
Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory
2.1. System Contributions to Band Broadening
2.2. Injector Contributions
2.3. Exponential Decay Flow Paths
2.4. Connecting Tubing
2.5. Detector Contributions
2.6. Calculating Peak Variance
3. Experimental Section
3.1. Reagents and Materials
3.2. Direct Measurement Instrumentation and Techniques
3.3. Flow Modeling Simulations
4. Results and Discussion
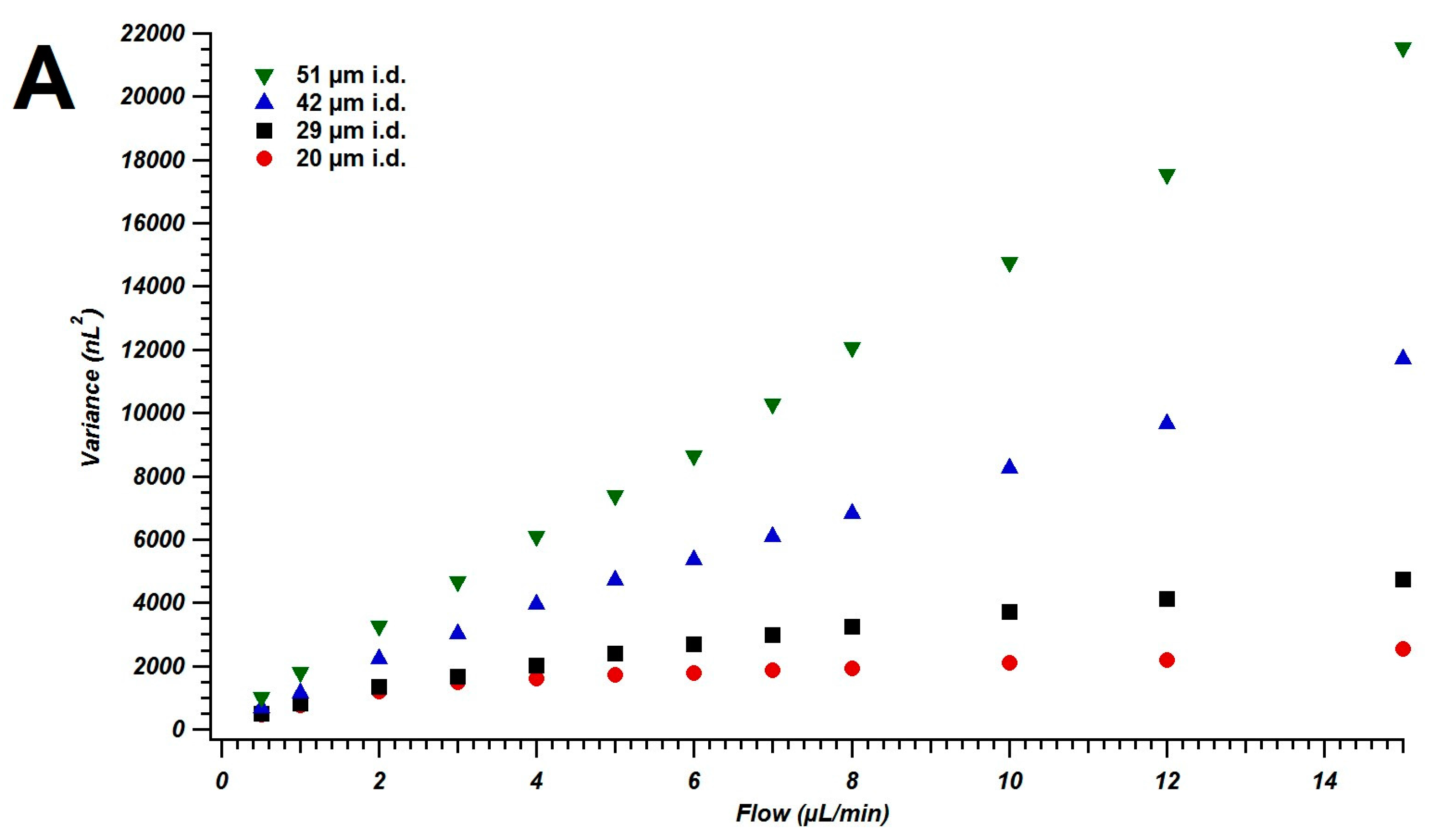
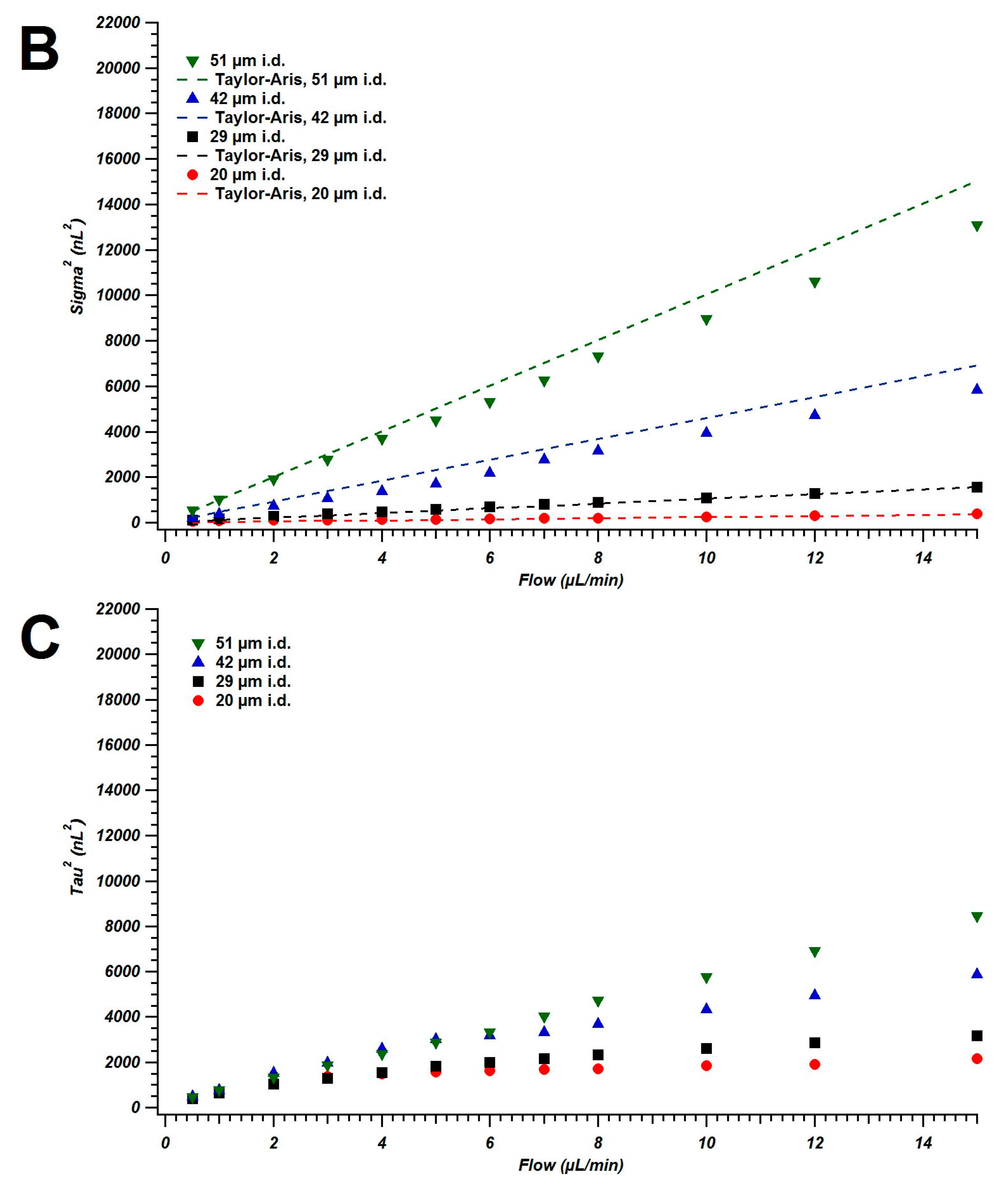
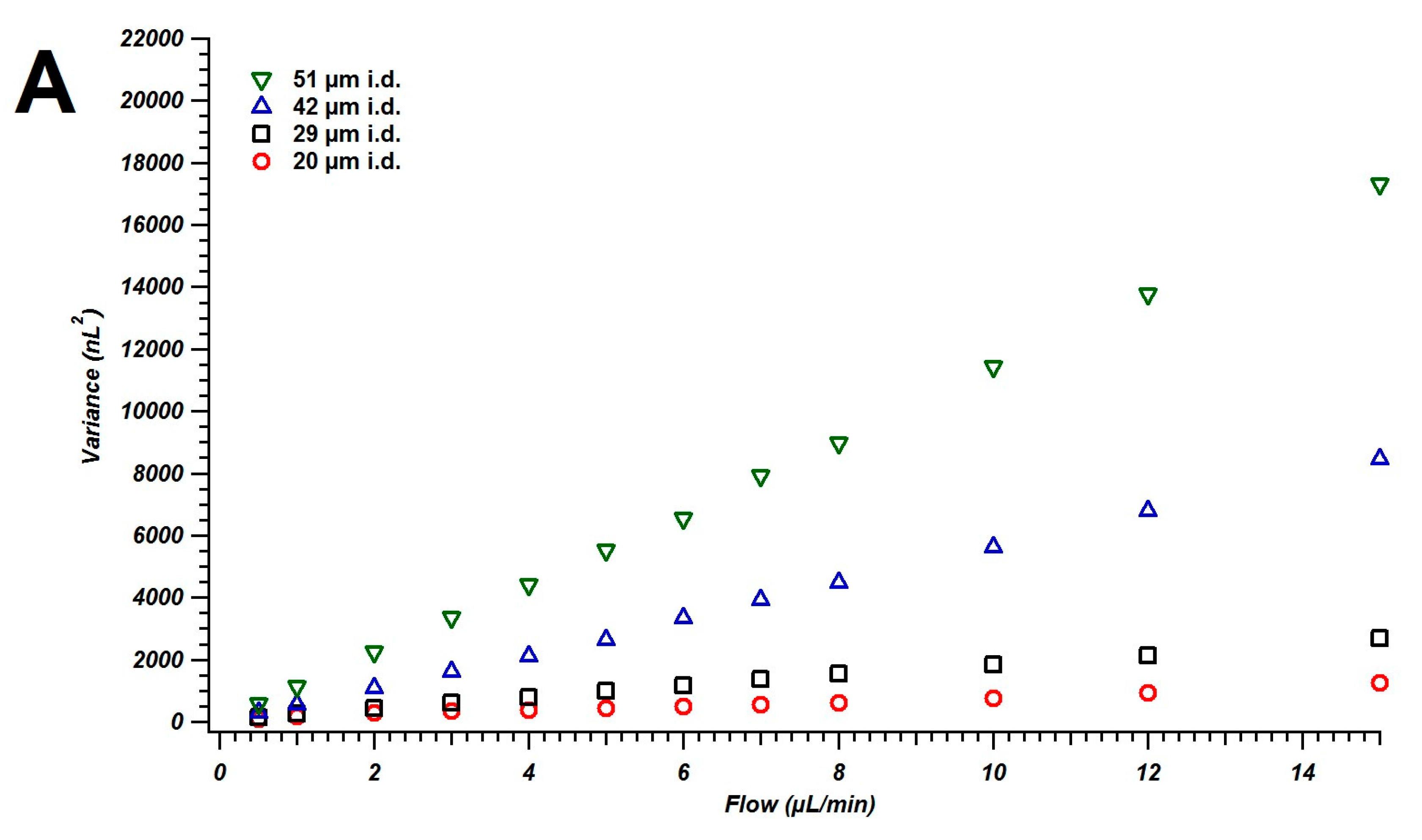
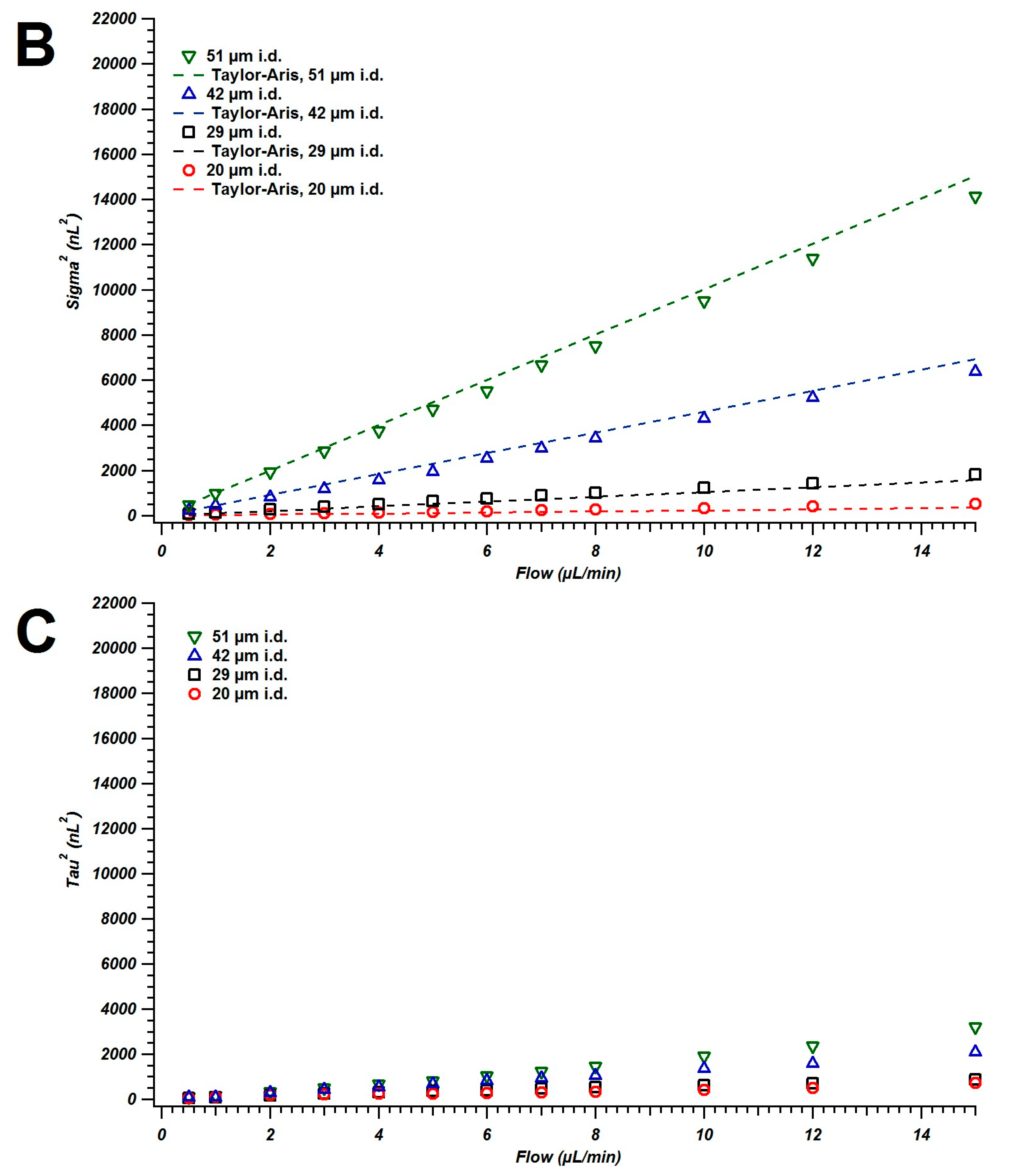
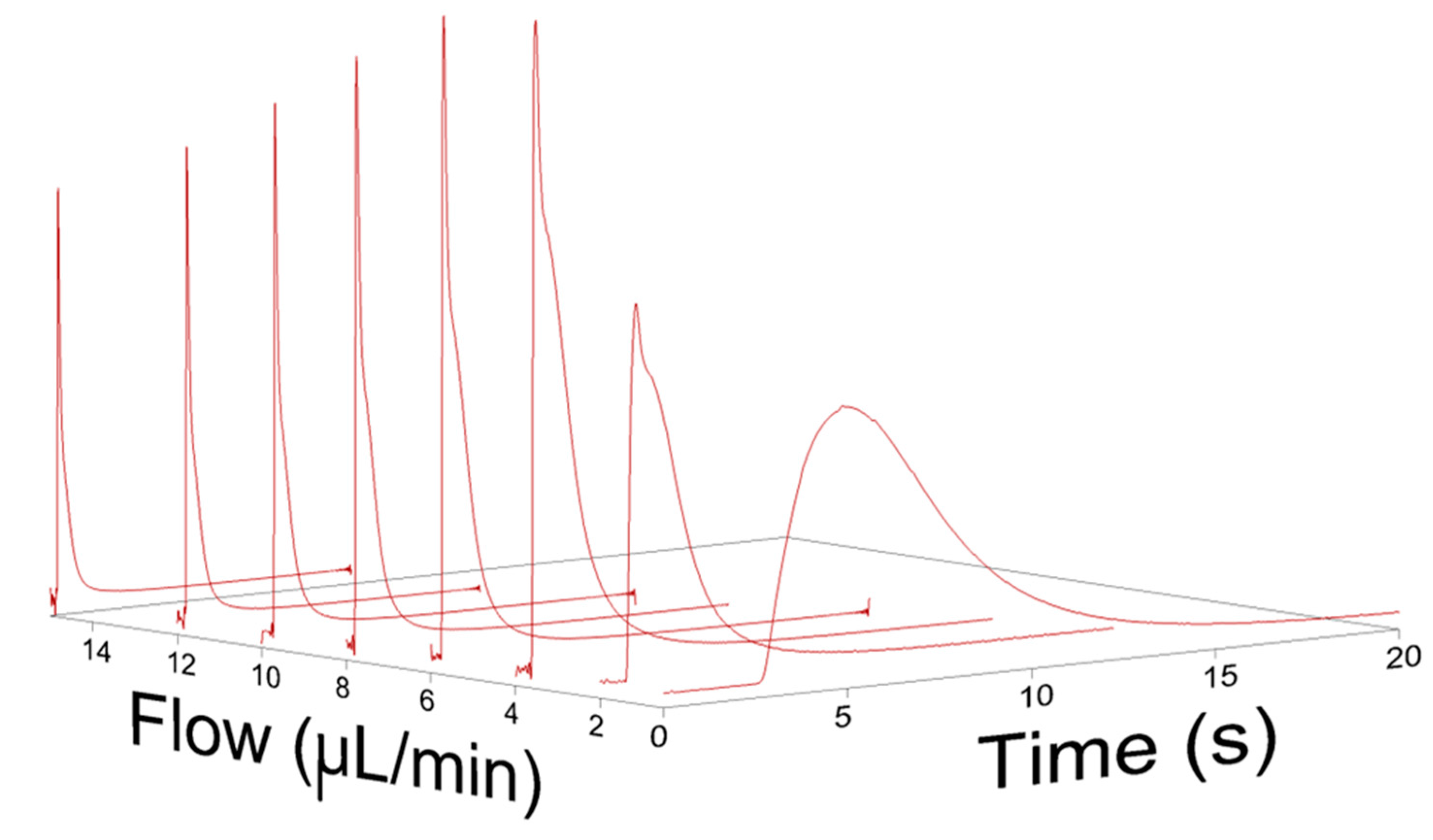
4.1. Broadening in Small Inner Diameter Connecting Capillaries


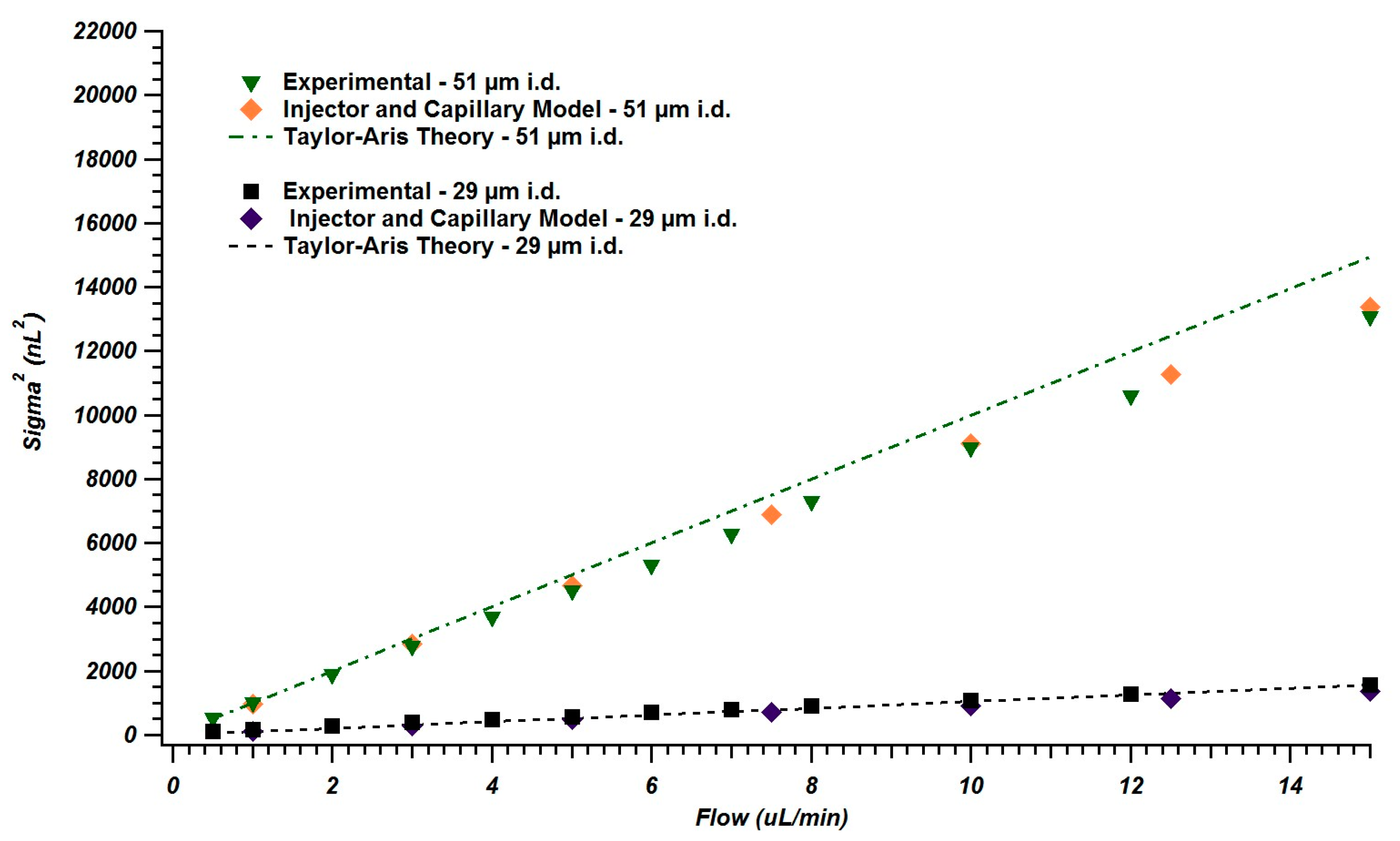
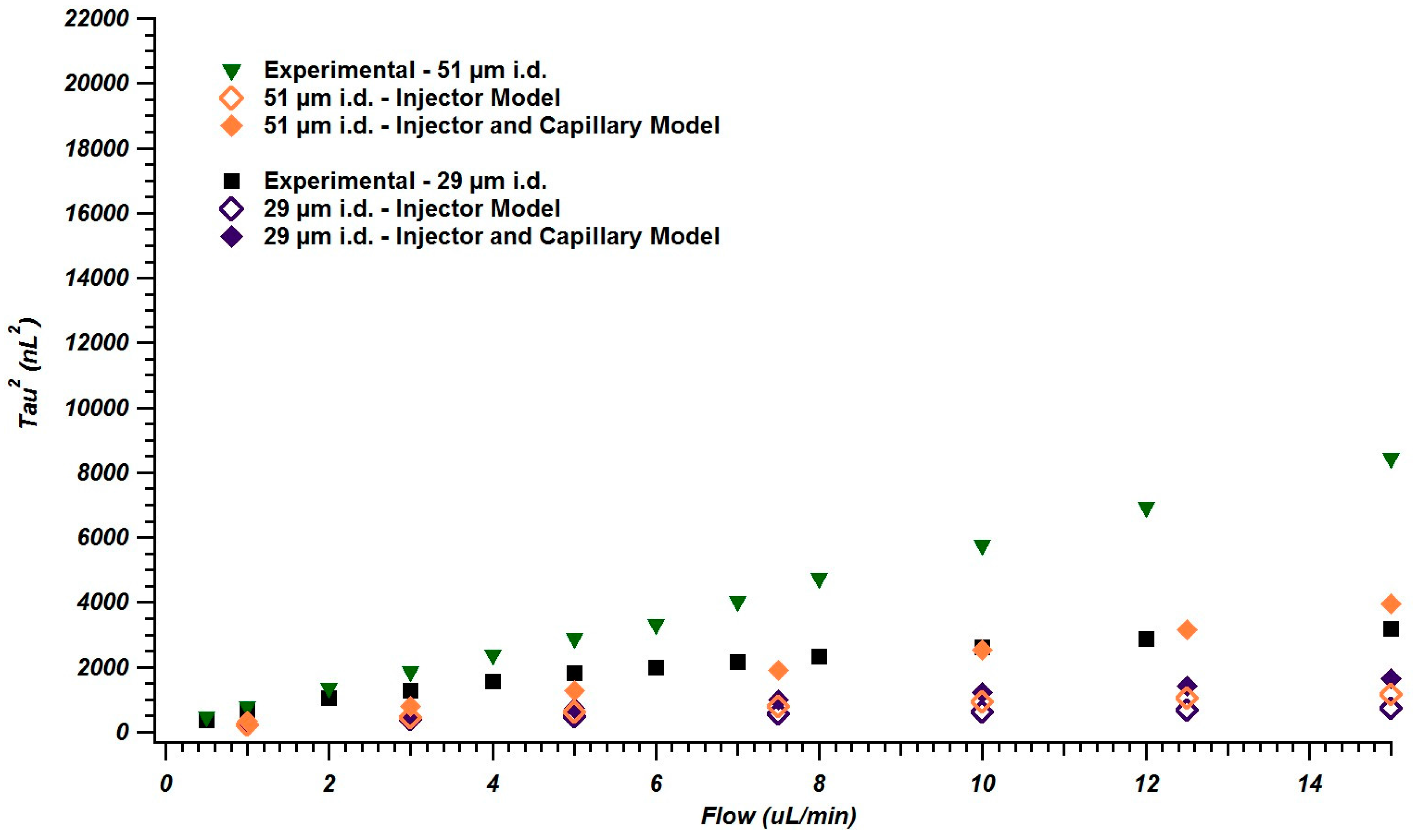
4.2. Simulations of Broadening in Injectors and Connecting Tubing


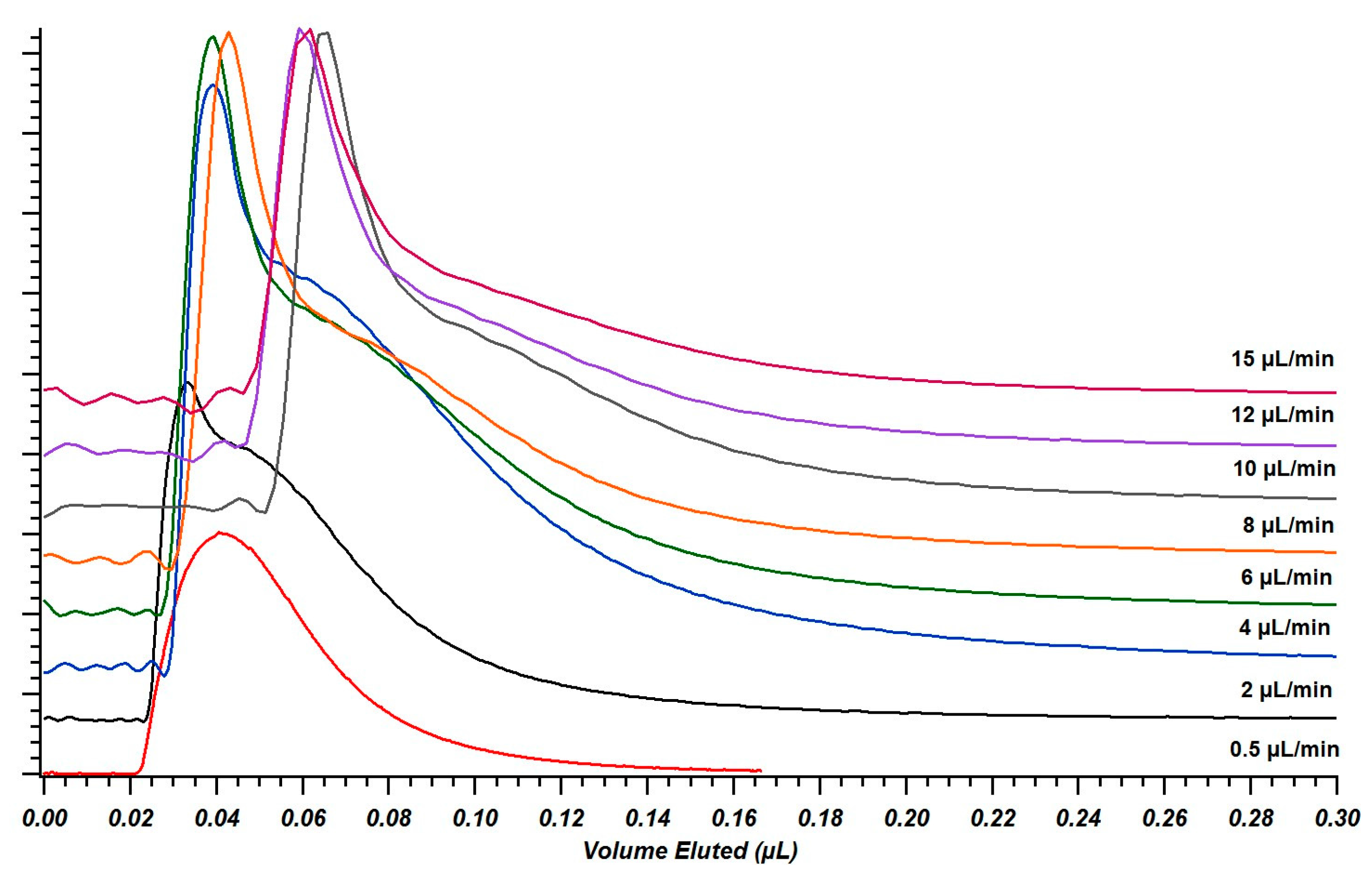
4.3. Reducing Tau-Type Broadening with Pinch Injections


4.4. Injector Elution Profiles


5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gritti, F.; Guiochon, G. On the extra-column band-broadening contributions of modern, very high pressure liquid chromatographs using 2.1 mm I.D. columns packed with sub-2 μm particles. J. Chromatogr. A 2010, 1217, 7677–7689. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Bradley, A.C.; Welch, C.J.; Zhang, L. Effect of extra-column volume on practical chromatographic parameters of sub-2-μm particle-packed columns in ultra-high pressure liquid chromatography. J. Sep. Sci. 2012, 35, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Sanchez, C.A.; Farkas, T.; Guiochon, G. Achieving the full performance of highly efficient columns by optimizing conventional benchmark high-performance liquid chromatography instruments. J. Chromatogr. A 2010, 1217, 3000–3012. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Fekete, J. The impact of extra-column band broadening on the chromatographic efficiency of 5 cm long narrow-bore very efficient columns. J. Chromatogr. A 2011, 1218, 5286–5291. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Leonardis, I.; Shock, D.; Stevenson, P.; Shalliker, A.; Guiochon, G. Performance of columns packed with the new shell particles, Kinetex-C18. J. Chromatogr. A 2010, 1217, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, S.; Desmet, G.; Clicq, D.; Rocca, J.-L. Kinetic plot equations for evaluating the real performance of the combined use of high temperature and ultra-high pressure in liquid chromatography: Application to commercial instruments and 2.1 and 1 mm I.D. columns. J. Chromatogr. A 2008, 1203, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Lestremau, F.; Wu, D.; Szücs, R. Evaluation of 1.0 mm i.d. column performances on ultra high pressure liquid chromatography instrumentation. J. Chromatogr. A 2010, 1217, 4925–4933. [Google Scholar] [CrossRef] [PubMed]
- Omamogho, J.O.; Hanrahan, J.P.; Tobin, J.; Glennon, J.D. Structural variation of solid core and thickness of porous shell of 1.7 μm core-shell silica particles on chromatographic performance: Narrow bore columns. J. Chromatogr. A 2011, 1218, 1942–1953. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Bradley, A.C. Effect of column dimension on observed column efficiency in very high pressure liquid chromatography. J. Chromatogr. A 2012, 1261, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. Rapid development of core-shell column technology: Accurate measurements of the intrinsic column efficiency of narrow-bore columns packed with 4.6 down to 1.3 µm superficially porous particles. J. Chromatogr. A 2014, 1333, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Buckenmaier, S.; Miller, C.A.; van de Goor, T.; Dittmann, M.M. Instrument contributions to resolution and sensitivity in ultra high performance liquid chromatography using small bore columns: Comparison of diode array and triple quadrupole mass spectrometry detection. J. Chromatogr. A 2015, 1377, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Eon, C.; Guiochon, G. Study of the pertinency of pressure in liquid chromatography: II. Problems in equipment design. J. Chromatogr. A 1975, 108, 229–241. [Google Scholar] [CrossRef]
- Reese, C.E.; Scott, R.P.W. Microbore columns—Design, construction, and operation. J. Chromatogr. Sci. 1980, 18, 479–486. [Google Scholar] [CrossRef]
- Fountain, K.J.; Neue, U.D.; Grumbach, E.S.; Diehl, D.M. Effects of extra-column band spreading, liquid chromatography system operating pressure, and column temperature on the performance of sub-2-μm porous particles. J. Chromatogr. A 2009, 1216, 5979–5988. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. On the minimization of the band-broadening contributions of a modern, very high pressure liquid chromatograph. J. Chromatogr. A 2011, 1218, 4632–4648. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.J.; Waeghe, T.J.; Himes, K.W.; Tomasella, F.P.; Hooker, T.F. Modifying conventional high-performance liquid chromatography systems to achieve fast separations with Fused-Core columns: A case study. J. Chromatogr. A 2011, 1218, 5456–5469. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. The current revolution in column technology: How it began, where is it going? J. Chromatogr. A 2012, 1228, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. Accurate measurements of the true column efficiency and of the instrument band broadening contributions in the presence of a chromatographic column. J. Chromatogr. A 2014, 1327, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. Kinetic performance of narrow-bore columns on a micro-system for high performance liquid chromatography. J. Chromatogr. A 2012, 1236, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Prüß, A.; Kempter, C.; Gysler, J.; Jira, T. Extracolumn band broadening in capillary liquid chromatography. J. Chromatogr. A 2003, 1016, 129–141. [Google Scholar] [CrossRef]
- Chervet, J.P.; Ursem, M.; Salzmann, J.P. Instrumental requirements for nanoscale liquid chromatography. Anal. Chem. 1996, 68, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- MacNair, J.E.; Lewis, K.C.; Jorgenson, J.W. Ultrahigh-pressure reversed-phase liquid chromatography in packed capillary columns. Anal. Chem. 1997, 69, 983–989. [Google Scholar] [CrossRef] [PubMed]
- MacNair, J.E.; Patel, K.D.; Jorgenson, J.W. Ultrahigh-pressure reversed-phase capillary liquid chromatography: Isocratic and gradient elution using columns packed with 1.0-micron particles. Anal. Chem. 1999, 71, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.D.; Jerkovich, A.D.; Link, J.C.; Jorgenson, J.W. In-depth characterization of slurry packed capillary columns with 1.0-microm nonporous particles using reversed-phase isocratic ultrahigh-pressure liquid chromatography. Anal. Chem. 2004, 76, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Mellors, J.S.; Jorgenson, J.W. Use of 1.5-μm Porous Ethyl-Bridged Hybrid Particles as a Stationary-Phase Support for Reversed-Phase Ultrahigh-Pressure Liquid Chromatography. Anal. Chem. 2004, 76, 5441–5450. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.A.; Xin, B.; Wu, N.; Lee, M.L. Fast ultrahigh-pressure liquid chromatography: On-column UV and time-of-flight mass spectrometric detection. J. Microcol. Sep. 1999, 11, 631–643. [Google Scholar] [CrossRef]
- Wu, N.; Lippert, J.A.; Lee, M.L. Practical aspects of ultrahigh pressure capillary liquid chromatography. J. Chromatogr. A 2001, 911, 1–12. [Google Scholar] [CrossRef]
- Gritti, F.; Felinger, A.; Guiochon, G. Influence of the errors made in the measurement of the extra-column volume on the accuracies of estimates of the column efficiency and the mass transfer kinetics parameters. J. Chromatogr. A 2006, 1136, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. Mass transfer kinetics, band broadening and column efficiency. J. Chromatogr. A 2012, 1221, 2–40. [Google Scholar] [CrossRef] [PubMed]
- Freebairn, K.W.; Knox, J.H. Dispersion measurements on conventional and miniaturised HPLC systems. Chromatographia 1984, 19, 37–47. [Google Scholar] [CrossRef]
- Gritti, F.; Guiochon, G. Accurate measurements of peak variances: Importance of this accuracy in the determination of the true corrected plate heights of chromatographic columns. J. Chromatogr. A 2011, 1218, 4452–4461. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, M.; Liekens, A.; Desmet, G. Accurate determination of extra-column band broadening using peak summation. J. Sep. Sci. 2012, 35, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; Gao, H.; Gritti, F.; Guiochon, G. Removing the ambiguity of data processing methods: optimizing the location of peak boundaries for accurate moment calculations. J. Sep. Sci. 2013, 36, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; Gritti, F.; Guiochon, G. Automated methods for the location of the boundaries of chromatographic peaks. J. Chromatogr. A 2011, 1218, 8255–8263. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; Guiochon, G. Effect of the pressure on pre-column sample dispersion theory, experiments, and practical consequences. J. Chromatogr. A 2014, 1352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kok, W.T.; Brinkman, U.A.T.; Frei, R.W.; Hanekamp, H.B.; Nooitgedacht, F.; Poppe, H. Use of conventional instrumentation with microbore column in high-performance liquid chromatography. J. Chromatogr. A 1982, 237, 357–369. [Google Scholar] [CrossRef]
- Claessens, H.A.; Cramers, C.A.; Kuyken, M.A.J. Estimation of the band broadening contribution of HPLC equipment to column elution profiles. Chromatographia 1987, 23, 189–194. [Google Scholar] [CrossRef]
- Dezaro, D.A.; Dvorn, D.; Horn, C.; Hartwick, R.A. Time optimization for routine separations, using high-speed microbore HPLC. Chromatographia 1985, 20, 87–96. [Google Scholar] [CrossRef]
- Claessens, H.A.; Hetem, M.J.J.; Leclercq, P.A.; Cramers, C.A. Estimation of the instrumental band broadening contribution to the column elution profiles in open tubular capillary liquid chromatography. J. High Res. Chromatogr. 1988, 11, 176–180. [Google Scholar] [CrossRef]
- Rebscher, H.; Pyell, U. A method for the experimental determination of contributions to band broadening in electrochromatography with packed capillaries. Chromatographia 1994, 38, 737–743. [Google Scholar] [CrossRef]
- Prüß, A.; Kempter, C.; Gysler, J.; Jira, T. Evaluation of packed capillary liquid chromatography columns and comparison with conventional-size columns. J. Chromatogr. A 2004, 1030, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O. On the estimation of extra-column contributions to band broadening through measurements on an authentic chromatogram. J. High Res. Chromatogr. 1979, 2, 605–608. [Google Scholar] [CrossRef]
- Wright, N.A.; Villalanti, D.C.; Burke, M.F. Fourier transform deconvolution of instrument and column band broadening in liquid chromatography. Anal. Chem. 1982, 54, 1735–1738. [Google Scholar] [CrossRef]
- Beisler, A.T.; Schaefer, K.E.; Weber, S.G. Simple method for the quantitative examination of extra column band broadening in microchromatographic systems. J. Chromatogr. A 2003, 986, 247–251. [Google Scholar] [CrossRef]
- Hupe, K.-P.; Jonker, R.J.; Rozing, G. Determination of band-spreading effects in high-performance liquid chromatographic instruments. J. Chromatogr. A 1984, 285, 253–265. [Google Scholar] [CrossRef]
- Reijn, J.M.; Van der Linden, W.E.; Poppe, H. Some theoretical aspects of flow injection analysis. Anal. Chim. Acta 1980, 114, 105–118. [Google Scholar] [CrossRef]
- Reijn, J.M.; Van der Linden, W.E.; Poppe, H. Transport phenomena in flow injection analysis without chemical reaction. Anal. Chim. Acta 1980, 126, 1–13. [Google Scholar] [CrossRef]
- Kristensen, E.W.; Wilson, R.L.; Wightman, R.W. Dispersion in flow injection analysis measured with microvoltammetric electrodes. Anal. Chem. 1986, 58, 986–988. [Google Scholar] [CrossRef]
- Knecht, L.A.; Guthrie, E.J.; Jorgenson, J.W. On-column electrochemical detector with a single graphite fiber electrode for open-tubular liquid chromatography. Anal. Chem. 1984, 56, 479–482. [Google Scholar] [CrossRef]
- Foley, J.P.; Dorsey, J.G. Equations for calculation of chromatographic figures of merit for ideal and skewed peaks. Anal. Chem. 1983, 55, 730–737. [Google Scholar] [CrossRef]
- Foley, J.P.; Dorsey, J.G. A Review of the Exponentially Modified Gaussian (EMG) Function: Evaluation and Subsequent Calculation of Universal Data. J. Chromatogr. Sci. 1984, 22, 40–46. [Google Scholar] [CrossRef]
- Jeansonne, M.S.; Foley, J.P. Review of the Exponentially Modified Gaussian (EMG) Function Since 1983. J. Chromatogr. Sci. 1991, 29, 258–266. [Google Scholar] [CrossRef]
- Foster, M.D.; Arnold, M.A.; Nichols, J.A.; Bakalyar, S.R. Performance of experimental sample injectors for high-performance liquid chromatography microcolumns. J. Chromatogr. A 2000, 869, 231–241. [Google Scholar] [CrossRef]
- Sternberg, J.C. Extracolumn Contributions to Chromatographic Band Broadening. In Advances in Chromatography; Giddings, J., Keller, R.A., Eds.; Marcel Dekker: New York, NY, USA, 1966; Volume 2, pp. 205–270. [Google Scholar]
- Poppe, H. Column liquid chromatography. In Journal of Chromatography Library; Heftmann, E., Ed.; Elsevier: New York, NY, USA, 1992; Volume 51A, pp. A151–A225. [Google Scholar]
- Vissers, J.P.C.; Claessens, H.A.; Cramers, C.A. Microcolumn liquid chromatography: Instrumentation, detection and applications. J. Chromatogr. A 1997, 779, 1–28. [Google Scholar] [CrossRef]
- Vissers, J.P.C. Recent developments in microcolumn liquid chromatography. J. Chromatogr. A 1999, 856, 117–143. [Google Scholar] [CrossRef]
- Colin, H.; Martin, M.; Guiochon, G. Extra-column effects in high-performance liquid chromatography: I. Theoretical study of the injection problem. J. Chromatogr. A 1979, 185, 79–95. [Google Scholar] [CrossRef]
- Schmid, A. Sample injection in liquid chromatography. Chromatographia 1979, 12, 825–829. [Google Scholar] [CrossRef]
- Lauer, H.H.; Rozing, G.P. The selection of optimum conditions in HPLC I. The determination of external band spreading in LC instruments. Chromatographia 1981, 14, 641–647. [Google Scholar] [CrossRef]
- Claessens, H.A.; Burcinova, A.; Cramers, C.A.; Mussche, P.; van Tilburg, C.C.E. Evaluation of injection systems for open tubular liquid chromatography. J. Microcol. Sep. 1990, 2, 132–137. [Google Scholar] [CrossRef]
- Bakalyar, S.R.; Phipps, C.; Spruce, B.; Olsen, K. Choosing sample volume to achieve maximum detection sensitivity and resolution with high-performance liquid chromatography columns of 1.0, 2.1 and 4.6 mm I.D. J. Chromatogr. A 1997, 762, 167–185. [Google Scholar] [CrossRef]
- Stankovich, J.J.; Gritti, F.; Stevenson, P.G.; Guiochon, G. The impact of column connection on band broadening in very high pressure liquid chromatography. J. Sep. Sci. 2013, 36, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Lucy, C.A.; Giavina, L.L.M.; Cantwell, F.F. A Laboratory Experiment on Extracolumn Band Broadening in Liquid Chromatography. J. Chem. Educ. 1995, 72, 367–374. [Google Scholar] [CrossRef]
- Taylor, G. Dispersion of soluble matter in solvent flowing slowly through a tube. P. Roy. Soc. A 1953, 219, 186–203. [Google Scholar] [CrossRef]
- Taylor, G. Conditions under which dispersion of a solute in a stream of solvent can be used to measure molecular diffusion. P. Roy. Soc. A 1954, 225, 473–477. [Google Scholar] [CrossRef]
- Aris, R. On the dispersion of a solute in a fluid flowing through a tube. P. Roy. Soc. A 1956, 235, 67–77. [Google Scholar] [CrossRef]
- Shankar, A.; Lenhoff, A.M. Dispersion in round tubes and its implications for extracolumn dispersion. J. Chromatogr. A 1991, 556, 235–248. [Google Scholar] [CrossRef]
- Liu, G.; Svenson, L.; Djordjevic, N.; Erni, F. Extra-column band broadening in high-temperature open-tubular liquid chromatography. J. Chromatogr. A 1993, 633, 25–30. [Google Scholar] [CrossRef]
- Golay, M.J.E.; Atwood, J.G. Early phases of the dispersion of a sample injected in poiseuille flow. J. Chromatogr. A 1979, 186, 353–370. [Google Scholar] [CrossRef]
- Atwood, J.G.; Golay, M.J.E. Dispersion of peaks by short straight open tubes in liquid chromatography systems. J. Chromatogr. A 1981, 218, 97–122. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. Dispersion in laminar tube flow at low Peclet numbers or short times. AIChE J. 1988, 34, 1423–1430. [Google Scholar] [CrossRef]
- Broeckhoven, K.; Desmet, G. Numerical and analytical solutions for the column length-dependent band broadening originating from axisymmetrical trans-column velocity gradients. J. Chromatogr. A 2009, 1216, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.P.W.; Kucera, P. Mode of operation and performance characteristics of microbore columns for use in liquid chromatography. J. Chromatogr. A 1979, 169, 51–72. [Google Scholar] [CrossRef]
- Kahle, V.; Krejčí, M. Role of extra-column volume in the efficiency of high-speed liquid chromatography. J. Chromatogr. A 1985, 321, 69–79. [Google Scholar] [CrossRef]
- Brooks, H.B.; Thrall, C.; Tehrani, J. High-performance liquid chromatography system for packed capillary columns. J. Chromatogr. A 1987, 385, 55–64. [Google Scholar] [CrossRef]
- Chervet, J.P.; Ursem, M.; Salzmann, J.P.; Vanoort, R.W. Ultra-sensitive UV detection in micro separation. J. High Res. Chromatogr. 1989, 12, 278–281. [Google Scholar] [CrossRef]
- Kamahori, M.; Watanabe, Y.; Miura, J.; Taki, M.; Miyagi, H. High-sensitivity micro ultraviolet absorption detector for high-performance liquid chromatography. J. Chromatogr. A 1989, 465, 227–232. [Google Scholar] [CrossRef]
- Bruin, G.J.M.; Stegeman, G.; van Asten, A.C.; Xu, X.; Kraak, J.C.; Poppe, H. Optimization and evaluation of the performance of arrangements for UV detection in high-resolution separations using fused-silica capillaries. J. Chromatogr. A 1991, 559, 163–181. [Google Scholar] [CrossRef]
- Yang, F.J. On-column detection using a fused silica column. J. High Res. Chromatogr. 1981, 4, 83–85. [Google Scholar] [CrossRef]
- Walbroehl, Y.; Jorgenson, J.W. On-column UV absorption detector for open tubular capillary zone electrophoresis. J. Chromatogr. A 1984, 315, 135–143. [Google Scholar] [CrossRef]
- Kientz, C.; Verweij, A. Construction of a UV cell for micro-liquid chromatography. J. High Res. Chromatogr. 1988, 11, 294–296. [Google Scholar] [CrossRef]
- Vindevogel, J.; Schuddinck, G.; Dewaele, C.; Verzele, M. Simple instrument modification for packed fused silica capillary or micro-LC: Evaluation on the use of tubular UV-detection cells. J. High Res. Chromatogr. 1988, 11, 317–321. [Google Scholar] [CrossRef]
- Kucera, P.; Umagat, H. Design of a post-column fluorescence derivatization system for use with microbore columns. J. Chromatogr. A 1983, 255, 563–579. [Google Scholar] [CrossRef]
- Guthrie, E.J.; Jorgenson, J.W. On-column fluorescence detector for open-tubular capillary liquid chromatography. Anal. Chem. 1984, 56, 483–486. [Google Scholar] [CrossRef]
- Rose, D.J., Jr.; Jorgenson, J.W. Post-capillary fluorescence detection in capillary zone electrophoresis using o-phthaldialdehyde. J. Chromatogr. A 1988, 447, 117–131. [Google Scholar] [CrossRef]
- Nickerson, B.; Jorgenson, J.W. Characterization of a post-column reaction—laser-induced fluorescence detector for capillary zone electrophoresis. J. Chromatogr. A 1989, 480, 157–168. [Google Scholar] [CrossRef]
- Manz, A.; Simon, W. Picoliter Cell Volume Potentiometric Detector for Open-Tubular Column LC. J. Chromatogr. Sci. 1983, 21, 326–330. [Google Scholar] [CrossRef]
- White, J.G.; St. Claire, R.L., III; Jorgenson, J.W. Scanning on-column voltammetric detector for open-tubular liquid chromatography. Anal. Chem. 1986, 58, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Manz, A.; Simon, W. Potentiometric detector for fast high-performance open-tubular column liquid chromatography. Anal. Chem. 1987, 59, 74–79. [Google Scholar] [CrossRef]
- Baur, J.E.; Kristensen, E.W.; Wightman, R.W. Radial dispersion from commercial high-performance liquid chromatography columns investigated with microvoltammetric electrodes. Anal. Chem. 1988, 60, 2334–2338. [Google Scholar] [CrossRef] [PubMed]
- Felinger, A.; Kilár, A.; Boros, B. The myth of data acquisition rate. Anal. Chim. Acta 2015, 854, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Grushka, E.; Myers, M.N.; Schettler, P.D.; Giddings, J.C. Computer characterization of chromatographic peaks by plate height and higher central moments. Anal. Chem. 1969, 41, 889–892. [Google Scholar] [CrossRef]
- Hsieh, S.; Jorgenson, J.W. Preparation and Evaluation of Slurry-Packed Liquid Chromatography Microcolumns with Inner Diameters from 12 to 33 μm. Anal. Chem. 1996, 68, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, T.J.; Thompson, J.W.; Mellors, J.S.; Jorgenson, J.W. Capillary-Based Instrument for the Simultaneous Measurement of Solution Viscosity and Solute Diffusion Coefficient at Pressures up to 2000 bar and Implications for Ultrahigh Pressure Liquid Chromatography. Anal. Chem. 2009, 81, 2860–2868. [Google Scholar] [CrossRef] [PubMed]
- Noga, M.; Sucharski, F.; Suder, P.; Silberring, J. A practical guide to nano-LC troubleshooting. J. Sep. Sci. 2007, 30, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, J.; Edström, L.; Forssén, P.; Fornstedt, T. Injection profiles in liquid chromatography. I. A fundamental investigation. J. Chromatogr. A 2010, 1217, 4306–4312. [Google Scholar] [CrossRef] [PubMed]
- Gritti, F.; McDonald, T.; Gilar, M. Accurate measurement of dispersion data through short and narrow tubes used in very high-pressure liquid chromatography. J. Chromatogr. A 2015, 1410, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; McDonald, T.S.; Johnson, J.S.; Murphy, J.P.; Jorgenson, J.W. Wide injection zone compression in gradient reversed-phase liquid chromatography. J. Chromatogr. A 2015, 1390, 86–94. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinias, J.P.; Bunner, B.; Gilar, M.; Jorgenson, J.W. Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography. Chromatography 2015, 2, 669-690. https://doi.org/10.3390/chromatography2040669
Grinias JP, Bunner B, Gilar M, Jorgenson JW. Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography. Chromatography. 2015; 2(4):669-690. https://doi.org/10.3390/chromatography2040669
Chicago/Turabian StyleGrinias, James P., Bernard Bunner, Martin Gilar, and James W. Jorgenson. 2015. "Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography" Chromatography 2, no. 4: 669-690. https://doi.org/10.3390/chromatography2040669