
Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling

Abstract
:1. Introduction
- (i)
- Two monomers react and form a singlet diradical
- (ii)
- The singlet diradical then undergoes intersystem crossing to form a triplet diradical:
- (iii)
- The triplet diradical finally reacts with a third monomer, leading to the formation of two mono-radicals:
2. Mathematical Modeling
2.1. Reaction Mechanisms
2.2. Rate Equations
2.3. Batch Reactor Model
3. Experimental and Analytical Procedures
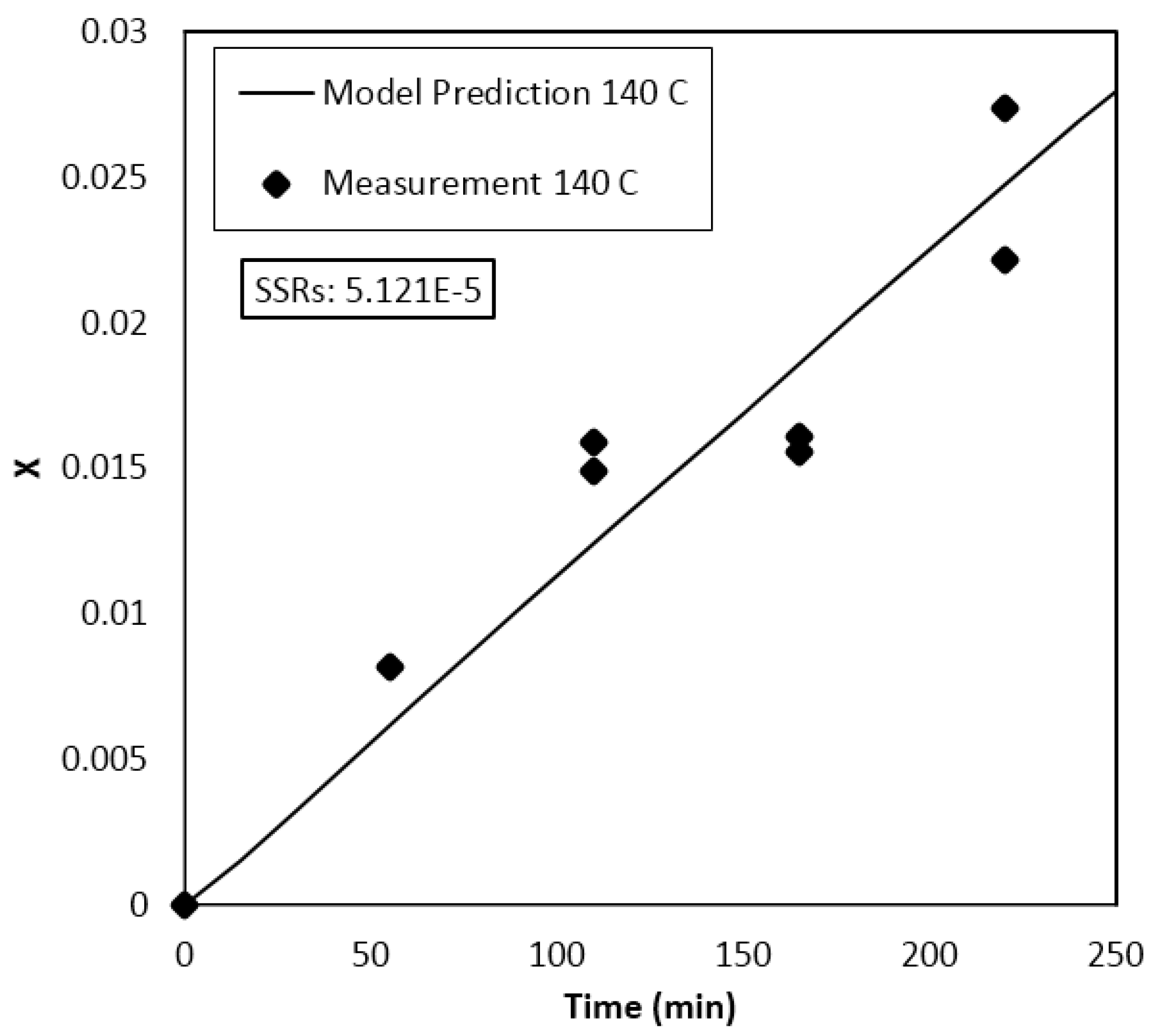
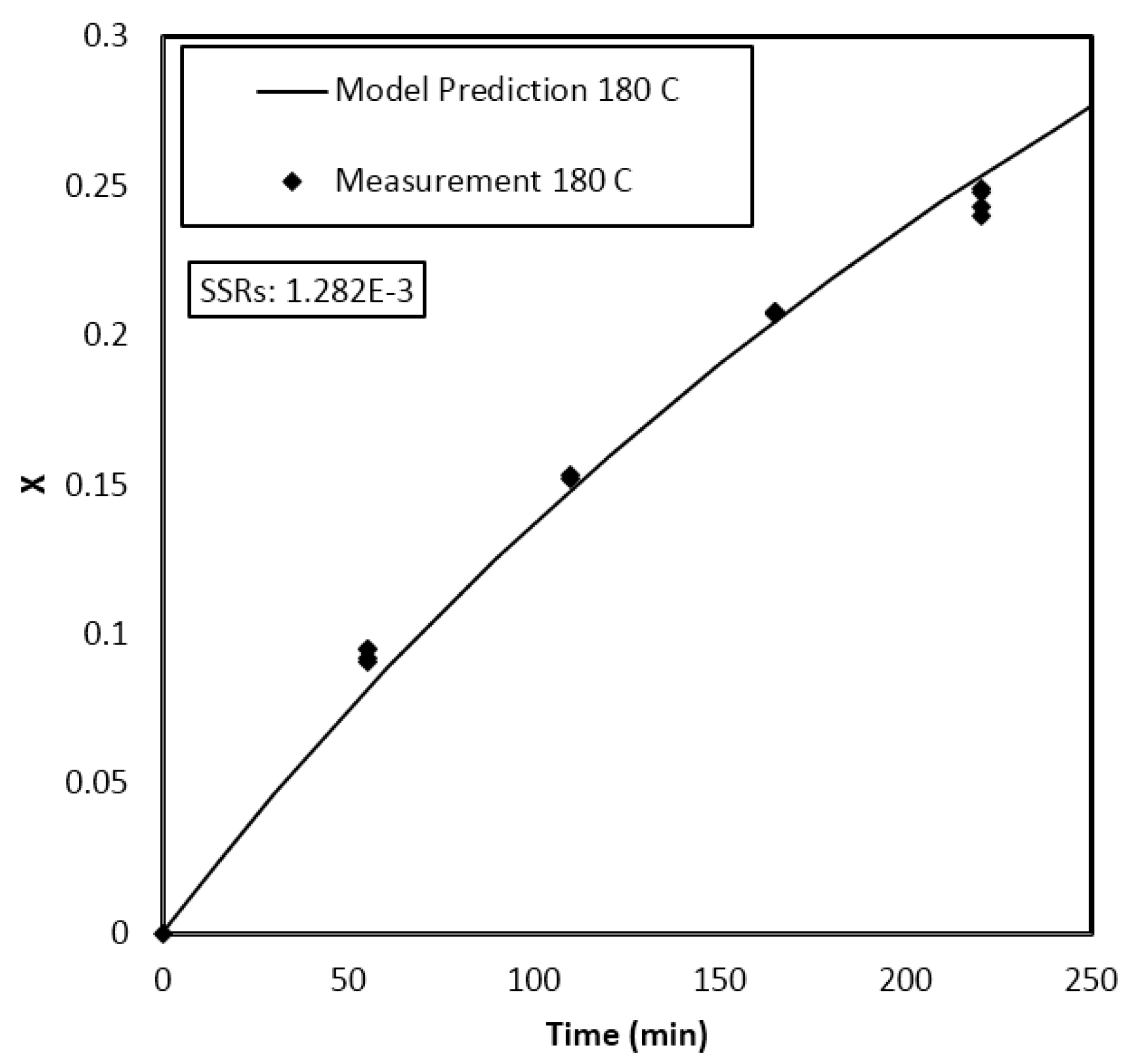
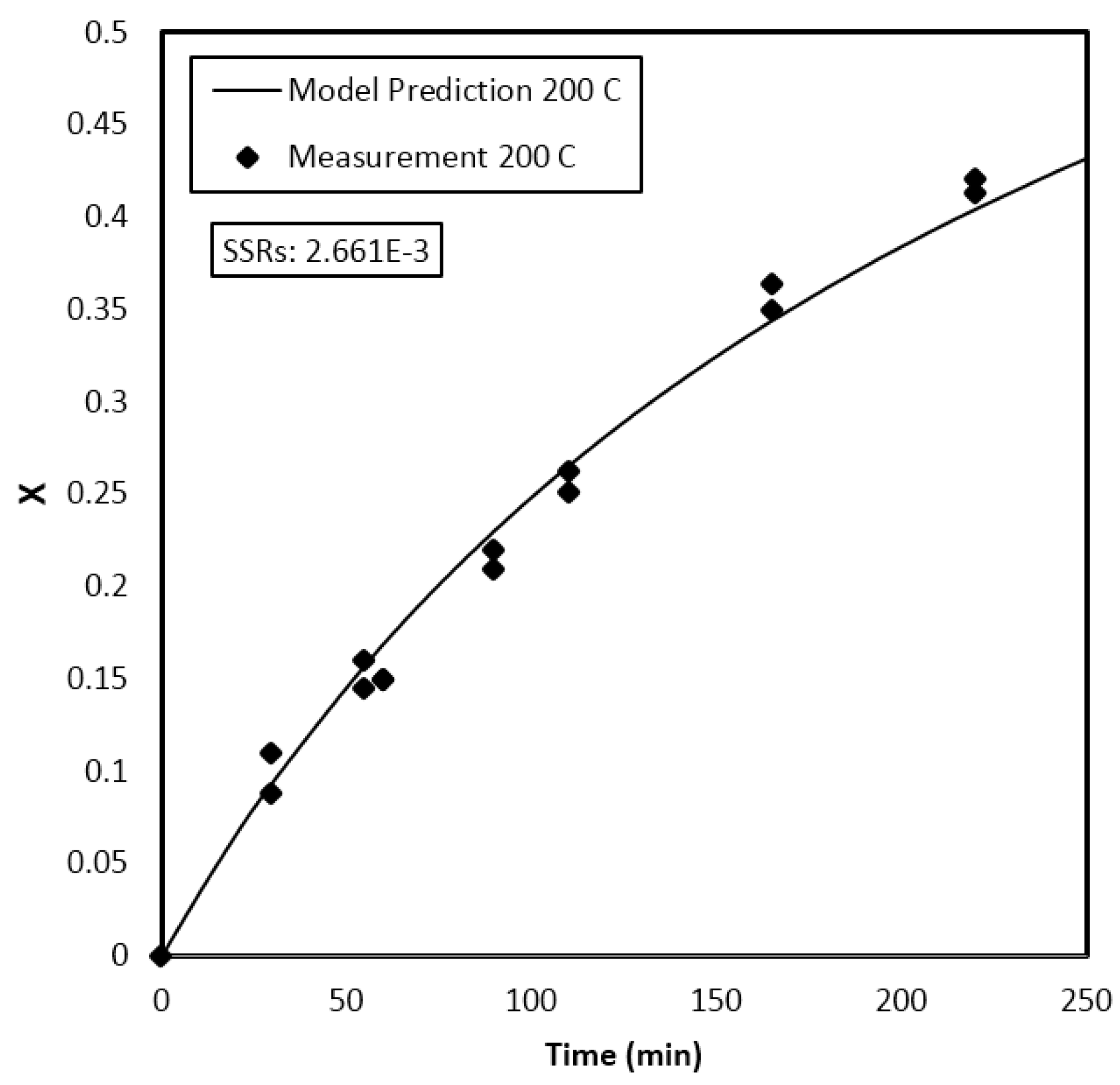
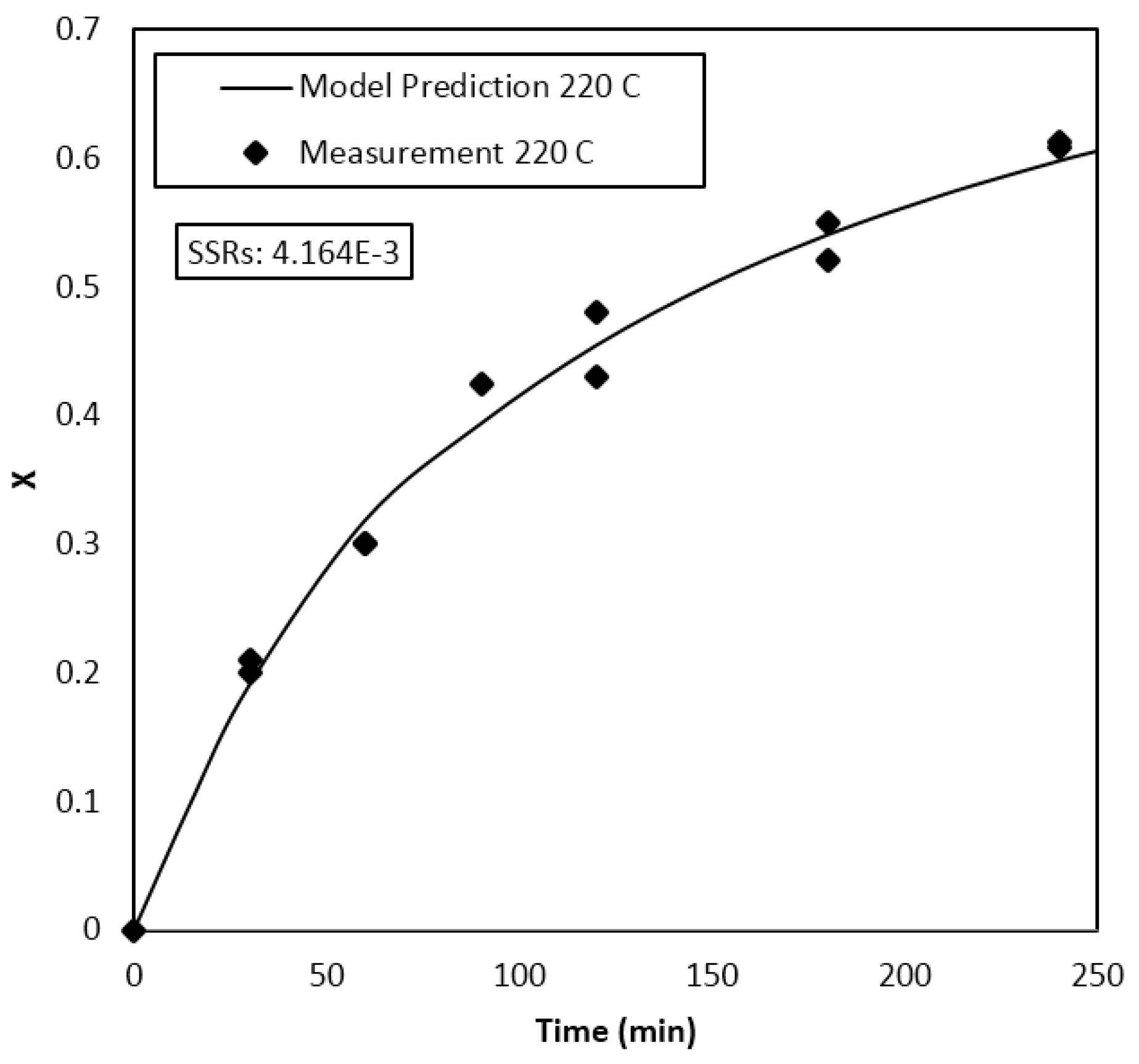
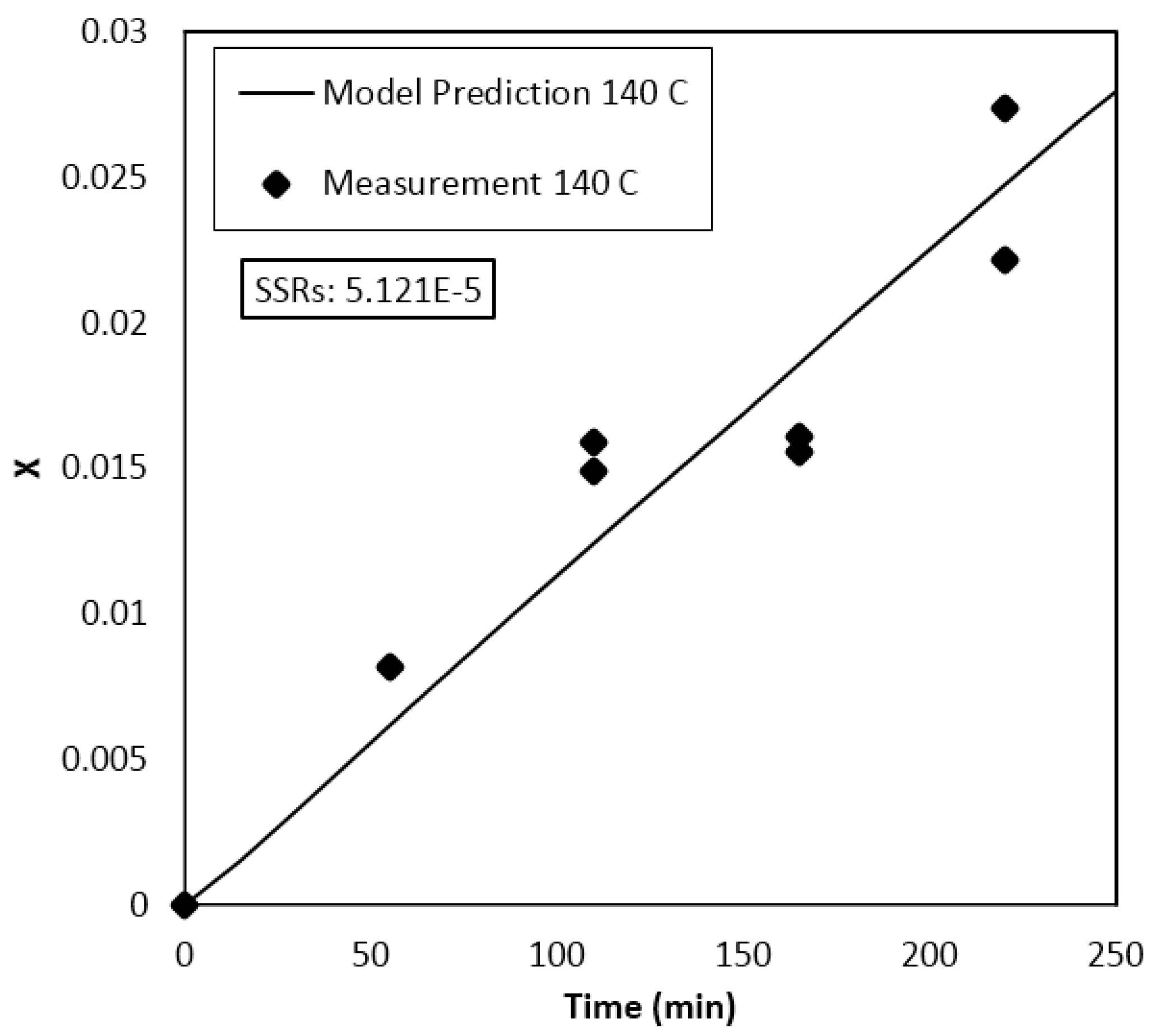
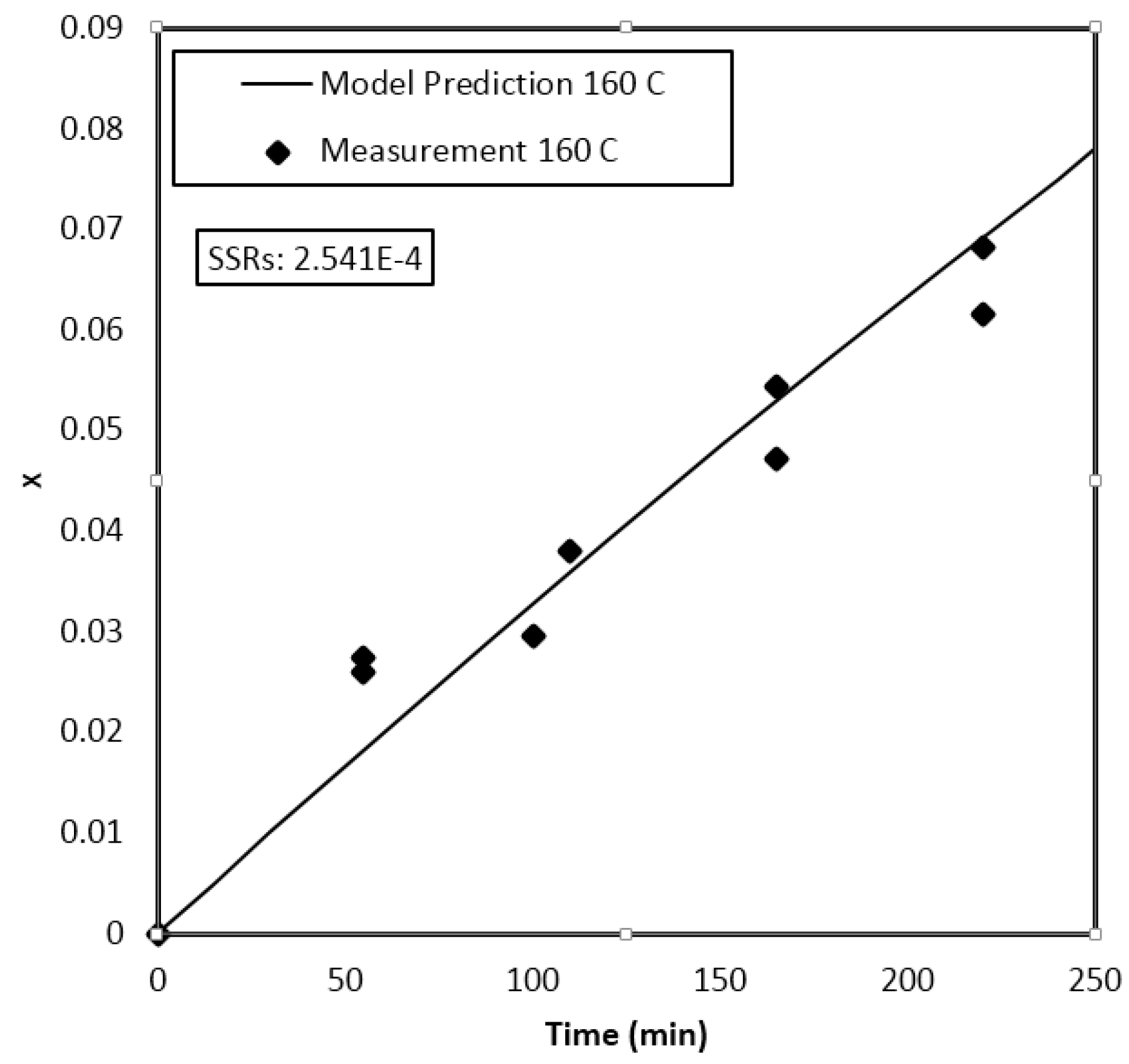
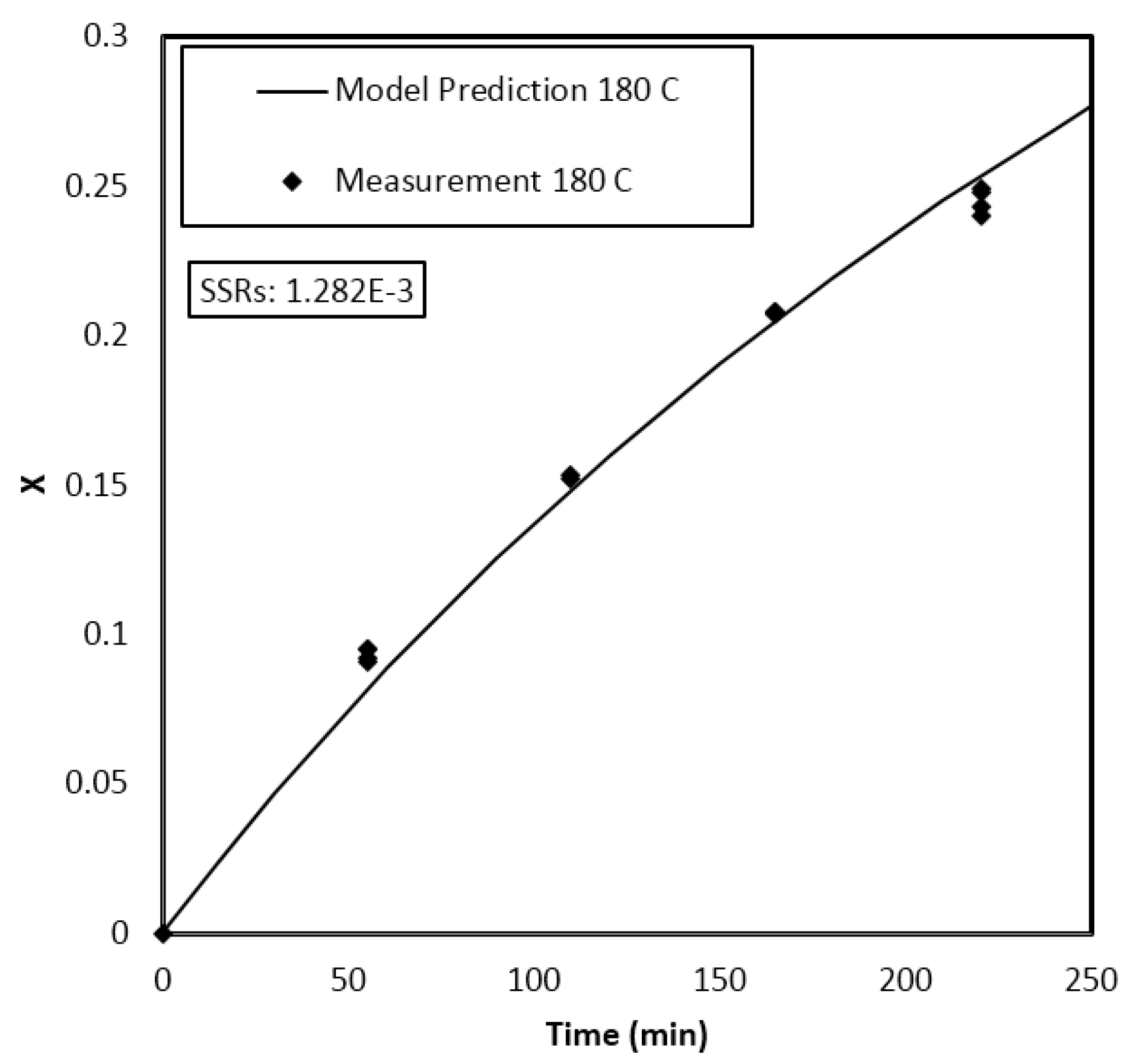
4. Results and Discussion
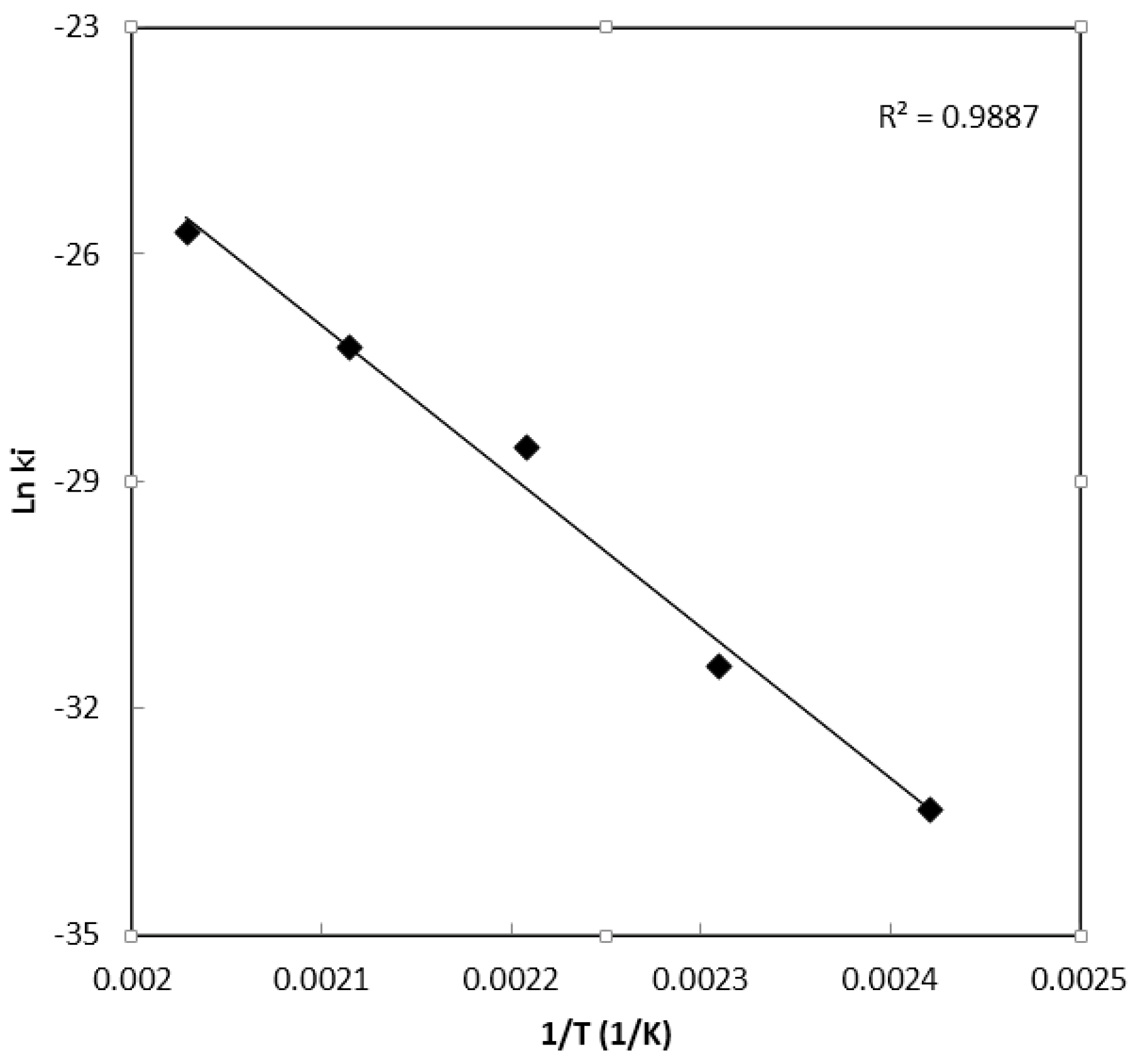
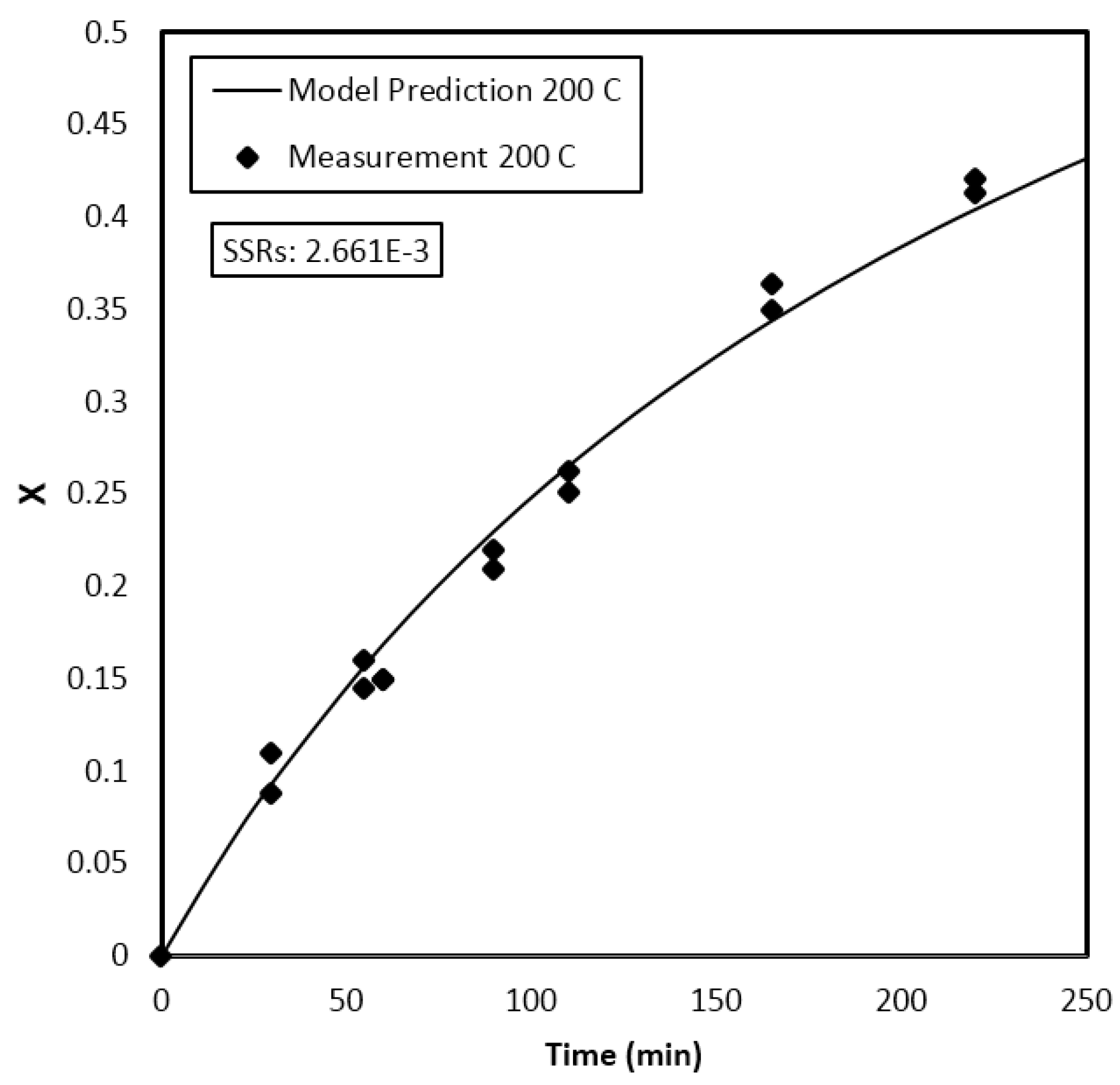
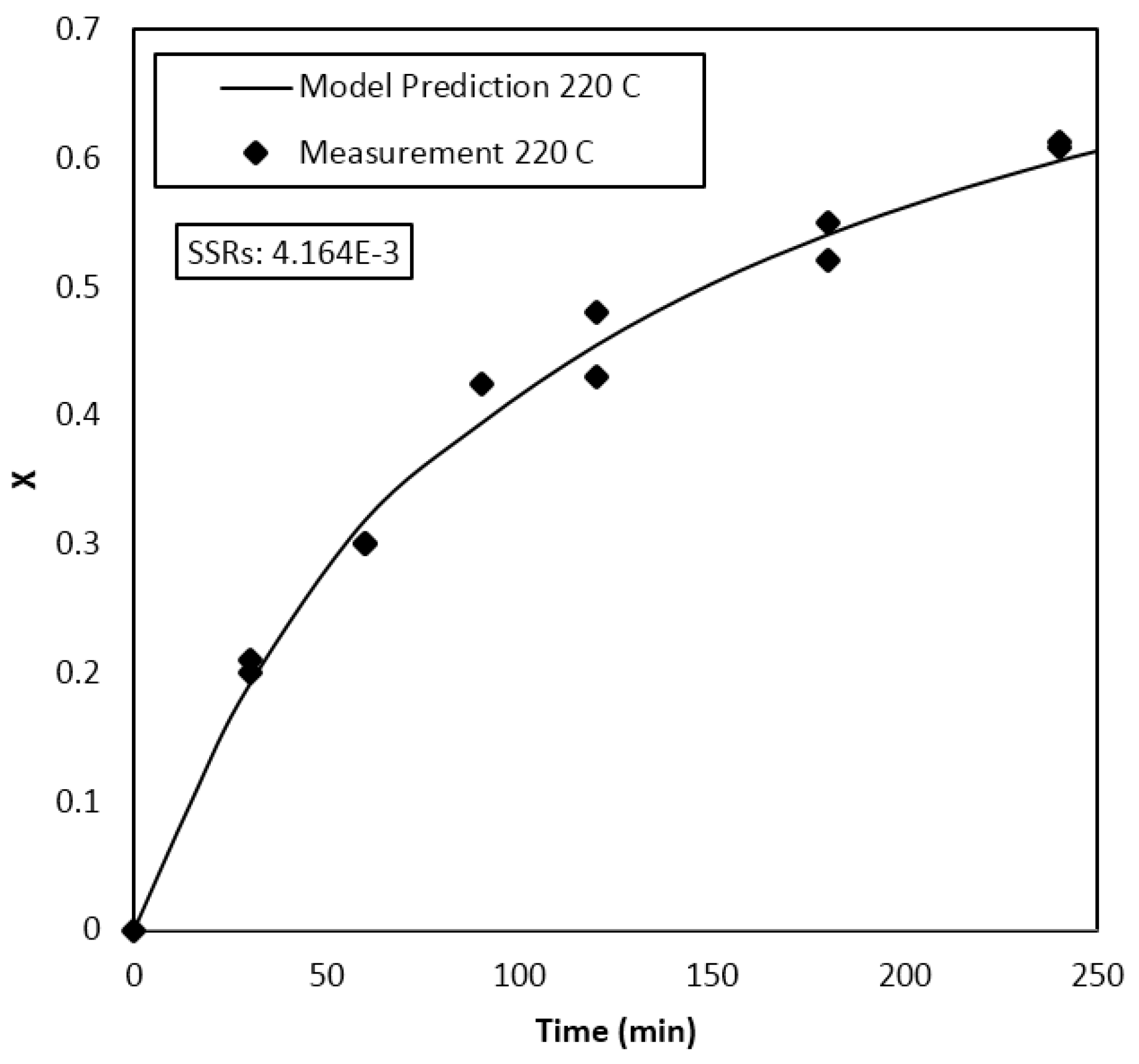
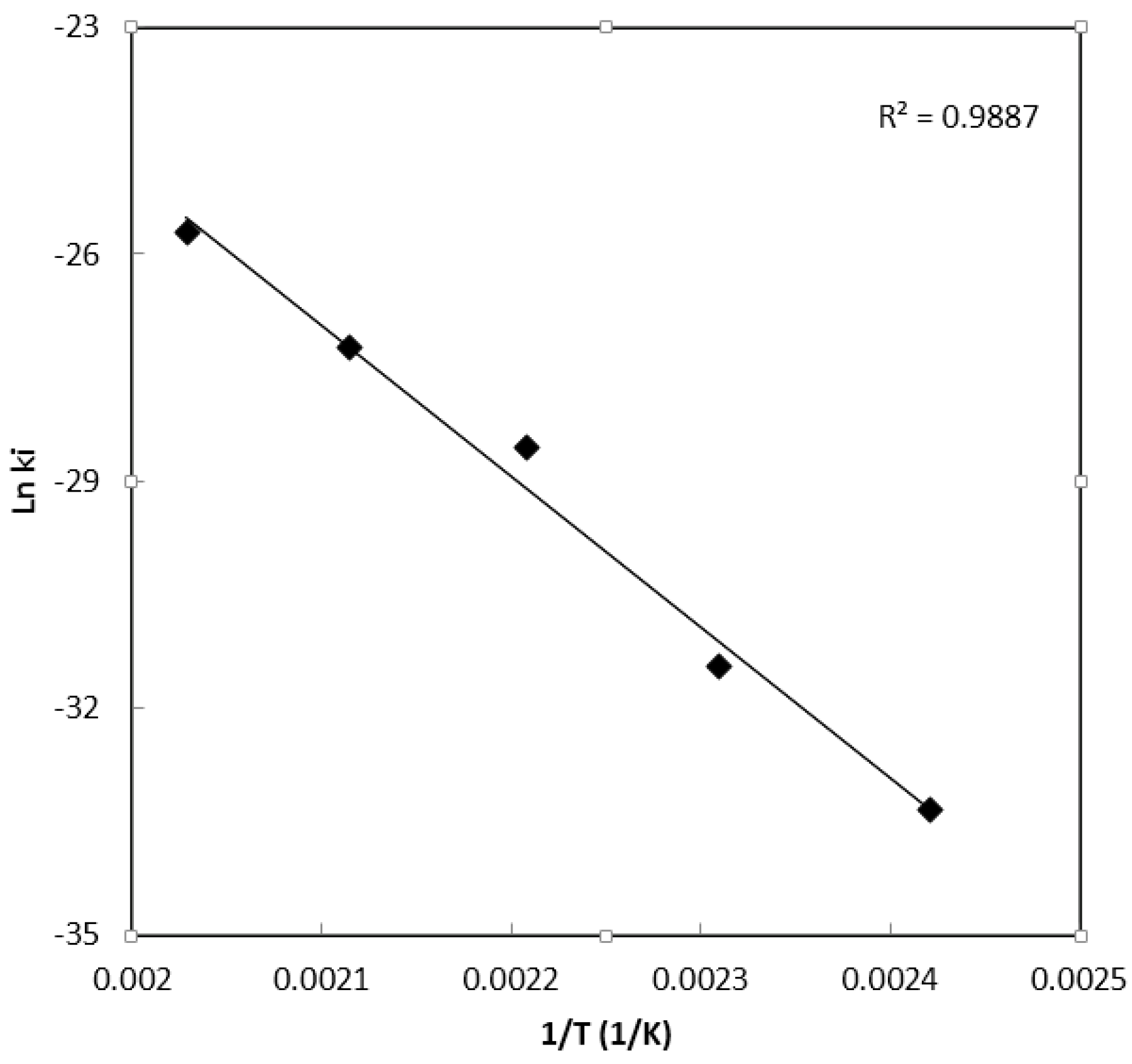
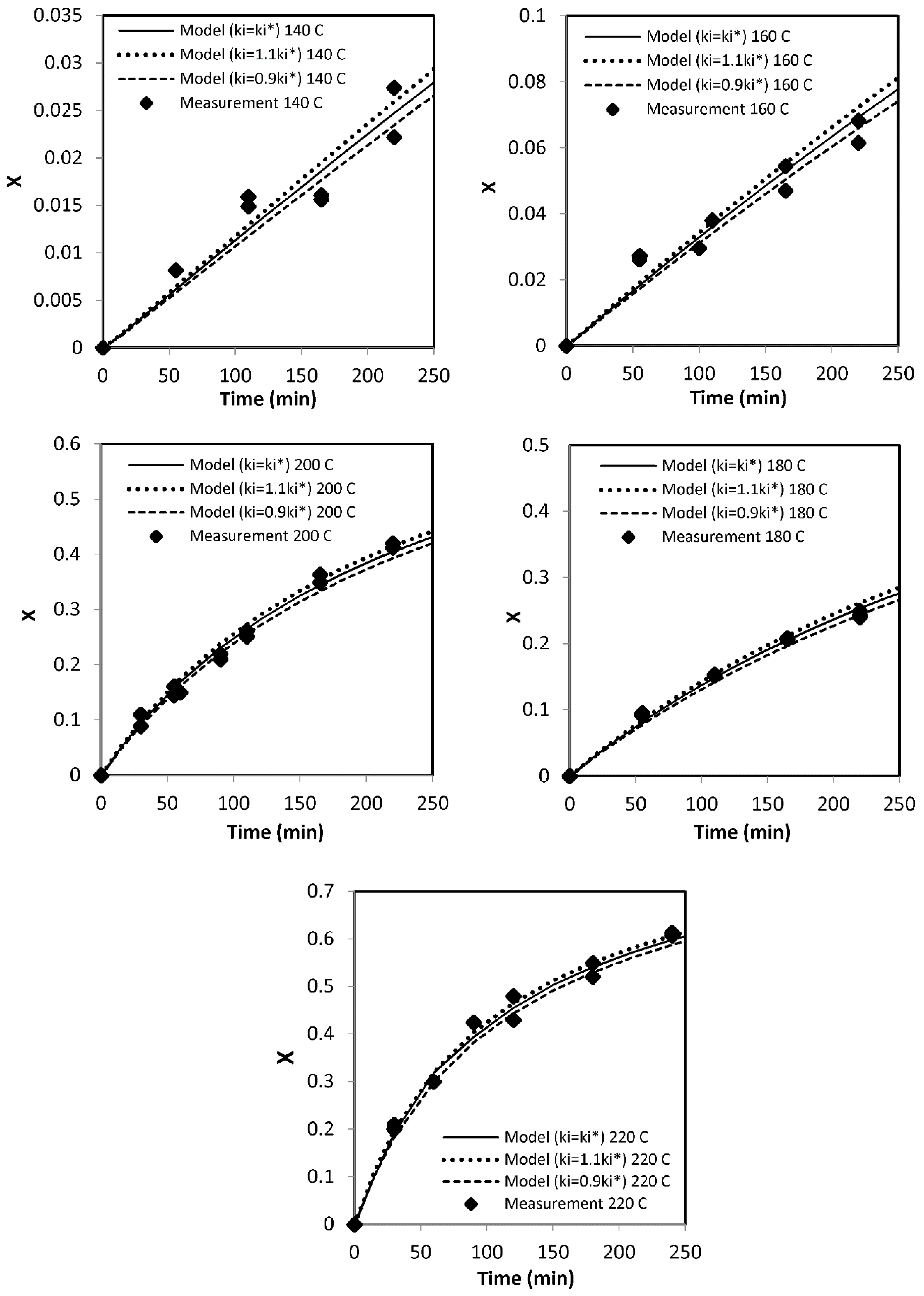
Parameter Estimation
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix: Reaction Rate Equations
References
- Consumer and Commercial Products, Group IV: Control Techniques Guidelines in Lieu of Regulations for Miscellaneous Metal Products Coatings, Plastic Parts Coatings, Auto and Light-Duty Truck Assembly Coatings, Fiberglass Boat Manufacturing Materials, and Miscellaneous Industrial Adhesives; EPA-HQ-OAR-2008-0411; FRL-8725-9; Environmental Protection Agency: Washington, DC, USA, 2008; Volume 73, No. 195; pp. 58481–58490.
- Economic Impact and Regulatory Flexibility Analyses of the Final Architectural Coatings VOC Rule; EPA-452/R-98-002; United States Environmental Protection Agency, Office of Air Quality Planning and Standards: Washington, DC, USA, 1998.
- Campbell, J.D.; Teymour, F.; Morbidelli, M. High temperature free radical polymerization. 1. Investigation of continuous styrene polymerization. Macromolecules 2003, 36, 5491–5501. [Google Scholar] [CrossRef]
- Wang, W.; Hutchinson, R.A. Recent advances in the study of high-temperature free radical acrylic solution copolymerization. Macromol. React. Eng. 2008, 2, 199–214. [Google Scholar] [CrossRef]
- Grady, M.C.; Simonsick, W.J.; Hutchinson, R.A. Studies of higher temperature polymerization of n-butyl methacrylate and n-butyl acrylate. Macromol. Symp. 2002, 182, 149–168. [Google Scholar] [CrossRef]
- Soroush, M.; Grady, M.C.; Kalfas, G.A. Free-radical polymerization at higher temperatures: Systems impacts of secondary reactions. Comput. Chem. Eng. 2008, 32, 2155–2167. [Google Scholar] [CrossRef]
- Quan, C.L.; Soroush, M.; Grady, M.C.; Hansen, J.E.; Simonsick, W.J. High-temperature homopolymerization of ethyl acrylate and n-butyl acrylate: Polymer characterization. Macromolecules 2005, 38, 7619–7628. [Google Scholar] [CrossRef]
- Chiefari, J.; Jeffery, J.; Mayadunne, R.T.A.; Moad, G.; Rizzardo, E.; Thang, S.H. Chain transfer to polymer: A convenient route to macromonomers. Macromolecules 1999, 32, 7700–7702. [Google Scholar] [CrossRef]
- Rantow, F.S.; Soroush, M.; Grady, M.C.; Kalfas, G.A. Spontaneous polymerization and chain microstructure evolution in high-temperature solution polymerization of n-butyl acrylate. Polymer 2006, 47, 1423–1435. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of the self-initiation mechanism in thermal polymerization of methyl acrylate. J. Phys. Chem. A 2009, 113, 10787–10794. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Self-initiation mechanism in spontaneous thermal polymerization of ethyl and n-butyl acrylate: A theoretical study. J. Phys. Chem. A 2010, 114, 7975–7983. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Tao, J.; Grady, M.C.; Soroush, M.; Rappe, A.M. Modeling Spin-Forbidden Monomer Self-initiation Reactions in Free-Radical Polymerization of Acrylates and Methacrylates. J. Phys. Chem. A 2014, 118, 9310–9318. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational Evidence for Self-Initiation in Spontaneous High-Temperature Polymerization of Methyl Methacrylate. J. Phys. Chem. A 2011, 115, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Kalfas, G.; Petkovska, V.I.; Bruni, C.; Grady, M.C.; Soroush, M. Experimental study of the spontaneous thermal homopolymerization of methyl and n-butyl acrylate. J. Appl. Polym. Sci. 2010, 118, 1898–1909. [Google Scholar] [CrossRef]
- Arzamendi, G.; Plessis, C.; Leiza, J.R.; Asua, J.M. Effect of the intramolecular chain transfer to polymer on PLP/SEC experiments of alkyl acrylates. Macromol. Theory Simul. 2003, 12, 315–324. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Buback, M.; Hesse, P. Determination of intramolecular chain transfer and midchain radical propagation rate coefficients for butyl acrylate by pulsed laser polymerization. Macromolecules 2007, 40, 8631–8641. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.D.; Manders, B.G.; Odriscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization. 1. Propagation rate coefficient for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Olaj, O.F.; Russell, G.T.; Schweer, J.; vanHerk, A.M. Critically evaluated rate coefficients for free-radical polymerization. 2. Propagation rate coefficients for methyl methacrylate. Macromol. Chem. Phys. 1997, 198, 1545–1560. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Buback, M.; Hesse, P. A novel approach for investigation of chain transfer events by pulsed laser polymerization. Macromol. Chem. Phys. 2011, 212, 699–707. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Gunzler, F.; Junkers, T. Pushing the Limit: Pulsed Laser Polymerization of n-Butyl Acrylate at 500 Hz. Macromolecules 2008, 41, 8971–8973. [Google Scholar] [CrossRef]
- Lyons, R.A.; Hutovic, J.; Piton, M.C.; Christie, D.I.; Clay, P.A.; Manders, B.G.; Kable, S.H.; Gilbert, R.G. Pulsed-laser polymerization measurements of the propagation rate coefficient for butyl acrylate. Macromolecules 1996, 29, 1918–1927. [Google Scholar] [CrossRef]
- Beuermann, S.; Paquet, D.A.; McMinn, J.H.; Hutchinson, R.A. Determination of free-radical propagation rate coefficients of butyl, 2-ethylhexyl, and dodecyl acrylates by pulsed-laser polymerization. Macromolecules 1996, 29, 4206–4215. [Google Scholar] [CrossRef]
- Busch, M.; Wahl, A. The significance of transfer reactions in pulsed laser polymerization experiments. Macromol. Theory Simul. 1998, 7, 217–224. [Google Scholar] [CrossRef]
- Ahmad, N.M.; Heatley, F.; Lovell, P.A. Chain transfer to polymer in free-radical solution polymerization of n-butyl acrylate studied by NMR spectroscopy. Macromolecules 1998, 31, 2822–2827. [Google Scholar] [CrossRef]
- Peck, A.N.F.; Hutchinson, R.A. Secondary reactions in the high-temperature free radical polymerization of butyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Heatley, F.; Lovell, P.A.; Yamashita, T. Chain transfer to polymer in free-radical solution polymerization of 2-ethylhexyl acrylate studied by NMR spectroscopy. Macromolecules 2001, 34, 7636–7641. [Google Scholar] [CrossRef]
- Stickler, M.; Meyerhoff, G. The spontaneous thermal polymerization of mthyl-methacrylate: 5. Experimental-study and computer-simulation of the high conversion reaction at 130 °C. Polymer 1981, 22, 928–933. [Google Scholar] [CrossRef]
- Blavier, L.; Villermaux, J. Free-radical polymerization engineering: 2. Modeling of homogeneous polymerization of styrene in a batch reactor, influence of initiator. Chem. Eng. Sci. 1984, 39, 101–110. [Google Scholar] [CrossRef]
- Rier, T.; Srinivasan, S.; Soroush, M.; Kalfas, G.A.; Grady, M.C.; Rappe, A.M. Macroscopic mechanistic modeling and optimization of a self-initiated high-temperature polymerization reactor. In Proceedings of the 2011 American Control Conference, San Francisco, CA, USA, 29 June 2011–1 July 2011; pp. 3071–3076.
- Asua, J.M.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Gilbert, R.G.; Hutchinson, R.A.; Leiza, J.R.; Nikitin, A.N. Critically evaluated rate coefficients for free-radical polymerization, 5—Propagation rate coefficient for butyl acrylate. Macromol. Chem. Phys. 2004, 205, 2151–2160. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A. Effect of intramolecular transfer to polymer on stationary free radical polymerization of alkyl acrylates, 2—Improved consideration of termination. Macromol. Theory Simul. 2006, 15, 128–136. [Google Scholar] [CrossRef]
- Wang, W.; Nikitin, A.N.; Hutchinson, R.A. Consideration of macromonomer reactions in n-butyl acrylate free radical polymerization. Macromol. Rapid Commun. 2009, 30, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, A.N.; Hutchinson, R.A.; Kalfas, G.A.; Richards, J.R.; Bruni, C. The effect of intramolecular transfer to polymer on stationary free-radical polymerization of alkyl acrylates, 3-Consideration of solution polymerization up to high conversions. Macromol. Theory Simul. 2009, 18, 247–258. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Wang, W.; Kalfas, G.A.; Richards, J.R.; Bruni, C. Effect of intramolecular transfer to polymer on stationary free-radical polymerization of alkyl acrylates, 5-Consideration of solution polymerization up to high temperatures. Macromol. React. Eng. 2010, 4, 691–706. [Google Scholar] [CrossRef]
- Yu, X. Kinetic Modeling of Acrylate Polymerization at High Temperature. Ph.D. Thesis, Northwestern University, Evanston, IL, USA, 2008. [Google Scholar]
- Kruse, T.M.; Woo, O.S.; Broadbelt, L.J. Detailed mechanistic modeling of polymer degradation: Application to polystyrene. Chem. Eng. Sci. 2001, 56, 971–979. [Google Scholar] [CrossRef]
- Zhu, S.P. Modeling of molecular weight development in atom transfer radical polymerization. Macromol. Theory Simul. 1999, 8, 29–37. [Google Scholar] [CrossRef]
- Villermaux, J.; Blavier, L. Free-radical polymerization engineering. 1. A new method for modeling free-radical homogeneous polymerization reactions. Chem. Eng. Sci. 1984, 39, 87–99. [Google Scholar] [CrossRef]
- Kim, D.M.; Iedema, P.D. Modeling of branching density and branching distribution in low-density polyethylene polymerization. Chem. Eng. Sci. 2008, 63, 2035–2046. [Google Scholar] [CrossRef]
- Benyahia, B.; Latifi, M.A.; Fonteix, C.; Pla, F.; Nacef, S. Emulsion copolymerization of styrene and butyl acrylate in the presence of a chain transfer agent. Part 1: Modelling and experimentation of batch and fedbatch processes. Chem. Eng. Sci. 2010, 65, 850–869. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.C. Model discrimination and selection in evolutionary optimization of batch processes with tendency models. Comput. Aided Chem. Eng. 2005, 20, 463–468. [Google Scholar]
- Fotopoulos, J.; Georgakis, C.; Stenger, H.G. Use of tendency models and their uncertainty in the design of state estimators for batch reactors. Chem. Eng. Process. 1998, 37, 545–558. [Google Scholar] [CrossRef]
- Fotopoulos, J.; Georgakis, C.; Stenger, H.G. Effect of process-model mismatch on the optimization of the catalytic epoxidation of oleic acid using tendency models. Chem. Eng. Sci. 1996, 51, 1899–1908. [Google Scholar] [CrossRef]
- Filippi, C.; Greffe, J.L.; Bordet, J.; Villermaux, J.; Barnay, J.L.; Bonte, P.; Georgakis, C. Tendency modeling of semibatch reactors for optimization and control. Chem. Eng. Sci. 1986, 41, 913–920. [Google Scholar] [CrossRef]
- Martinez, E.C.; Cristaldi, M.D.; Grau, R.J. Design of dynamic experiments in modeling for optimization of batch processes. Ind. Eng. Chem. Res. 2009, 48, 3453–3465. [Google Scholar] [CrossRef]
- Mueller, P.A.; Richards, J.R.; Congalidis, J.P. Polymerization reactor modeling in industry. Macromol. React. Eng. 2011, 5, 261–277. [Google Scholar] [CrossRef]
- Maeder, S.; Gilbert, R.G. Measurement of transfer constant for butyl acrylate free-radical polymerization. Macromolecules 1998, 31, 4410–4418. [Google Scholar] [CrossRef]
- Moad, G.; Solomon, D.H. The Chemistry of Free Radical Polymerization; Elsevier: Oxford, UK, 1995. [Google Scholar]
- Derboven, P.; D’hooge, D.R.; Reyniers, M.-F.; Marin, G.B.; Barner-Kowollik, C. The long and the short of radical polymerization. Macromolecules 2015, 48, 492–501. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M.; Hesse, P.; Sergeeva, T. Termination and transfer kinetics of butyl acrylate radical polymerization studies via SP-PLP-EPR. Macromolecules 2010, 43, 4023–4031. [Google Scholar] [CrossRef]
- Hulburt, H.M.; Katz, S. Some problems in particle technology. Chem. Eng. Sci. 1964, 19, 555–574. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
| Parameter | Frequency Factor | Activation Energy (kJ.mol−1) | Ref. | |
|---|---|---|---|---|
| 2.21 × 107 | L·mol−1·s−1 | 17.9 | [30] | |
| 1.20 × 106 | L·mol−1·s−1 | 28.6 | [49] | |
| 7.41 × 107 | s−1 | 32.7 | [33] | |
| 2.90 × 105 | L·mol−1·s−1 | 32.6 | [47] | |
| 3.89 × 109 | L·mol−1·s−1 | 8.4 | [31] | |
| 5.30 × 109 | L·mol−1·s−1 | 19.6 | [34] | |
| 1.49 × 109 | s−1 | 63.9 | [34] | |
| 4.01 × 103 | L·mol−1·s−1 | 29.0 | [15] | |
| 1.07 × 102 | 35.4 | [34] | ||
| Temperature | This Work | Theoretical [11] | Theoretical [12] |
| 413 | 3.30 × 10−15 | 2.81 × 10−18 | 1.04 × 10−14 |
| 433 | 2.20 × 10−14 | 2.86 × 10−17 | 4.72 × 10−14 |
| 453 | 4.00 × 10−13 | 2.37 × 10−16 | 1.95 × 10−13 |
| 473 | 1.50 × 10−12 | 1.64 × 10−15 | 7.11 × 10−13 |
| 493 | 6.80 × 10−12 | 9.74 × 10−15 | 2.34 × 10−12 |
| Parameter | This Work | Theoretical [11] | Theoretical [12] |
| 165.51 ± 4.52 | 172.50 | 115.00 | |
| 14.86 ± 1.20 | 9.68 | 1.38 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arabi Shamsabadi, A.; Moghadam, N.; Srinivasan, S.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling. Processes 2016, 4, 15. https://doi.org/10.3390/pr4020015
Arabi Shamsabadi A, Moghadam N, Srinivasan S, Corcoran P, Grady MC, Rappe AM, Soroush M. Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling. Processes. 2016; 4(2):15. https://doi.org/10.3390/pr4020015
Chicago/Turabian StyleArabi Shamsabadi, Ahmad, Nazanin Moghadam, Sriraj Srinivasan, Patrick Corcoran, Michael C. Grady, Andrew M. Rappe, and Masoud Soroush. 2016. "Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling" Processes 4, no. 2: 15. https://doi.org/10.3390/pr4020015