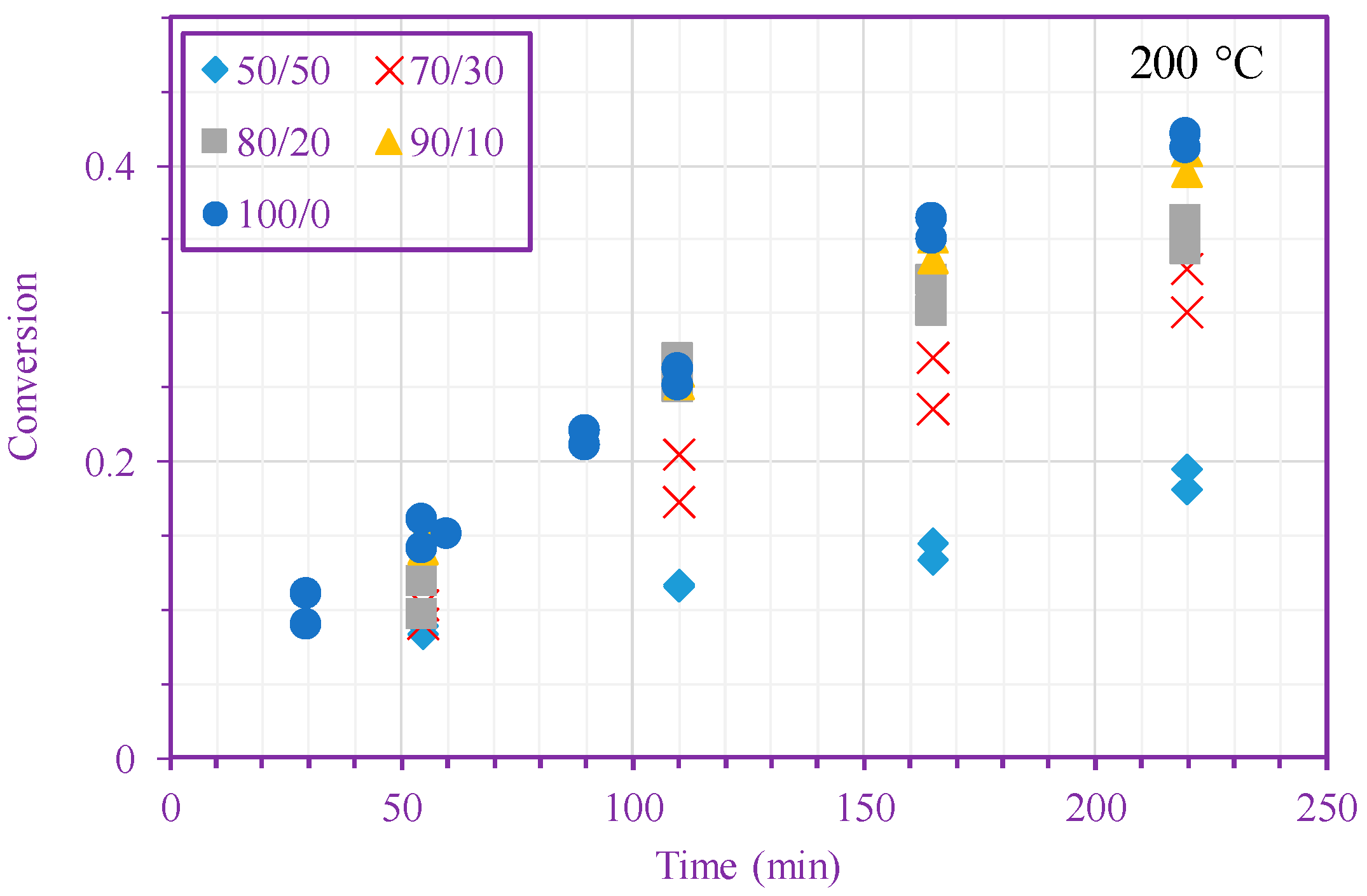
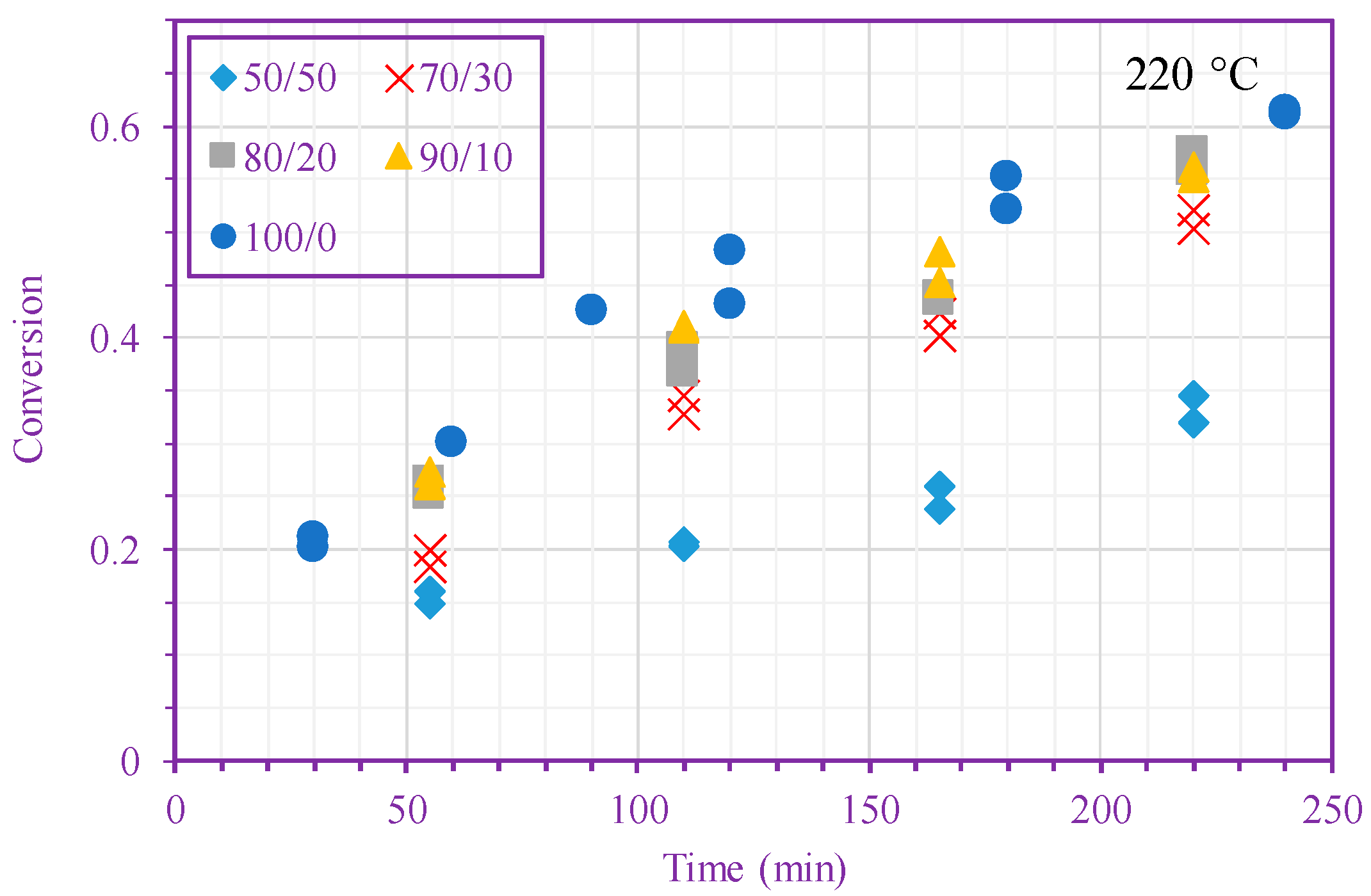
Figure 3.
Measured and predicted n-BA conversion at 200 °C (n-BA/n-heptane volume ratios 50/50 and 70/30 (top), and 80/20, 90/10, and 100/0 (bottom)).
Figure 3.
Measured and predicted n-BA conversion at 200 °C (n-BA/n-heptane volume ratios 50/50 and 70/30 (top), and 80/20, 90/10, and 100/0 (bottom)).
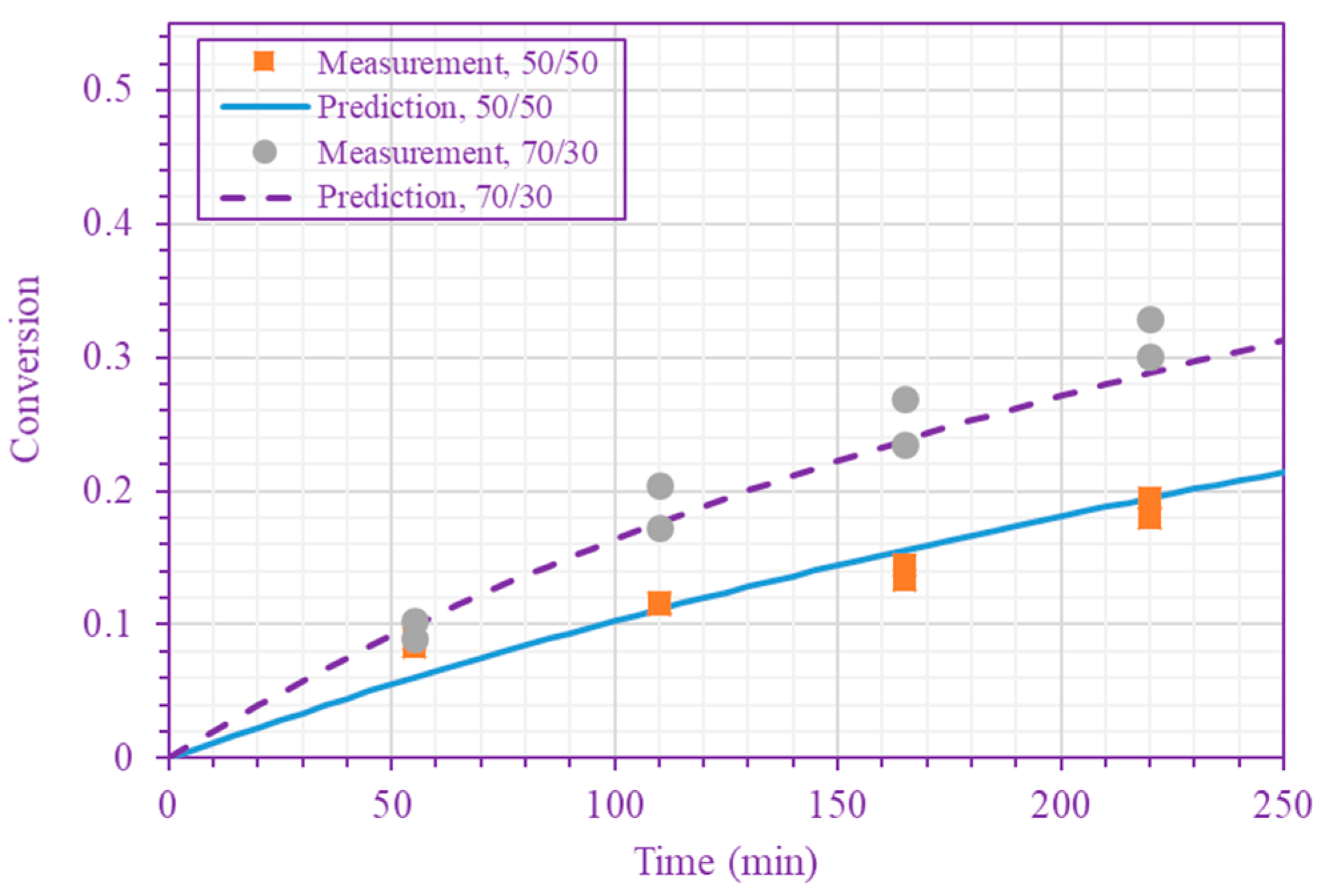
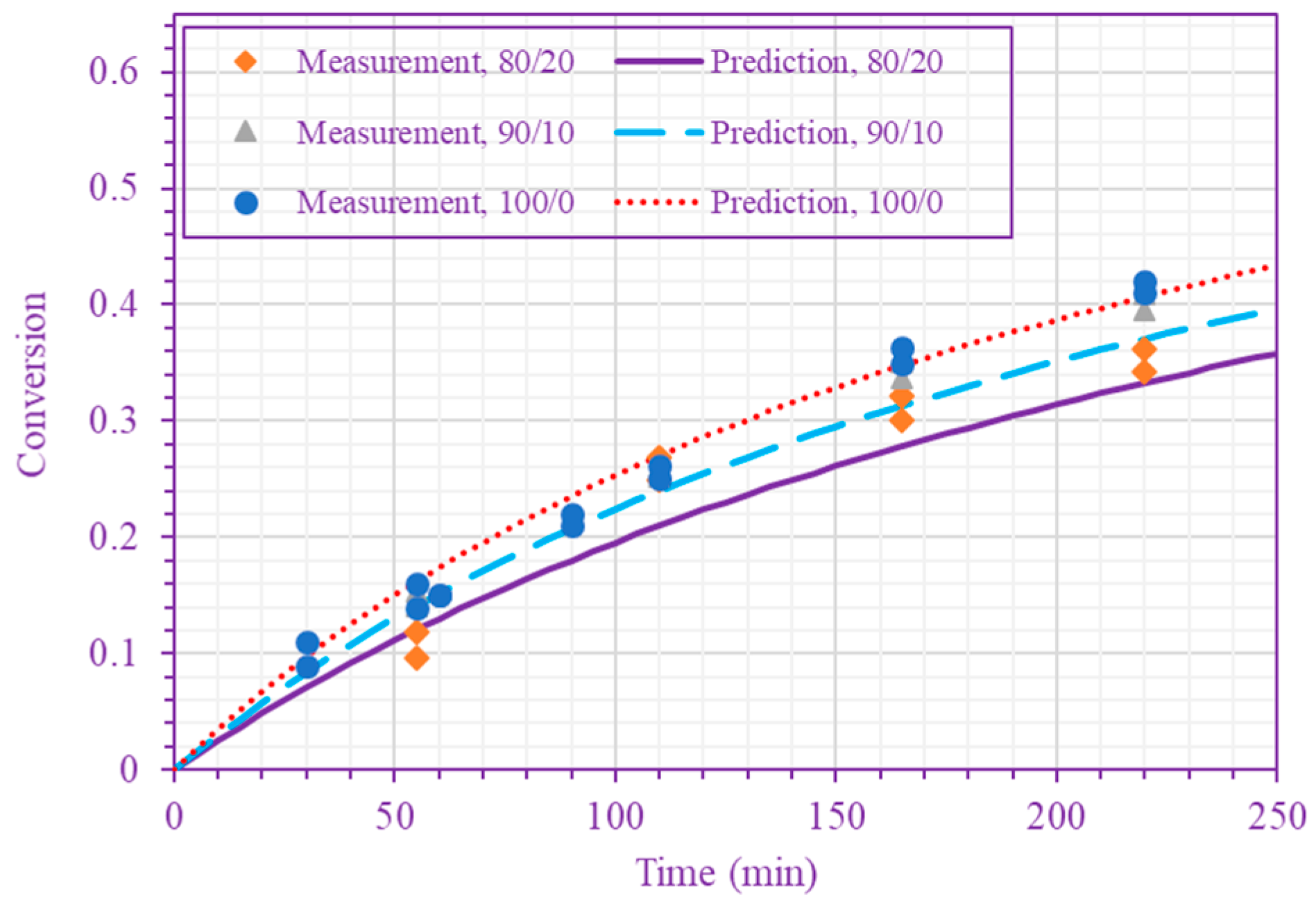
Figure 4.
Measured and predicted n-BA conversion at 220 °C ( n-BA/n-heptane volume ratios 50/50 and 70/30 (top), and 80/20, 90/10, and 100/0 (bottom)).
Figure 4.
Measured and predicted n-BA conversion at 220 °C ( n-BA/n-heptane volume ratios 50/50 and 70/30 (top), and 80/20, 90/10, and 100/0 (bottom)).
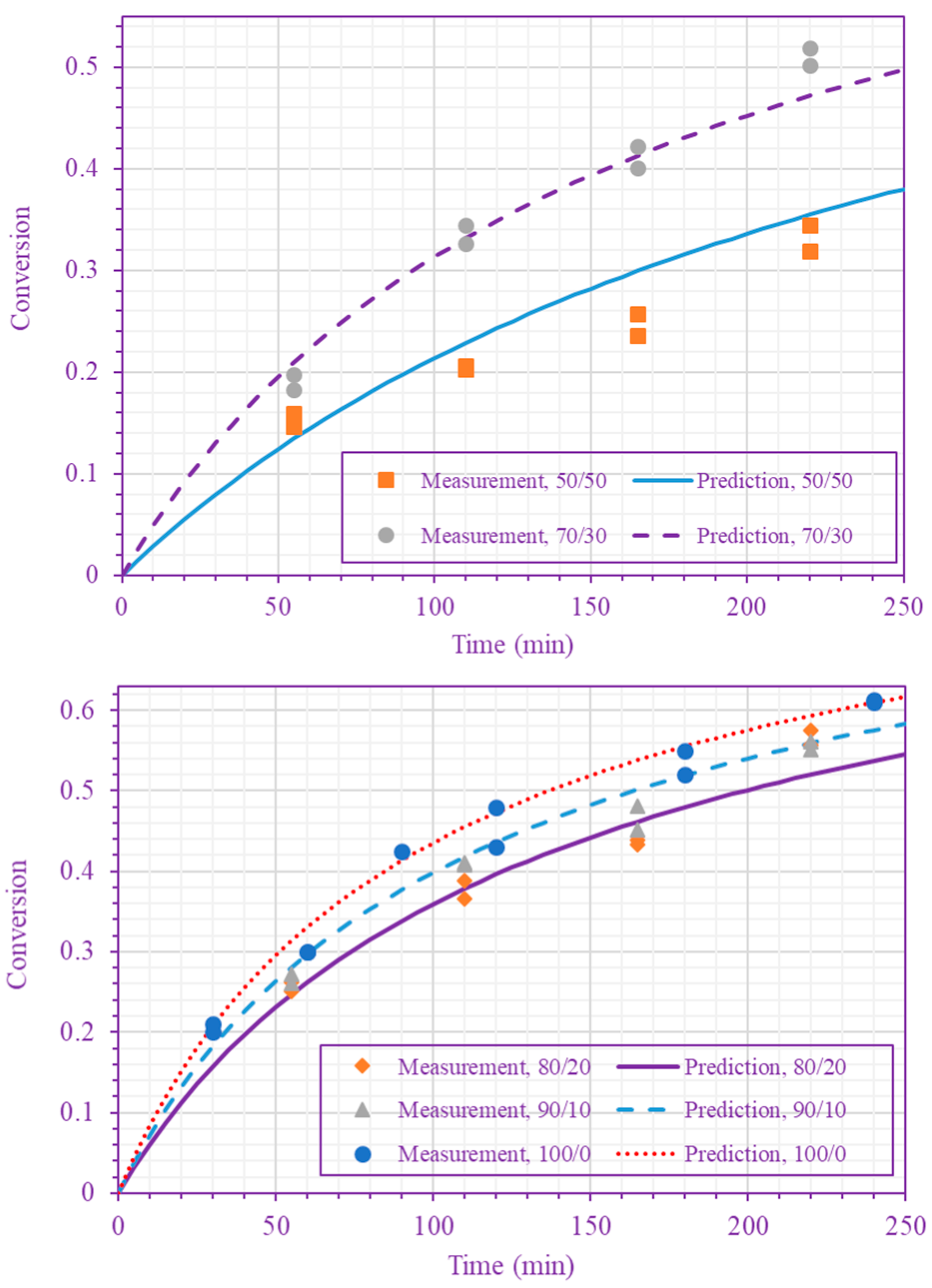
Figure 5.
Sensitivity of the model predictions to changes in n (top figure), Z (middle figure), and
(bottom figure) (50/50 n-BA/n-heptane ratio; 220 °C). , and .
Figure 5.
Sensitivity of the model predictions to changes in n (top figure), Z (middle figure), and
(bottom figure) (50/50 n-BA/n-heptane ratio; 220 °C). , and .
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Riazi, H.; Shamsabadi, A.A.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes 2018, 6, 3. https://doi.org/10.3390/pr6010003
AMA Style
Riazi H, Shamsabadi AA, Corcoran P, Grady MC, Rappe AM, Soroush M. On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes. 2018; 6(1):3. https://doi.org/10.3390/pr6010003
Chicago/Turabian StyleRiazi, Hossein, Ahmad Arabi Shamsabadi, Patrick Corcoran, Michael C. Grady, Andrew M. Rappe, and Masoud Soroush. 2018. "On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization" Processes 6, no. 1: 3. https://doi.org/10.3390/pr6010003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.